
Abstract
Mitochondrial cytochrome c translocation to the cytosol initiates the mitochondrial-dependent apoptotic pathway. This event has not been previously reported in traumatic brain injury (TBI). The authors determined the expression of cytochrome c in cytosolic and mitochondrial fractions after severe TBI produced by the controlled cortical impact model in the mouse. One hour after trauma there was an increase in cytosolic cytochrome c immunoreactivity. The increases in cytosolic cytochrome c preceded DNA fragmentation, which started at 4 hours. Western blots of mitochondrial and cytosolic fractions confirmed that there was a translocation of cytochrome c from the mitochondria after TBI. Mice deficient in manganese superoxide dismutase (MnSOD) showed an increased loss of mitochondrial cytochrome c after trauma, but less apoptotic cell death 4 and 24 hours after injury compared with wild-type control mice. However, the overall cell death was increased in MnSOD mice, as illustrated by a larger cortical lesion in these animals. The results show that cytochrome c is released from the mitochondria after severe TBI partly by a free radical–dependent mechanism, and that massive mitochondrial cytochrome c release is a predictor of necrotic cell death rather than apoptosis.
Apoptotic cell death has been demonstrated after experimental traumatic brain injury (TBI) in several studies (Rink et al., 1995; Clark et al., 1997; Skoglösa et al., 1999; Lewén et al., 2001), including electron microscopy observations (Colicos and Dash, 1996). Two major apoptotic pathways have been identified in mammalian cells, the Fas/TNF-R receptor pathway and the mitochondrial pathway. The exact mechanisms by which TBI initiates different apoptotic pathways are not well known. In general, the release of cytochrome c from the mitochondrial inner membrane into the cytosol initiates the mitochondrial-dependent apoptosis pathway (Liu et al., 1996; Yang and Cortopassi, 1998). In the cytosol, cytochrome c binds to apoptosis activating factor-1, caspase-9, and deoxyadenosine triphosphate (Li et al., 1997b), leading to a subsequent activation of caspase-3, followed by cleavage of a substrate, such as poly(ADP-ribose) polymerase, and activation of endonucleases, finally resulting in DNA degradation (Enari et al., 1998). Therefore, studies of mechanisms initiating mitochondrial cytochrome c release in cellular injury are important for understanding apoptosis after acute brain injury. Increases in cytosolic cytochrome c have been demonstrated in cerebral hypoxia (Araya et al., 1998), transient focal cerebral ischemia (Fujimura et al., 1998), global cerebral ischemia (Sugawara et al., 1999), cerebral cold injury (Morita-Fujimura et al., 1999), and in a model of subarachnoid hemorrhage (Matz et al., 2001). Mitochondrial release of cytochrome c has been described to occur in axons after TBI (Buki et al., 2000). In the current study, the authors sought to determine if cytochrome c is released from mitochondria within the cytoplasm of neuronal somata after experimental TBI and if this event precedes the downstream effects of caspase-3 activation such as DNA fragmentation. In addition, the authors wanted to test if such release is modulated by levels of mitochondrial oxidative stress. Release of mitochondrial cytochrome c after trauma was found and this release preceded the appearance of terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL)-positive cells. Mice deficient in manganese superoxide dismutase (MnSOD, SOD2) showed increased signs of cytochrome c release together with indirect evidence of increased levels of mitochondrial superoxide after trauma and an overall larger cortical lesion. However, assaying cytoplasmic DNA fragmentation as an early sign of apoptotic cell death showed that there was less apoptotic cell death in SOD2-deficient mice. The discrepancy among cytochrome c release, overall cell death, and apoptosis in SOD2-deficient mice is likely because of an insult that produces a more severe injury in these animals, leading to mitochondrial failure and adenosine triphosphate (ATP) depletion, which shifts the injury from apoptosis to necrosis.
