
Abstract
The proto-oncogene, BCL-2, has been suggested to participate in cell survival during development of, and after injury to, the CNS. Transgenic (TG) mice overexpressing human Bcl-2 (n = 21) and their wild-type (WT) littermates (n = 18) were subjected to lateral controlled cortical impact brain injury. Lateral controlled cortical impact brain injury resulted in the formation of a contusion in the injured cortex at 2 days, which developed into a well-defined cavity by 7 days in both WT and TG mice. At 7 days after injury, brain-injured TG mice had a significantly reduced cortical lesion (volume = 1.99 mm3) compared with that of the injured WT mice (volume = 5.1 mm3, P <0.01). In contrast, overexpression of BCL-2 did not affect the extent of hippocampal cell death after lateral controlled cortical impact brain injury. Analysis of motor function revealed that both brain-injured WT and TG mice exhibited significant right-sided deficits at 2 and 7 days after injury (P < 0.05 compared with the uninjured controls). Although composite neuroscores (sum of scores from forelimb and hind limb flexion, lateral pulsion, and inclined plane tests) were not different between WT and TG brain-injured mice, TG mice had a slightly but significantly reduced deficit in the inclined plane test (P < 0.05 compared to the WT mice). These data suggest that the cell death regulatory gene, BCL-2, may play a protective role in the pathophysiology of traumatic brain injury.
Long-term cognitive and neurologic motor dysfunction have been recognized as the most debilitating consequence of traumatic brain injury (TBI) in humans (Kurth et al., 1994; Klein et al., 1996). Although the anatomic substrates for posttraumatic behavioral dysfunction have yet to be defined, neuronal loss, primarily in the cortex, hippocampus, and thalamus, have been observed in the traumatically injured human brain (Adams et al., 1985; Kotapka et al., 1992; Ross et al., 1993). Animal models of TBI, such as fluid-percussion (FP) and controlled cortical impact (CCI), result in deficits in spatial learning, memory (retention), and neurologic motor function in the rat and mouse (McIntosh et al., 1989; Lyeth et al., 1990; Smith et al., 1991, 1995; Dixon et al., 1991; Hamm et al., 1992). The temporal and spatial patterns of neuronal loss after experimental brain injury have been extensively described (Sutton et al., 1993; Dietrich et al., 1994; Hicks et al., 1996; Colicos et al., 1996). Neuronal degeneration appears to be initiated acutely (minutes to hours) after experimental TBI, with injured (acidophilic) neurons exhibiting vacuolated cytoplasm, swollen mitochondria, and a general pyknotic appearance indicative of necrosis (Sutton et al., 1993; Dietrich et al., 1994). In addition, intensely silver-stained neurons were observed in the injured hemisphere as early as 10 minutes and up to 24 hours after either lateral FP or CCI brain injury (Hicks et al., 1996; Colicos et al., 1996). Morphologic evaluation of these dystrophic, argyrophilic neurons revealed condensed chromatin and cytoplasmic bodies suggestive of apoptosis (Colicos et al., 1996). Furthermore, previous studies have used in situ nick end-labeling, DNA agarose gel electrophoresis, and electron microscopy to demonstrate that apoptotic cell death is a part of the pathology of experimental TBI (Rink et al., 1995; Colicos and Dash, 1996; Conti et al., 1998).
The proto-oncogene BCL-2 has been suggested to participate in cell survival during development of the CNS (Martinou et al., 1994; Farlie et al., 1995; Greenlund et al., 1995). Multiple members of the BCL-2 family have been isolated and characterized and have been reported to either promote cell survival or cell death (Davies, 1995). Thus, although BCL-2, BCL-X
Regional alterations in immunoreactivity (IR) of BCL-2 and its homologs have been observed after injury to the nervous system, suggesting that these proteins may participate in the cellular response to injury. Increased BCL-2 IR was observed in endothelial cells and neurons surrounding the infarcted zone after focal ischemia (Chen et al., 1995) and trauma (Clark et al., 1997). Increased expression of BCL-2 has also been reported in spinal cord axons after trauma (Li et al., 1996) and in glial cells surrounding retinal neurons after axotomy (Grosche et al., 1995). Recently, we observed acute decreases in cellular IR of Bcl-2 in neurons that appear to be destined to die after lateral FP brain injury (Raghupathi and McIntosh, 1995). Collectively, the above data suggest that BCL-2 (and its homologs) may have a potential neuroprotective role in trauma-induced pathology. To further elucidate the role of BCL-2 in the pathophysiology of TBI, we evaluated the neurobehavioral and histologic response to lateral CCI brain injury in TG mice overexpressing the human BCL-2 ygene.
METHODS
Generation of Bcl-2 transgenic mice and verification of transgene expression
A 1.9-kilobase (kb) EcoR1 fragment containing the human BCL-2 coding sequence was ligated into a modified pcDNA1 plasmid (Invitrogen, San Diego, CA, U.S.A.) downstream of the 4.5-kb rat synapsin I promoter fragment (Howland et al., 1991; Hoesche et al., 1993) and the 127-bp SV40t intron splice (Fig. 1A). To prepare DNA for microinjection, the transgenic plasmid was digested with Sal I and Nco I, and the 6.6-kb insert was recovered from a 1% agarose gel using Gene-Clean Reagent (Bio-101, Vista, CA, U.S.A.), further purified over Qiagen (Qiagen, Santa Clarita, CA, U.S.A.) resin to remove residual impurities, precipitated with isopropanol, and resuspended in Tris-EDTA buffer to a concentration of 2 ng/µL. Transgenic mice were generated by pronuclear injection of fertilized C57B6/SJL ova (DNX Technologies, Princeton, NJ, U.S.A.) using standard procedures (Hogan et al., 1982). G3 transgenic and their wild-type (WT) littermates (base strain: C57SJL) derived from this line have been used for the experiments described in this report.
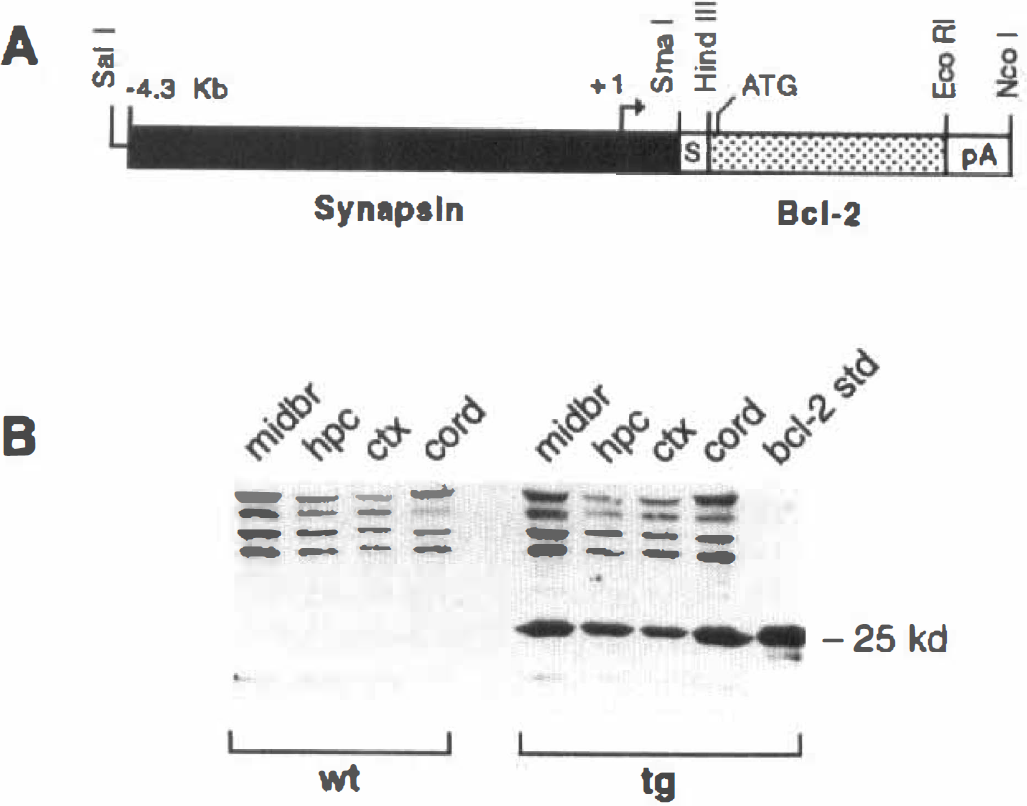
Structure and expression of the Syn-Bcl-2 transgene.