MATERIALS AND METHODS
Animals and production of trauma
All animals were treated in accordance with Stanford University guidelines and an animal care protocol approved by Stanford's Administrative Panel on Laboratory Animal Care. Ninety-one male mice weighing 35 to 42 g, with free access to food and water, were used as follows: for immunohistochemistry at 1, 8, and 24 hours (n = 3 each time point; sham, n = 2 at 8 hours); Western blots at 1, 4, and 8 hours (each time point, n = 3; sham, n = 3 at 4 hours); and for DNA laddering at 24 hours (sham, n = 1; TBI, n = 3), CD1 mice were used (Charles River Laboratories, Wilmington, MA, U.S.A.). Furthermore, SOD2 heterozygous knockout (KO) mutants or their wild-type (WT) littermates were used for Western blots at 8 hours (WT, n = 4; SOD2KO, n = 5) in a blinded, random fashion. For the cell death assay, animals were killed 4 and 24 hours after injury (6 SOD2KO, 6 WT at each time point, and 2 naive animals). For the MnSOD activity assay, three each of SOD2KO and WT animals were used. For the aconitase assay, animals were killed 24 hours after injury (6 SOD2KO, 5 WT). For lesion volume, 6 SOD2KO and 6 WT were killed at 8 hours and 7 days after injury, respectively. The genetic background of the SOD2KO mutants has been described elsewhere (Murakami et al., 1998). Briefly, SOD2KO mice with a CD1/SV129 background were backcrossed with CD1 mice for five generations. The animals were anesthetized with 2.5% isoflurane and a nitrous oxide/oxygen mixture (70%/30%) and put on a heating pad. During surgery, rectal temperature was monitored and kept at 37.5°C. Mice were placed in a stereotaxic frame and a craniotomy with a diameter of 5 mm was made over the right parietal cortex with its center between the bregma and lambda. Trauma was performed by a controlled cortical impact device (Dixon et al., 1991). Briefly, a pneumatically driven piston with a diameter of 4 mm was used. The piston compressed the brain at a speed of 2.5 m/s with a compression depth of 1 mm. After trauma, the bone flap was repositioned and the skin was closed with continuous sutures. Apart from not being hit by the piston, sham-operated animals were treated identically. After the experiments, the mice were moved back into their cages to rest on a heating pad that maintained the rectal temperature at 37.5°C until they fully recovered from anesthesia.
Cytochrome c immunohistochemistry
Before the animals were killed, they were sedated with isoflurane and perfused through the heart with 10 U/mL heparin in 0.9% saline, followed by 4% formaldehyde in phosphate-buffered saline. Brains were removed and postfixed in 4% formaldehyde up to 5 days and thereafter sectioned at 50 μm on a vibratome and processed for immunohistochemistry. The sections were incubated with 20% normal goat blocking serum and reacted with anti-cytochrome c polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) at a dilution of 1:100, as previously described (Fujimura et al., 1998).
Western blot analysis
The cortical lesion was cut out and homogenized on ice with a suspension buffer containing 20 mmol/L HEPES-KOH, pH 7.5, 250 mmol/L sucrose, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA, and a protease inhibitor cocktail (Sigma, St. Louis, MO, U.S.A.), and cytosolic and mitochondrial fractions were prepared as previously described (Fujimura et al., 1998). The protein content was measured by a commercial kit (BCA Protein Assay Kit; Pierce, Rockford, IL, U.S.A.). Five micrograms of protein from each cytosolic sample and each mitochondrial sample was loaded in a 12% Tris-glycine gel (Novex, San Diego, CA, U.S.A.). The primary antibodies were either 1:1000 of a monoclonal anti-cytochrome c antibody (65981A; BD PharMingen, San Diego, CA, U.S.A.), 1:5000 dilution of anti β-actin monoclonal antibody, or 1:1000 of a anti-mitochondrial hsp70 antibody (Affinity Bioreagents, Golden, CO, U.S.A.). Western blot analysis was performed with horseradish peroxidase-conjugated, anti-mouse immunoglobulin G using the Boehringer Mannheim (Indianapolis, IN, U.S.A.) chemiluminescence system. The film was scanned with a GS-700 imaging densitometer (Bio-Rad, Hercules, CA, U.S.A.) and the results were quantified using Multi-Analyst software (Bio-Rad). Densitometric values were corrected for local background and expressed as adjusted volume. For the mitochondrial fractions, the adjusted volumes of the cytochrome c bands were divided with the values for the internal controls. Thus, values are expressed as a fraction between cytochrome c and the internal standard.
Double labeling with cytochrome c and TUNEL
After immunostaining for cytochrome c, double staining of cytochrome c immunohistochemistry and TUNEL were performed as previously described (Namura et al., 1998), using nickel-diaminobenzidine as the chromogen for TUNEL.
DNA laddering
To verify the posttraumatic fragmentation of DNA, it was extracted from fresh brains 24 hours after trauma. The cortical lesion was rapidly cut out and the samples were homogenized in lysis buffer (0.5% sodium dodecyl sulfate, 10 mmol/L Tris-HCl pH 8.0, 0.1 mol/L EDTA), the DNA was extracted, and 1 μg of DNA was subjected to gel electrophoresis using a previously published method (Fujimura et al., 1999). The concentration of DNA was measured with TO-PRO-1 dye (Molecular Probes, Eugene, OR, U.S.A.).
Cell death assay
For quantification of apoptotic-related DNA fragmentation, the authors used a commercial enzyme immunoassay to determine cytoplasmic histone-associated DNA fragmentation (Roche Molecular Biochemicals, Mannheim, Germany), which has been shown to detect apoptotic but not necrotic cell death (Leist et al., 1998). Because of the extensive sensitivity of the cell death enzyme-linked immunosorbent assay and for practical reasons, the entire injured hemisphere was used for this analysis. Brains were rapidly removed after perfusion with an ice-cold, heparin-saline solution, were homogenized with a Teflon homogenizer in ice-cold buffer (50 mmol/L KH2 PO4, 0.1 mmol/L EDTA, pH 7.8), were spun for 10 minutes at 800×g. The supernatant was collected and spun for 10 minutes at 20,000×g. The resulting supernatant was collected and the protein concentration was determined. Following the manufacturer's protocol, a cytosolic volume containing 35 μg of protein was used for the enzyme-linked immunosorbent assay.
Manganese superoxide dismutase and aconitase activity assays
To verify that the SOD2KO animals had less mitochondrial SOD activity, the SOD2 activity was estimated in mitochondrial samples using the xanthine/xanthine oxidase and ferrycytochrome c method (Crapo et al., 1978). Residual cytosolic SOD was blocked with 1 mmol/L KCN. SOD activity was determined in triplicate. One unit of SOD was defined as the quantity of enzyme necessary to produce 50% inhibition of the rate of reduction of the ferrycytochrome c. Superoxide dismutase activity was expressed in U/mg soluble protein (Copin et al., 2000). To verify increased levels of mitochondrial oxidative stress in SOD2KO animals, an aconitase activity assay (Oxis Research, Portland, OR, U.S.A.) was performed on mitochondrial subcellular preparations. Mitochondrial aconitase is inactivated by superoxide and to a 10-fold lesser degree by peroxynitrite. The inactivation of this enzyme can therefore serve as a relative indicator of mitochondrial levels of superoxide (Castro et al., 1994; Hausladen and Fridovich, 1994). The animals were perfused with an ice-cold, heparin-saline solution. Because of a need for greater sample volumes for these assays, the cortical lesion, including the underlying hippocampus, was cut out and homogenized in 0.2 mmol/L ice-cold sodium citrate with subsequent mitochondrial isolation (see above), and the activity assay was performed according to the manufacturer's instructions.