To evaluate human BCL-2 transgene expression in TG mice, brain regions (midbrain, hippocampus, and cortex) and spinal cord were dissected and homogenized in 2× GIPA buffer (0.1 mol/L Tris-HCl, pH 8.0, 0.3 mol/L NaCl, 0.2% Triton-X-100, 0.02% sodium dodecylsulfate, 4 mmol/L EDTA, 2 mmol/L benzamidine) and centrifuged at 100,000g for 60 minutes. Clarified supernatants were normalized for protein levels and subjected to SDS-PAGE followed by electroblotting onto nitrocellulose membranes (Bio-Rad, Richmond, CA, U.S.A.). Blots were probed with the human Bcl-2–specific monoclonal antibody (6C8, PharMingen, San Diego, CA, U.S.A.; Hockenberry et al., 1990) diluted 1: 100 in Blotto. Filters were washed and probed with biotinylated goat anti-hamster secondary antibody followed by treatment with peroxidase-conjugated streptavidin (Caltag, San Francisco, CA, U.S.A.). Bands were visualized by detection with an enhanced chemiluminescence kit (Amersham, Arlington Heights, IL, U.S.A.) and compared to recombinant BCL-2 standards. In other TG mice, the synapsin I promoter has been reported to direct expression of transgene(s) only in neurons of the mouse brain (Hoesche et al., 1993; Howland et al., 1995).
Controlled cortical impact injury
All animal use procedures were in accordance with the NIH Guide for the Care and Use of Laboratory Animals (Pub. No. 85-23, 1985) and were approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Mice (25 to 40 g, n = 18 WT and 21 TG) were anesthetized with sodium pentobarbital (60 mg/kg, intraperitoneally) and placed in a stereotactic frame. The vertex of the skull was exposed and a plastic disc (diameter, 4 mm) was attached to the center of the left parietal bone to guide the trephine on the rounded mouse skull. After a circular groove was made with the trephine, the bone flap was removed gently with a spatula and curette, resulting in a 5-mm craniotomy. Forty-five minutes after administration of anesthesia, mice (n = 12 WT and 15 TG) were subjected to CCI brain injury using a pneumatic impactor previously described (Dixon et al., 1991) and modified for use in the mouse (Smith et al., 1995). The impactor tip had a diameter of 3 mm and a beveled edge. The rod was angled at 20° from the vertical such that the tip was perpendicular to the exposed dural surface. Injury was produced using a velocity of 4.9 to 5.1 m·s−1 with the impactor penetrating to a depth of 1.2 mm; the duration of the impact was kept constant at 100 msec. After injury, cranioplasty was performed over the craniotomy site and the scalp was sutured shut, and mice were allowed to recover on a heating pad maintained at 37°C. In the present study, all mice survived brain injury. Uninjured (sham-injured) control mice (n = 6 WT and 6 TG) were anesthetized and surgically prepared but received no impact.
Study protocol
All mice were trained in the rotorod task for two days before injury and their preinjury (baseline) neurologic motor function was also evaluated. Twenty-four hours after the last rotorod training trial, TG mice and their WT littermates were anesthetized, surgically prepared, and randomly assigned to receive injury or designated as sham-injured animals. Motor function (including rotorod) was tested in all animals at 2 days after injury. After testing, 6 brain-injured WT mice and 8 brain-injured TG mice were sacrificed for histologic evaluation of the brains. The remaining brain-injured mice (6 WT and 7 TG) and all sham-injured mice were tested again at 7 days for motor function and sacrificed for histologic analysis.
Motor function
Postinjury neuromotor function was evaluated using a battery of tests by a trained observed blinded to the genotype and injury status of the animal. The tests were adapted from those previously described for the rat (McIntosh et al., 1989; Hamm et al., 1994) and are described below.
Neuromotor function. In initial experiments, it was observed that mice subjected to lateral CCI over the left temporoparietal cortex exhibited a hemiparetic deficit on the right side. Therefore in the present study, mice were evaluated for right-sided deficits on a scale of 0 (severely injured) to 4 (preinjury control status) for the following reflex tasks: (1) forelimb contraflexion response during suspension by the tail (maximum score = 4), (2) hind limb flexion response when the hind limbs are lifted from the surface (maximum score = 4), and (3) decreased resistance to lateral pulsion (maximum score = 4). In addition, the ability of mice to stand on all four limbs on an inclined plane (angle board task) for at least 5 seconds in each of four directions—face up and vertical, face down and vertical, face right and lateral, and face left and lateral—was determined. The maximum angle at which each mouse was able to stand was determined before (baseline) and after brain injury. Angle board scores for each mouse were calculated based on a change from baseline in the maximum angle that the mouse stood, where 4 = 0° change from baseline, 3 = 2.5° change from baseline, 2 = 5° change from baseline, 1 = 7.5° change from baseline, and 0 = 10° or greater change from baseline. Each of the four directions were scored separately and these scores were averaged, reflecting a maximum score of 4. A composite neurologic score was created by adding the scores of all individual tests (maximum score = 16).
Rotorod. The rotorod test described by Hamm et al. (1994) was modified for use in mice. The time (latency) each animal spent on a rotating rod was recorded when the animal either fell off the rod or gripped the rod and spun around the lowest point. Two separate tests were performed: in the “fast” paradigm, the rotational velocity of the rotorod increased 1.75 rpm per second; in the “slow” paradigm, the velocity increased 0.5 rpm per second. Animals were tested four times in each of the two paradigms with 1-minute intervals between trials. The highest and lowest latencies were discarded and an average of the two remaining latencies for each paradigm was used as the rotorod score. Mice were acclimated on the device before injury and tested at 2 and 7 days after injury immediately after the evaluation of the neuromotor function.
Histologic analysis
After analysis of motor function, brain-injured mice were killed by transcardial perfusion with 4% paraformaldehyde either at 2 days or at 7 days after injury. Uninjured mice were killed at 1 week after injury. Brains were removed, fixed for an additional 24 hours by immersion, processed, and embedded in paraffin.
Lesion volume. For analysis of lesion volume, 6-µm coronal sections, taken every 0.5 mm between 0.2 mm and 4.5 mm posterior to bregma (Franklin and Paxinos, 1996), were stained with hematoxylin and eosin (H&E). The area of the lesion in each section included the cavity and surrounding tissue that was characterized by minimal staining with H&E and was measured directly using an image analysis system (MCID, Ontario, Canada). The total volume of the cortical lesion was calculated by integrating the area obtained from each section with the distance between each level. Brains from 5 WT and 6 TG injured mice and 5 WT and 5 TG injured mice were evaluated, respectively, at 2 and 7 days after injury.