Lesion volume
The brains were cut on a cryostat in 20-μm sections from the anterior to the posterior at 500-μm intervals. Sections were stained with hematoxylin and eosin. After staining, the sections were scanned with a GS-700 imaging densitometer (Bio-Rad). The cortical lesion was identified as a region with pallor or loss of hematoxylin and eosin staining, then was traced out. The area of the lesion was calculated using Multi-Analyst software (Bio-Rad), and the volume of the lesion was calculated by adding the lesion area from each section multiplied by the distance between the sections.
Statistical analysis
Densitometric measurements of Western blot films were subjected to analysis of variance and Fisher's protected least-significant difference post hoc test (StatView 4.51; SAS Institute, Cary, NC, U.S.A.). P < 0.05 was considered statistically significant. All data are presented as mean ± SD.
RESULTS
Macroscopic morphology
One hour after injury, there was a cortical contusion with intraparenchymal hemorrhages with a diameter of approximately 4 mm. There was edema in the lesion, in the adjacent cortical areas, and in the subcortical white matter beneath the impact. At 4 hours, the lesion had further developed and the center appeared necrotic. The tissue contained multiple intraparenchymal hemorrhages. After 24 hours, a cortical cavity developed in the center of the lesion in most cases.
Cytochrome c immunoreactivity
In normal control animals, the authors could only detect minimal cytoplasmic cytochrome c immunoreactivity throughout the cerebral cortex and other areas, which is consistent with the poor ability of the antibody to penetrate the mitochondrial membrane (Fujimura et al., 1998; Hortelano et al., 1999; Sugawara et al., 1999) (Fig. 1A, left). Beginning 1 hour after trauma, the authors observed increased immunostaining for cytochrome c in the cortical cells on the circumference of the lesion (Fig. 1A, left inset). The morphology of the cells resembled neurons. The cytosolic cytochrome c immunoreactivity peaked in the cortex 8 hours after injury (Fig. 1A, middle). Some stellate-shaped cells, likely to be astrocytes, also showed increased cytosolic cytochrome c immunostaining (Fig. 1A, middle inset). At this time there also was marked cytosolic cytochrome c immunoreactivity in the ipsilateral CA1 region and dentate gyrus of the hippocampus (not shown). Omission of the primary antibody resulted in no immunoreactivity (not shown).