Hippocampal neuronal damage. Sections (6 µm) from bregma −3.3 mm (Franklin and Paxinos, 1996) of brain-injured mice (6 WT and 7 TG) and 6 uninjured mice (3 WT and 3 TG) at 2 and 7 days after injury or surgery were stained with toluidine blue. Three sections, each 36 to 42 µm apart from each other, were examined using conventional light microscopy by one of us (SCF) who was blinded to the injury status and genotype of the mice. Neuronal loss in the CA3 region of the hippocampus was scored on a scale where 0 = contiguous cell loss in greater than 50% of the length of the CA3, 1 = patchy cell loss throughout the length of the CA3, and 2 = similar to control (sham) brains. A similar scale was used in the hilus, where 0 = no neurons visible, 1 = a few (1–10) neurons were seen, and 2 = similar to control brains. The score (mean ± SD and median) obtained for each brain represented the average score of the three sections. Because the extent of hippocampal cell loss observed at 2 days after injury in both WT and TG mice appeared to be similar to that observed at 7 days, only the evaluation from the 7-day tissue is discussed.
Statistical analysis
Because neurologic motor function scores (composite and individual tests) are nonparametric data, scores were analyzed by a Kruskal-Wallis analysis of all groups followed by a Mann-Whitney comparison between individual groups. Because multiple comparisons were made using the Mann-Whitney test, the stringency (to account for a potential alpha error) of the P values was verified by performing an analysis of variance (ANOVA) followed by a post-hoc t test with a Bonferroni correction. Scores from the individual tests were also compared by an ANOVA followed by the post-hoc Bonferroni t test. Latencies recorded from the rotorod paradigm were compared by an ANOVA followed by the Bonferroni t test. Lesion volume comparisons were performed using the unpaired Student's t test between WT and TG groups. A P value of less than 0.05 was considered significant in all analyses. Statistical outliers were calculated using the formulas described by Grubbs and Beck (1972), and outlier values that fell beyond two standard deviations from the mean, considered too “large” or “small” for normal data (Grubbs and Beck, 1972), were excluded in the final statistical analysis. Only one outlier was detected in the inclined plane test at 2 days after injury and was excluded from the analysis. However, it must be mentioned that the P values did not change appreciably when this outlier value was included in the statistical analysis.
RESULTS
Expression of human BCL-2 in CNS of transgenic mice
Seven founder animals were identified by Southern blot and polymerase chain reaction analysis (data not shown). G1 offspring from these mice were analyzed for RNA expression and one line, line 52, was found to exhibit relatively high levels of human BCL-2 mRNA (2.5-fold over the next highest expressing line). Western blot analysis revealed almost equivalent BCL-2 protein expression in cortex, hippocampus, midbrain, and spinal cord of TG mice (Fig. 1B).
Cortical lesion
By 2 days after injury, a contused area was evident in the injured cortex below the site of impact, which developed into a well-defined cavity by 7 days (Fig. 2). The brain parenchyma immediately surrounding the cavity exhibited pale staining (with H&E) and was characterized by a lack of neurons (data not shown). In brain-injured WT mice the contusion occupied a mean volume of 2.9 mm3 at 2 days after injury, and increased in size to 5.1 mm3 by 7 days after injury (fig. 3A, P = 0.01 between the two time points). At 2 days after injury, brain-injured TG mice exhibited a smaller cortical lesion (1.39 mm3) compared to their WT counterparts (P = 0.06, Fig. 3). However, by 7 days after injury, the cortical lesion in brain-injured mice overexpressing Bcl-2 (1.99 mm3) was observed to be significantly smaller than that observed in WT mice (P = 0.007 compared to brain-injured WT mice, Figs. 2 and 3A). In contrast to the WT mice, the cortical lesion in TG mice at 7 days was not significantly different from that observed at 2 days after injury, suggesting that Bc1-2 may also inhibit the progression of cortical damage. The contusion areas in all coronal sections examined at 7 days after injury were smaller in brain-injured TG mice compared to the WT mice (Fig. 3B).
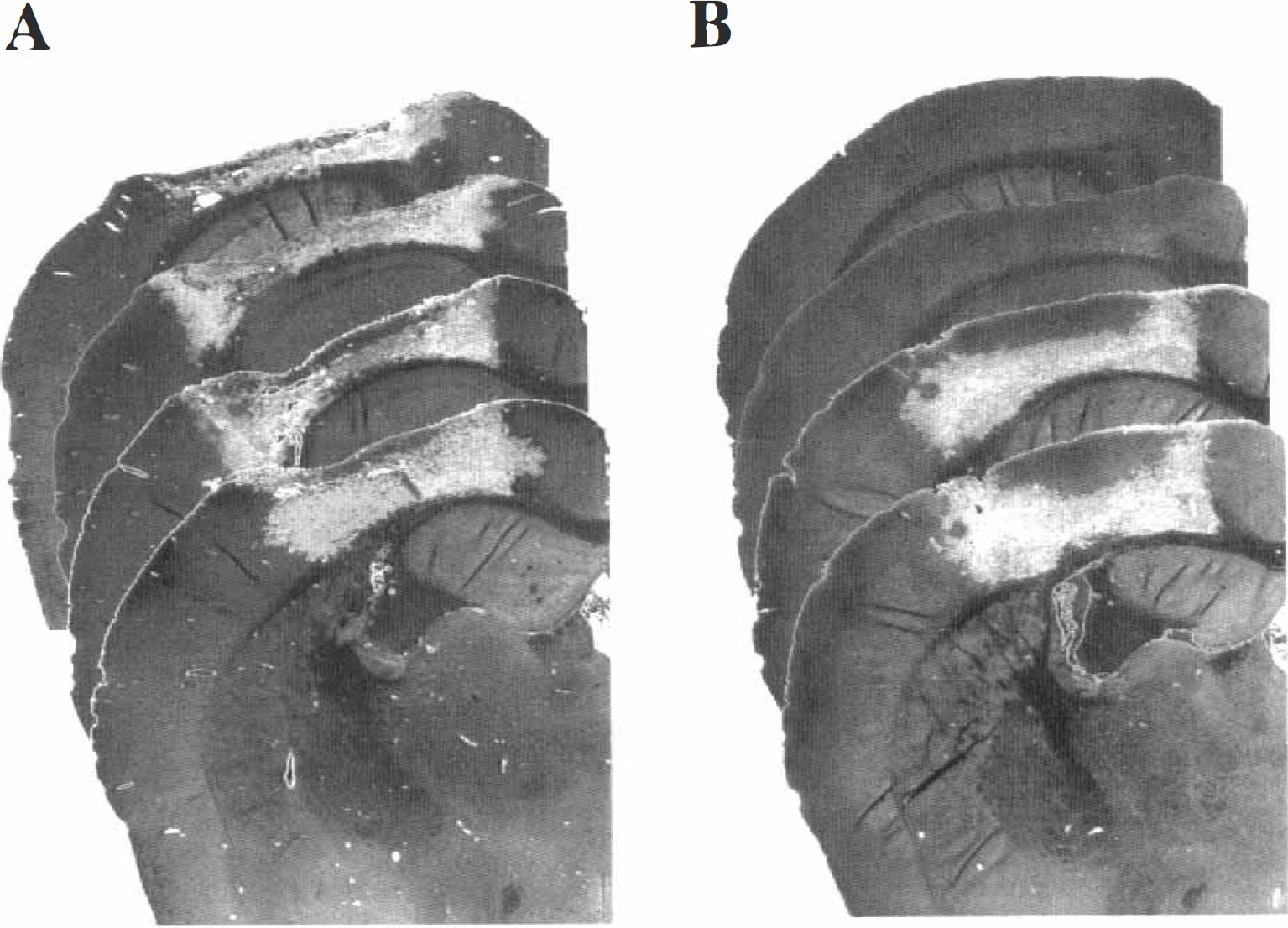
Contused cortex in
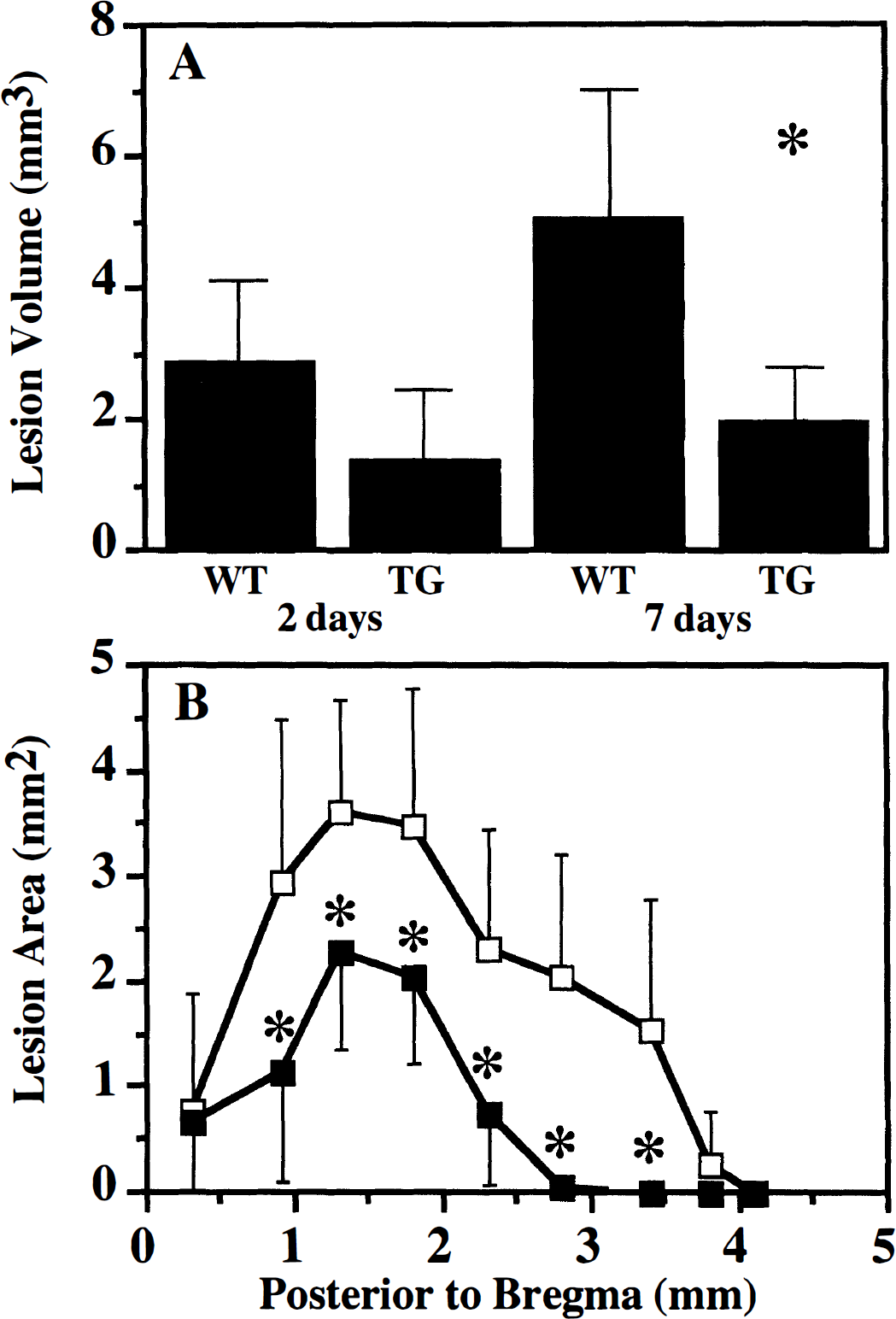
Quantitation of cortical lesion in WT and TG mice after lateral CCl brain injury.
Hippocampal neuronal loss
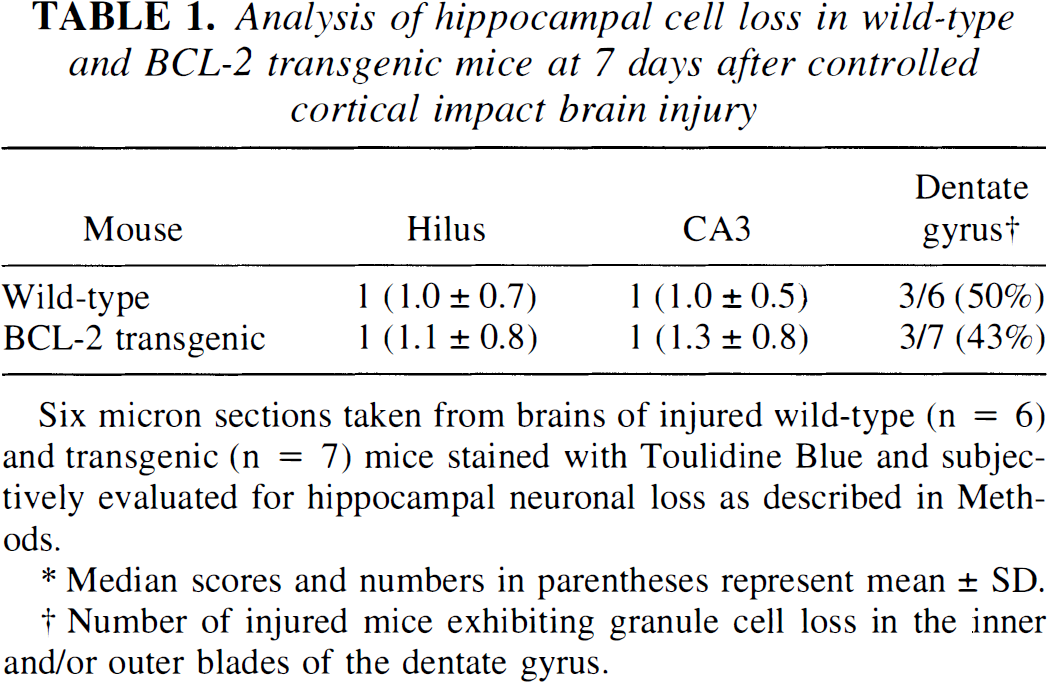
Lateral CCI brain injury in rodents is characterized by selective neuronal loss in area CA3 and the hilus of the hippocampus ipsilateral to the impact. In the mouse, neuronal loss in these regions of the hippocampus was observed at 2 days (data not shown) and 7 days (CA3, Fig. 4A; hilus, Fig. 4C) after lateral CCI brain injury. Overexpression of BCL-2 did not appear to affect injury-induced hippocampal neuronal death in either area CA3 (Fig. 4B and Table 1) or the dentate hilus (Fig. 4D and Table 1). In addition, in about 50% of the injured animals, occasional cell loss was observed in the outer blade of the dentate gyrus, which was also unaffected by overexpression of BCL-2 (Table 1).
Analysis of hippocampal cell loss in wild-type and BCL-2 transgenic mice at 7 days after controlled cortical impact brain injury
Six micron sections taken from brains of injured wild-type (n = 6) and transgenic (n = 7) mice stained with Toulidine Blue and subjectively evaluated for hippocampal neuronal loss as described in Methods.
Median scores and numbers in parentheses represent mean ± SD.
Number of injured mice exhibiting granule cell loss in the inner and/or outer blades of the dentate gyrus.