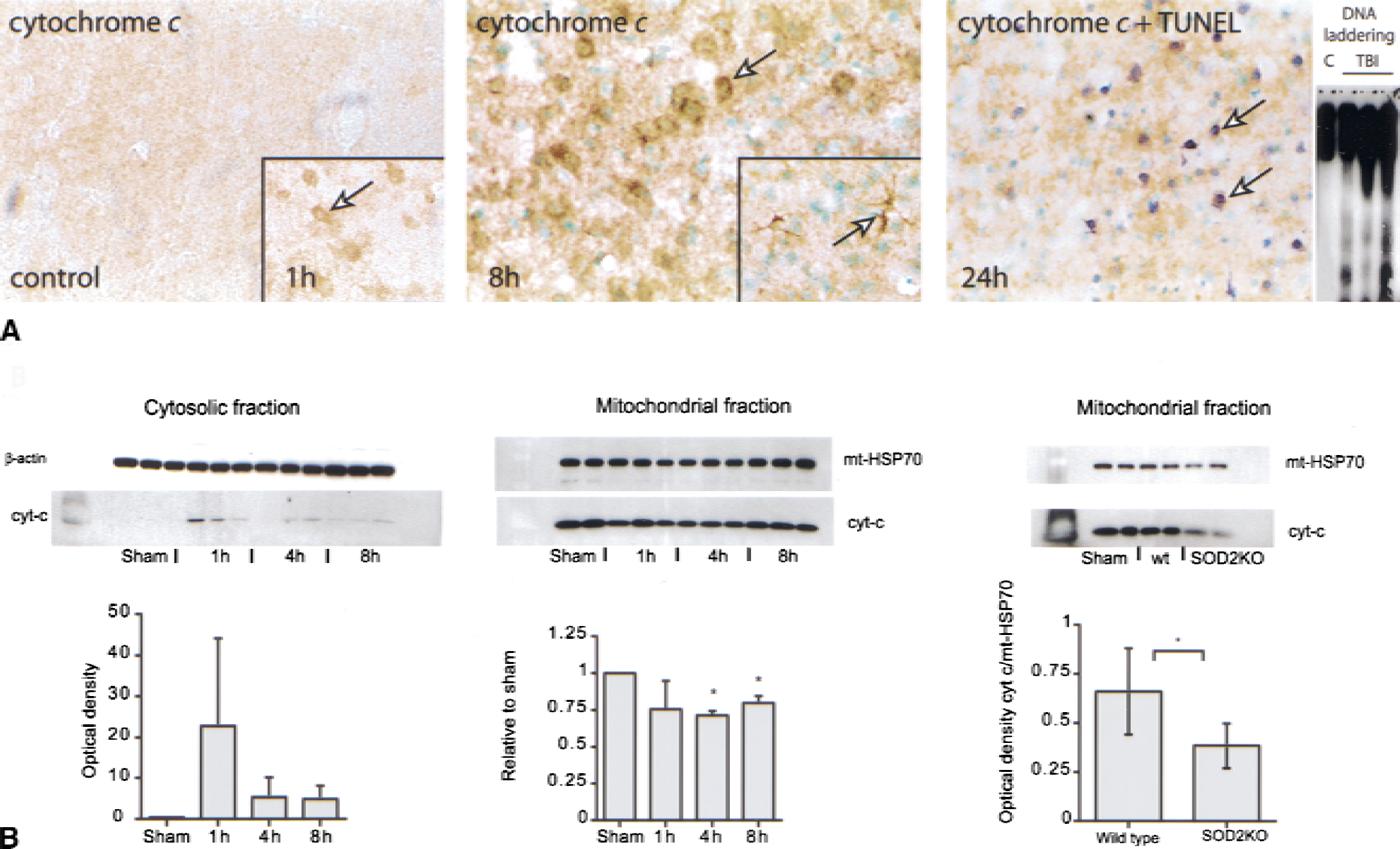
DNA fragmentation and cytochrome c release
To detect DNA fragmentation the authors performed the TUNEL assay, as well as DNA gel electrophoresis. Beginning 4 hours after trauma, there was a significant increase in TUNEL-positive cells in the cortex, CA1–4 regions, and in the dentate gyrus (data not shown). TUNEL-positive cells were identified by a condensed or fragmented nucleus, and 4 and 8 hours after injury most TUNEL cells showed such morphology. Some cells, however (especially at 24 hours), showed more diffuse and cytoplasmic TUNEL, characteristic of necrosis (not shown). Double staining with cytochrome c immunoreactivity and TUNEL revealed that most TUNEL cells were positive for cytoplasmic cytochrome c 24 hours after trauma (Fig. 1A, right). Because cytosolic cytochrome c appeared before the TUNEL labeling, the authors concluded that there was a positive correlation between DNA fragmentation and cytosolic cytochrome c. DNA gel electrophoresis showed evidence of DNA laddering 24 hours after injury, thus confirming the current TUNEL data (Fig. 1A, far right).
Cytochrome c immunoblotting
To investigate the source of the increase in cytosolic cytochrome c after trauma, immunoblots were performed from mitochondrial and cytosolic fractions. A trend toward an increase in cytochrome c immunoreactivity in the cytosolic fractions was found as early as 1 hour after injury (Fig. 1B, left). In the mitochondrial samples, there was a corresponding decrease in cytochrome c (Fig. 1B, middle). Quantification of the cytochrome c bands revealed a 29% reduction in mitochondrial cytochrome c 4 hours after injury. Samples were corrected for protein concentrations and the optical density of the bands was corrected toward mitochondrial hsp70, which served as an internal standard. Thus, these data support the idea that cytochrome c was translocated from the mitochondrial intermembrane space to the cytosol after TBI.