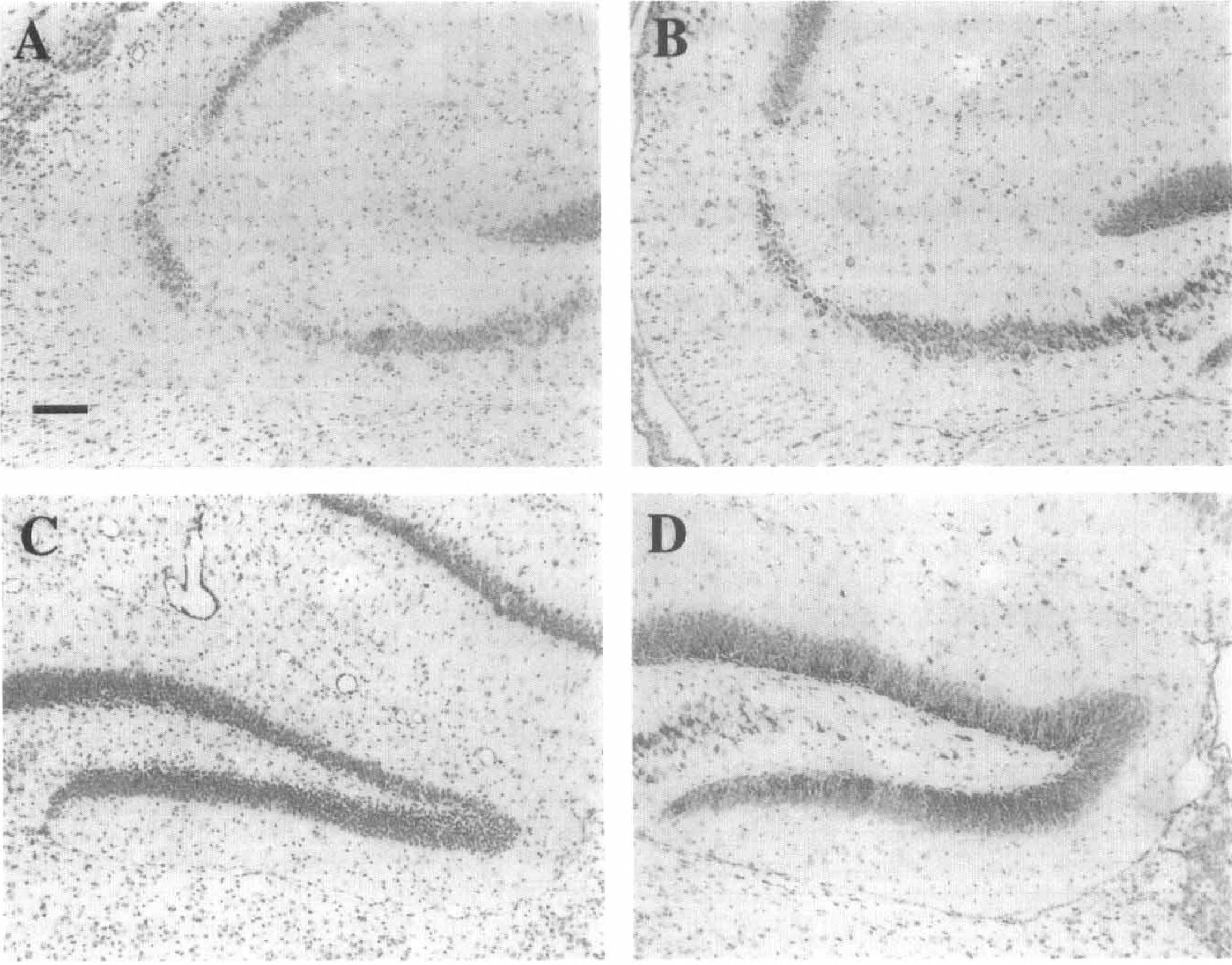
Hippocampal neurodegeneration after lateral CCI brain injury. Representative toluidine blue—stained sections illustrating neuronal loss in the hippocampus taken at 3.3 mm posterior to bregma of injured mice at 7 days after lateral CCI brain injury.
Neuromotor function
At 2 days, a significant deficit in composite neuromotor function was observed in both WT and TG brain-injured mice compared to sham-injured controls (Fig. 5A, P = 0.004, ANOVA; P = 0.013, Bonferroni corrected t test for injured WT or TG mice compared to sham-injured mice). Brain-injured mice exhibited a significant right-sided hemiparesis, with median composite neuroscores for right-sided tasks of 10.4 (out of a possible 16) compared to 15.4 for the left-sided tasks (data not shown). Overexpression of Bcl-2 appeared to have no effect on injury-induced motor dysfunction, because the composite neuroscore (right-sided tasks) for brain-injured WT and TG mice were not significantly different (10.4 and 11.5, respectively) (Fig. 5A). This lack of a transgene effect was also observed when the scores from three of the four individual tests that comprise the composite neuroscore (Fig. 6B—D) were compared. However, the performance of TG mice on the angle board task was slightly, but significantly, improved (median score = 3.75) when compared to injured WT mice (median score = 3.25, P = 0.034, Bonferroni correction for three comparisons) (Fig. 6A).
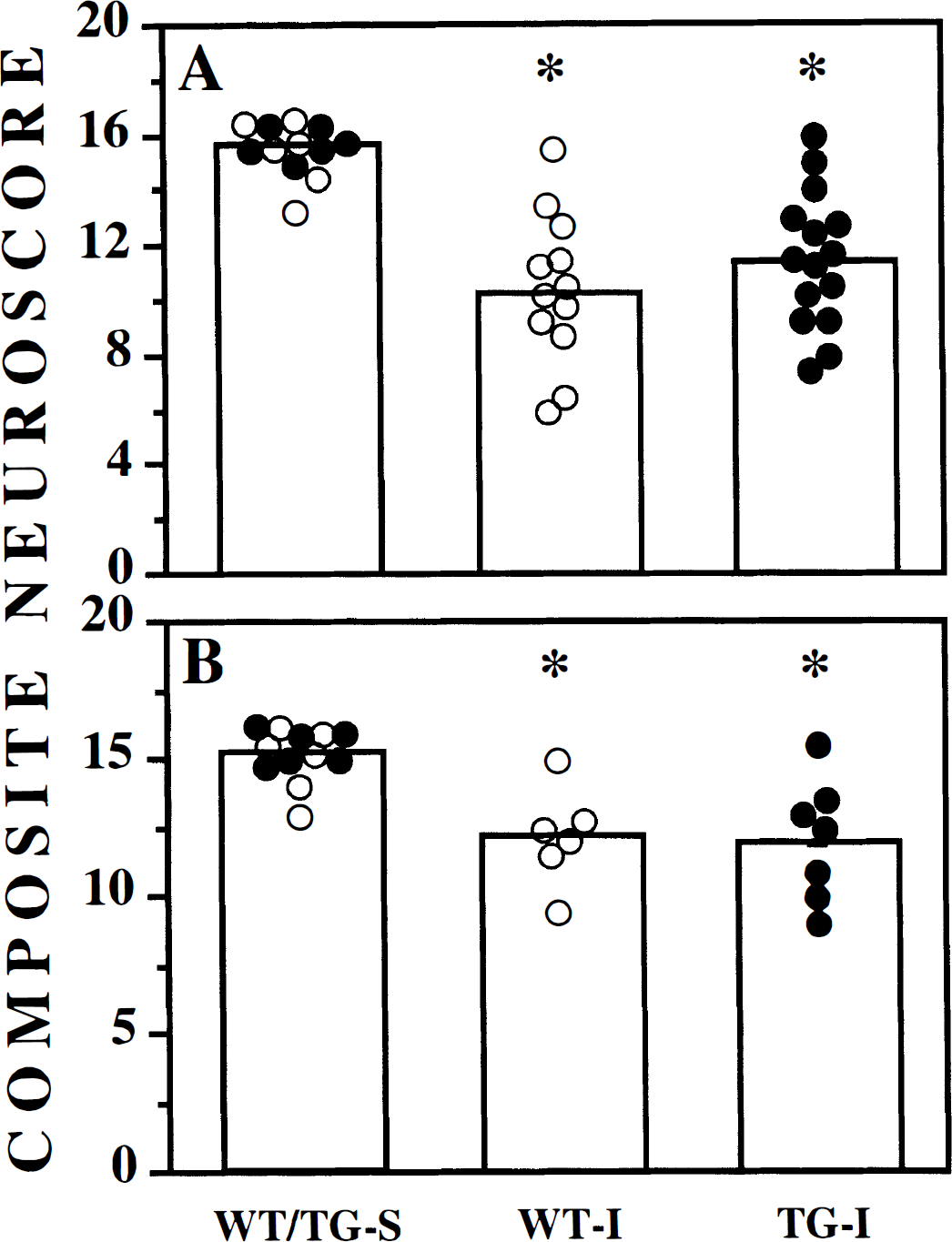
Neurologic motor deficits in wild-type (WT, open circles) and transgenic (TG, filled circles) mice after lateral CCl brain injury. Injured wild-type (WT-I) and transgenic (TG-I) mice were evaluated for composite neuromotor function as described in Methods at
The neuromotor deficits persisted up to at least 1 week after injury in all injured animals. As illustrated in Fig. 5B, both WT and TG mice subjected to lateral CCI brain injury had significantly lower composite neuroscores at 7 days after injury compared to the sham-injured group (P = 0.01, ANOVA; P = 0.023, Bonferroni corrected t test for injured WT or TG mice compared to sham-injured mice). However, injured TG mice were not significantly different compared to injured WT mice (median composite scores of 12.25 and 11.63, respectively) (Fig. 5B). Similarly, both brain-injured WT and TG mice remained significantly impaired compared to uninjured controls in three of the four individual tests (Figs. 7B—D). Only WT mice were significantly impaired in the inclined plane test compared to sham-injured controls, although the median score for injured TG mice was not significantly different from injured WT mice (Fig. 7A).