Mitochondrial superoxide–dependent decrease in cytochrome c after TBI
To investigate the role of mitochondrial oxidative stress in mediating cytochrome c release after TBI, the authors included studies on mitochondrial MnSOD heterozygous knockout animals. These animals are likely to have less capability to cope with increases in mitochondrial superoxide stress after injury and also show increased levels of intracellular superoxide after cerebral ischemia (Murakami et al., 1998). The authors found that SOD2KO mutants showed a 42% reduction in mitochondrial cytochrome c 8 hours after TBI compared with the injured WT littermates (Fig. 1B, right), suggesting that cytochrome c is released by a mitochondrial superoxide-dependent mechanism after TBI.
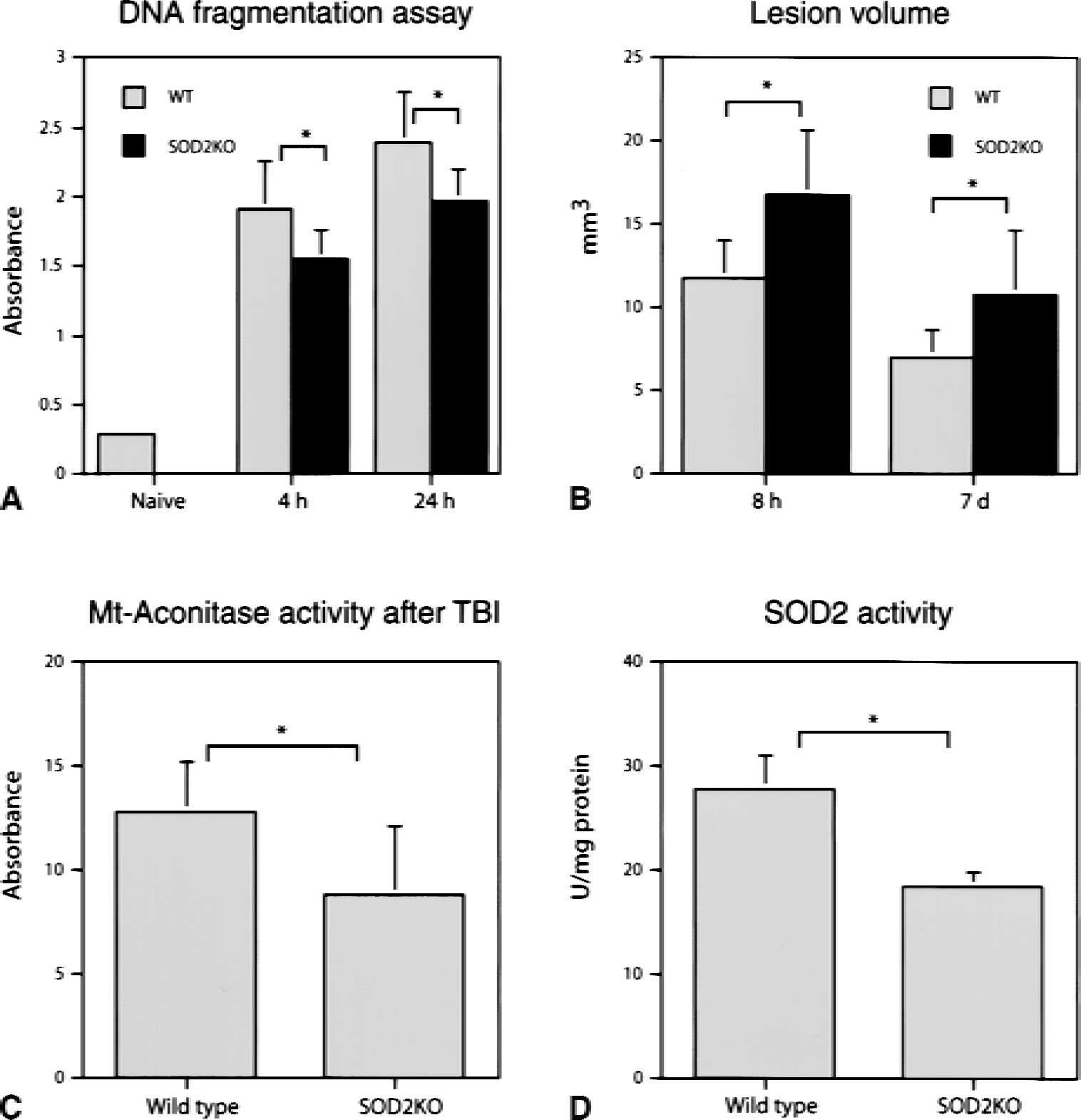
Oxidative stress and cellular injury after TBI in SOD2KO mice
To investigate whether increased cytochrome c release in SOD2KO animals was associated with increased apoptosis, the authors performed a DNA fragmentation assay. Both at 4 and 24 hours, SOD2KO animals showed less signs of DNA fragmentation after trauma compared with WT controls (Fig. 2A). However, SOD2KO animals had a significantly greater lesion 8 hours and 7 days after injury, indicating an increased overall cell death in these animals (Fig. 2B). To find whether SOD2KO animals were subjected to increased mitochondrial superoxide levels, the mitochondrial activity of aconitase, an enzyme which is inhibited by superoxide, was measured. The authors found that aconitase was less active in SOD2KO mice 24 hours after trauma compared with WT traumatized control mice (Fig. 2C), which can be explained by increased levels of superoxide in SOD2KO animals after injury. An obvious explanation for the increased levels of superoxide is that SOD2KO animals showed only one third of SOD2 activity in mitochondrial samples compared with WT control mice (Fig. 2D).
DISCUSSION