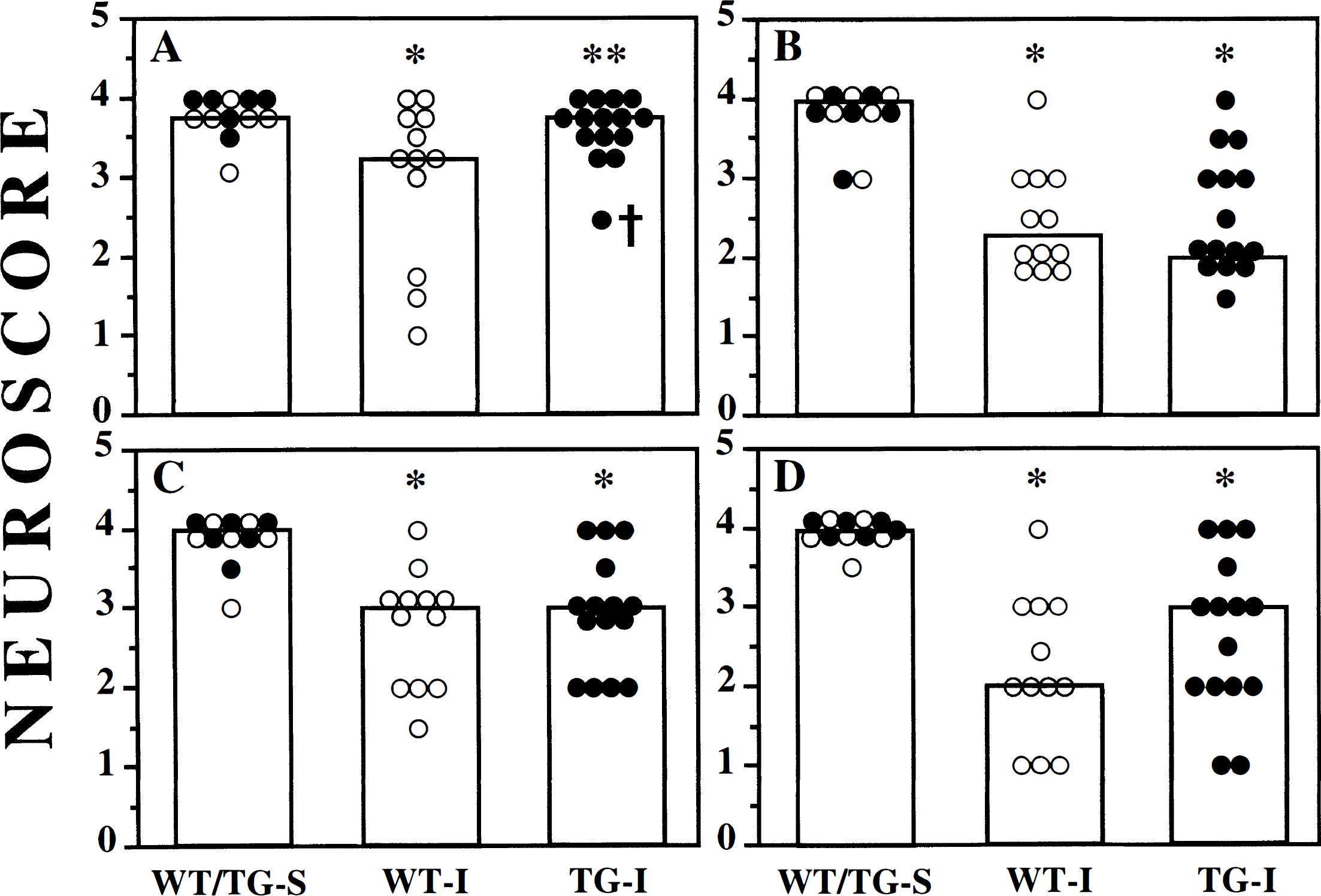
Performance in individual tests of motor function in WT (open circles) and TG (filled circles) mice at 2 days after lateral CCI brain injury. Injured wild-type (WT-I) and transgenic (TG-I) mice were tested in the
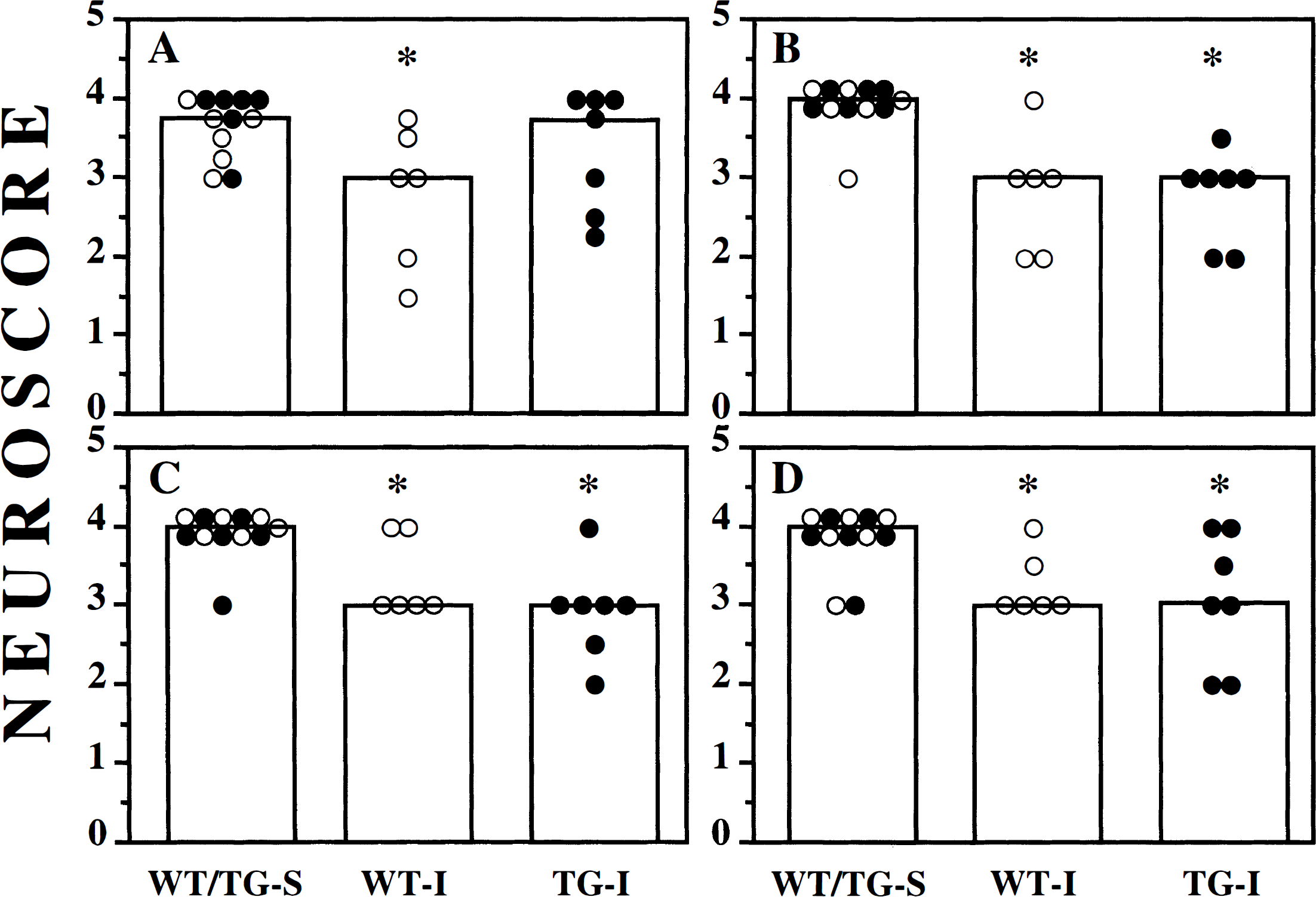
Performance in individual tests of motor function in WT (open circles) and TG (filled circles) mice at 7 days after lateral CCl brain injury. Injured wild-type (WT-I) and transgenic (TG-I) mice were tested in the
Rotorod performance
At 2 days after injury, brain-injured WT and TG mice showed significantly decreased latencies on the rotorod compared to their respective sham-injured controls (P = 0.003, Fig. 8). Latencies in the “fast” (Fig. 8A) and “slow” (Fig. 8B) paradigms were 35% to 50% shorter in brain-injured mice compared to the control mice. No significant differences were observed between brain-injured WT and TG mice in either the “fast” or “slow” paradigms. By 1 week after injury, performance of brain-injured mice on the rotorod was similar to uninjured control mice (data not shown).
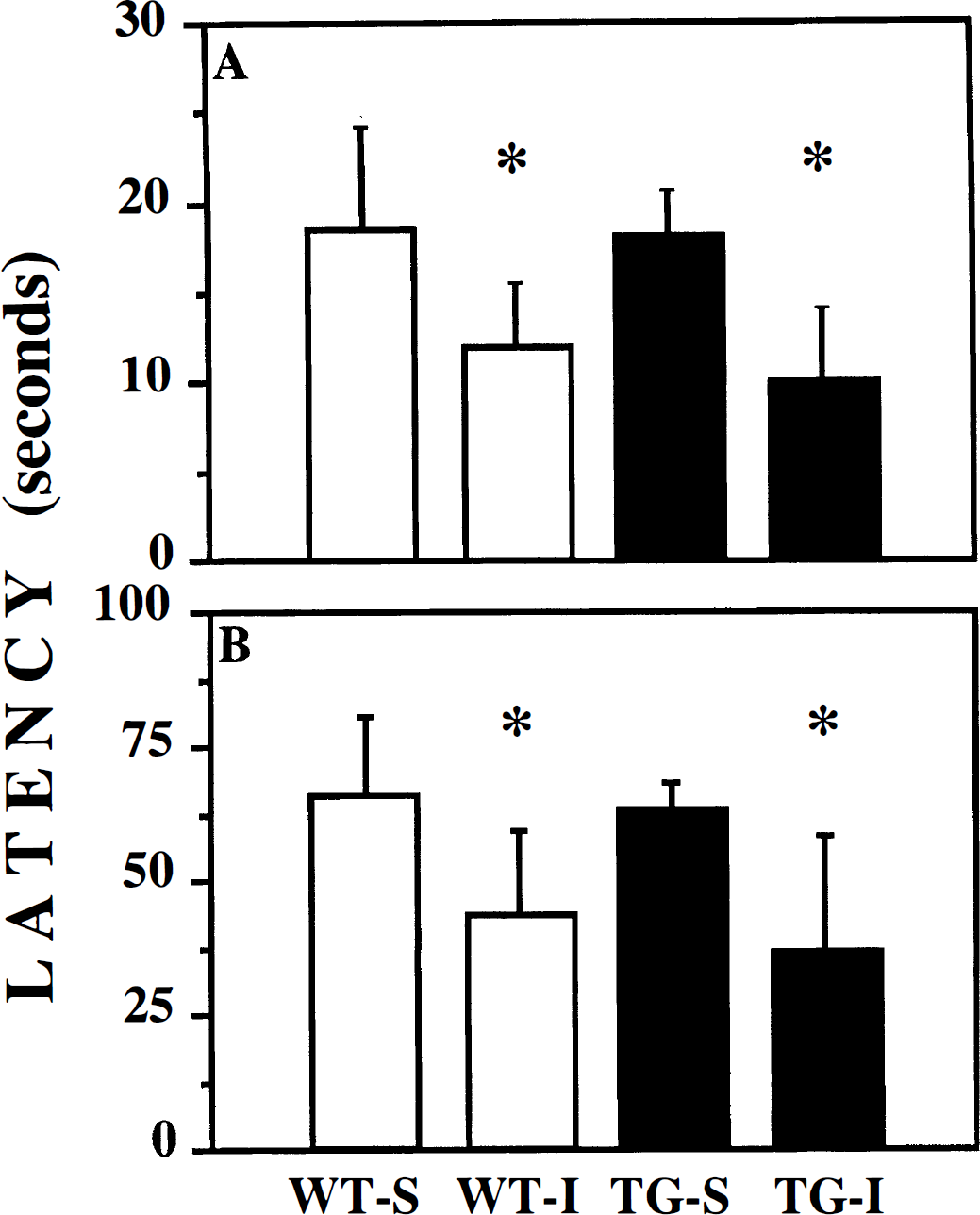
Rotorod performance at 2 days after lateral CCl brain injury in WT (open bars) and TG (filled bars) mice. Injured WT (WT-I) and TG (TG-I) mice were tested in the rotorod task in the
DISCUSSION
In the present study, mice overexpressing the anti-cell death protein, BCL-2, exhibited significantly attenuated cell death in the motor and somatosensory regions of the cortex by 1 week after CCI brain injury. In contrast, the extent of trauma-induced hippocampal cell death did not appear to be affected in brain-injured TG mice. Although significant neuroprotection was observed in regions implicated, in part, as the anatomic substrates for severe posttraumatic neurologic motor deficits (Soblosky et al., 1996), TG mice were as impaired as their WT counterparts in three of four tests of motor function and exhibited an improvement in the ability to stand on an angle board (inclined plane test) only at 2 days after injury.