The authors have shown that from 1 hour after severe TBI, there was a translocation of cytochrome c from the mitochondria to the cytosol, which preceded DNA fragmentation, debuting at 4 hours. The cytochrome c translocation was correlated by the gene dosage of SOD2, suggesting that the release of cytochrome c after TBI is mediated in part by mitochondrial oxidative stress-dependent mechanisms. The data are supported by similar results in studies in ischemic brain injury (Fujimura et al., 1998, 1999, 2000; Sugawara et al., 1999), including studies showing higher levels of intracellular superoxide after ischemia in the same strain of MnSOD-deficient mice that were used in this study (Murakami et al., 1998. Previous data have supported a role for cytochrome c release as the initiator of the mitochondrial apoptotic pathway (Li et al., 1997a; Liu et al., 1996). The current study supports that observation; however, there was a stronger correlation between cytochrome c release and overall cell death than between cytochrome c release and apoptosis, which is explained if cytochrome c is released from the mitochondria in response to the severity of the insult. In SOD2KO animals, the insult would likely lead to more severe mitochondrial damage (shown by increased cytochrome c release), causing severe secondary mitochondrial failure, and conceivably, more ATP depletion than in WT control mice, which may shift the insult from ATP-dependent apoptosis toward passive necrosis (Leist et al., 1997, 1998; Nicotera et al., 1998). An alternative explanation behind the increased overall cell death noticed in MnSOD-deficient mice is that those animals have an altered inflammatory response, which also could contribute to and/or exacerbate cell death. Alternatively, if there is a different response within different cell types, especially microglia and macrophages, that may contribute to inflammation and subsequent neuronal necrosis between WT animals and MnSOD knockouts, it remains to be studied.