It has been suggested that the neuronal degeneration after experimental brain injury may be mediated by a combination of primary mechanisms (shearing forces) and activation of secondary mechanisms, such as excitotoxicity, increase in intracellular calcium, and release of free radicals (McIntosh, 1994; Gennarelli, 1997). In addition, axonal injury caused by the initial traumatic injury leading to secondary axotomy has been proposed to mediate neuronal degeneration (Povlishock et al., 1992). A progressive increase in the size of the cortical lesion was observed in brain-injured WT mice during the first 7 days after injury, with no apparent increase observed in cortical lesion volume in brain-injured TG mice during the same period, suggesting that BCL-2 may prevent or delay the manifestation or progression of post-traumatic secondary cell death cascade. Axotomy of sciatic, optic, or facial nerves results in degeneration of the cell bodies, which is prevented in mice overexpressing BCL-2 (Martinou et al., 1994; Farlie et al., 1995; de BilBao and Dubois-Dauphin, 1996). These observations suggest that BCL-2 may function in a similar manner to prevent the ongoing neuronal degeneration after TBI. However, the significant attenuation of neuronal cell loss in the cortex of injured TG mice was not reflected in an improvement in their motor function, which was no different from that of injured WT mice. This apparent discrepancy may be explained by the fact that other regions of the brain, such as the basal ganglia, thalamus, and cerebellum (not evaluated in the present study), may contribute to the motor function deficits in brain-injured mice.
In contrast to the cortex, overexpression of BCL-2 had no effect on posttraumatic hippocampal cell loss. A unique feature of TBI in both man and animals (also observed in this study) is the selective neuronal cell loss in area CA3 and the hilus of the dentate gyrus in the hippocampus (Kotapka et al., 1992; Hicks et al., 1996; Lowenstein et al., 1992; Dietrich et al., 1994; Baldwin et al., 1997). Unlike models of ischemia in which hippocampal neuronal death is delayed, posttraumatic neurodegeneration in the hippocampus appears to be rapidly activated (within hours after injury) and appears to be maximal by 24 hours (Hicks et al., 1996; Baldwin et al., 1997). It is tempting to speculate that the combination of primary and secondary mechanisms may potentially overwhelm the protective action(s) of BCL-2.
Increased expression of BCL-2 appears to regulate neuronal apoptosis during development (Davies, 1995; Greenlund et al., 1995, Martinou et al., 1994; Farlie et al., 1995). In addition, accumulating evidence points to a significant role for BCL-2 and its family of cell death-regulating proteins in CNS injury and neurodegenerative diseases (Bredesen, 1995). Increased BCL-2 IR has been observed in postmortem tissue from patients with Alzheimer's disease (Su et al., 1996) or amyotrophic lateral sclerosis (Troost et al., 1995). An increase in BCL-2 IR was observed at 24 hours in cortical neurons and endothelial cells surrounding the infarct after focal ischemia (Chen et al., 1995) and in neurons that surround the cortical contusion at 72 hours after experimental CCI brain injury (Clark et al., 1997). Conversely, decreased expression of Bcl-2 protein has been observed in selectively vulnerable neuronal populations in the acute period (2 hours) after ischemia (Gillardon et al., 1996) or lateral FP brain injury in the rat (Raghupathi and McIntosh, 1995), suggesting that neuronal survival may be associated with their ability to maintain homeostatic levels of BCL-2. Experimental brain injury results in both necrotic and apoptotic forms of cell death (Dietrich et al., 1994; Rink et al., 1995; Hicks et al., 1996; Colicos and Dash, 1996; Conti et al., 1998). Although BCL-2 has been reported to function primarily as an antiapoptotic protein, evidence suggests that BCL-2 can also prevent necrotic cell death (Kane et al., 1995). Although not specifically evaluated, the attenuation of cortical cell death in brain-injured TG mice observed in the present study may, therefore, be reflective of a general neuroprotective effect of BCL-2 from both necrotic an apoptotic mechanisms.
Direct evidence for a neuroprotective role for BCL-2 in CNS injury comes from reports in which increased expression of BCL-2 in vitro or in vivo appears to protect cells from a variety of insults. In vitro, neural cells overexpressing BCL-2 appear to be more resistant to hypoxia-induced apoptotic cell death (Jacobson and Raff, 1995) and cell death induced by free radicals (Kane et al., 1993), calcium ionophores, glutamate, and lipid peroxidation (Zhong et al., 1993; Kane et al., 1995). Overexpression of BCL-2 in vivo has been reported to prevent neuronal cell loss after ischemia (Martinou et al., 1994; Linnik et al., 1995; Lawrence et al., 1996). In TG mice overexpressing human BCL-2 under control of the neuron-specific enolase promoter, the volume of the cortical infarct after focal ischemia was markedly reduced (Martinou et al., 1994). Preischemic infection of BCL-2 in neurons using herpes simplex virus has been reported to reduce neuronal death after the ischemic insult (Linnik et al., 1995; Lawrence et al., 1996). In addition, neurotoxin-induced neuronal death in the hippocampus was ameliorated by BCL-2 overexpression (Lawrence et al., 1996). Recent evidence suggests that increased cellular BCL-2 levels after CNS injury can prevent excitotoxic cell death in vivo (Jia et al., 1996) and ischemic cell death in vivo (Lawrence et al., 1997).
The specific cellular mechanism by which BCL-2 prevents cell death is unclear, and a number of potentially different pathways have been suggested. By virtue of its intracellular localization in the mitochondrial membrane, BCL-2 has been suggested to improve the ability of the mitochondria to buffer increases in intracellular calcium (Murphy et al., 1996). Increases in intracellular calcium after experimental brain injury have been described (Fineman et al., 1993) and have been suggested to participate in the process of posttraumatic neuronal damage (Young, 1992). BCL-2 has been reported to prevent oxidative damage, which has been implicated in the pathology of TBI (Kane et al., 1993; Chan et al., 1995). BCL-2 has also been suggested to regulate cell death by forming dimers with, and thereby neutralizing the effect of, the proapoptotic protein, BAX (Reed, 1994). We have recently reported that both BAX mRNA and protein were upregulated in the hippocampus after experimental brain injury (Strauss et al., 1996). BCL-2 and its homologs have been postulated to act upstream of and thereby inhibit interleukin-1 β–converting enzyme (caspase-1) and its homologs such as caspase-3 and caspase-2 (Reed 1994); the activation of caspase-3 after experimental brain injury has been reported (Yakovlev et al., 1997) suggesting that BCL-2 may exert its protective function in TBI via the inhibition of such proteases.
In this report, we provide direct evidence of a neuroprotective role for an endogenous protein involved in the regulation of cell death. It must be noted that BCL-2 is not the only protein that participates in the regulation of cell death, nor is it universally neuroprotective: BCL-2 overexpression does not protect cells from ciliary neuronotropic factor withdrawal—induced death (Allsopp et al., 1995). Our observations that overexpression of BCL-2 did not effectively attenuate TBI-induced behavioral deficits or hippocampal cell death, and the recent report of histologic and behavioral protection in brain-injured Cu, Zn-superoxide dismutase TG mice (Mikawa et al., 1996), suggest that multiple cellular pathways may be simultaneously associated with the pathophysiologic sequelae of experimental brain injury.
Footnotes
Acknowledgments
The authors thank Dr. Jesse Berlin from the Department of Biostatistics and Epidemiology, University of Pennsylvania for help with the statistical analyses performed in this study, and Ms. Laura Meehan for careful preparation of the manuscript.