Recently, reactive oxygen species (ROS) have been shown to inhibit caspase activity in cultured cells, probably by oxidative modification of the proteases (Samali et al., 1999). Whether this is also the case in the brain in vivo remains to be studied. Results from the current study are in line with the authors' recent data showing that treatment with a free radical scavenger reduced the size of the lesion after TBI, an effect that was followed by increased apoptosis (Lewén et al., 2001. Thus, it is conceivable that a severe injury with marked ROS stress leads to necrotic cell death, whereas milder ROS stress leads to an apoptotic type of cell death after TBI. The authors' group has previously shown that cytochrome c release is aggravated in SOD2KO animals after ischemia and cerebral hemolysate exposure, together with increased DNA fragmentation (Fujimura et al., 1999; Matz et al., 2001). The discrepancy in the relation between cytochrome c release and subsequent DNA fragmentation between these studies and the current study is likely because of differences in pathophysiology between TBI and cerebral ischemia. Traumatic brain injury, in the current setting with a penetrating contusion type of lesion, is likely more severe than both hemolysate exposure and transient focal ischemia, which is shown by the time course of TUNEL-positive cells that appear after 4 hours in TBI, but which first appear after 24 hours of ischemia and after hemolysate exposure (Fujimura et al., 1998; Matz et al., 2000).
An important issue that remains is the signaling events leading to the release of mitochondrial cytochrome c after cerebral injury. The authors' group has previously shown that this release is modulated by oxygen free radical stress—that is, that overexpression of cytosolic copper-zinc SOD attenuates cytochrome c release after cerebral ischemia—whereas there is an exacerbation after loss of the mitochondrial antioxidant MnSOD, or SOD2 (Fujimura et al., 1999, 2000). These data give evidence that increases in cellular superoxide, cytosolic or mitochondrial, can serve as the signaling event for cytochrome c release after injury. The release of cytochrome c may result in further ROS production by inhibition of the respiratory chain (Cai and Jones, 1998). These events promote a vicious cycle of increased cytochrome c release followed by increased mitochondrial ROS production, which may be maintained.
What other factors trigger the initial cytochrome c release after brain injury? Cytosolic superoxide may modulate other cytosolic factors such as Bax, Bid, or caspase-8, all of which have been linked to the release of cytochrome c and the initiation of apoptosis (Jurgensmeier et al., 1998; Narita et al., 1998). Bax mRNA has been shown to be up-regulated after TBI, suggesting a role for this molecule in apoptosis after brain injury (O'Dell et al., 2000). Conversely, cytosolic superoxide per se may alter the mitochondrial membrane either by opening of the transition pore (Cassarino et al., 1999; Ghafourifar et al., 1999) or by compromising membrane integrity, resulting in the release of the relatively loosely attached cytochromes. Another interesting mechanism is whether there are specific nuclear factors released after injury that can be translocated to the mitochondria, resulting in cytochrome c release. A pathway like this was recently demonstrated in LNCaP human prostate cancer cells (Li et al., 2000), where the protein Nur77 (also called TR3, NAK1) was translocated from the nucleus to the mitochondria. This translocation preceded mitochondrial cytochrome c release and apoptosis (Li et al., 2000). Further studies are needed to clarify if such nuclear messengers, or triggers, also play a role in inducing the apoptotic cascade in acute and chronic brain injury.
Conclusion
This study shows that mitochondrial cytochrome c is translocated from mitochondria to the cytosol after TBI, where it can initiate the mitochondrial apoptotic cell death pathway. In MnSOD-deficient mice, this translocation was aggravated. However, the amount of apoptosis was less pronounced in these animals. This finding together with an increase in the overall cell death in SOD2KO animals suggest that cytochrome c release after TBI is correlated to ROS-mediated mitochondrial damage and that severe mitochondrial cytochrome c release is a predictor of necrotic cell death.
Footnotes
Acknowledgments:
The authors acknowledge Dr. Kirk Maples and Dr. Mingshan Cheng at Centaur Pharmaceuticals, Inc. for use of the controlled cortical impact device.