
Abstract
Reduced infarct volume in TLR2-knockout mice compared with C57Bl/6 wild-type mice has recently been shown in experimental stroke and confirmed in this study. We now also show a significant decrease of CD11b-positive cell counts and decreased neuronal death in the ischemic hemispheres of TLR2-deficient mice compared with C57Bl/6wt mice 2 days after transient focal cerebral ischemia. To examine the potential benefit of intravascular TLR2 inhibition, C57Bl/6wt mice were treated intraarterially with TLR2-blocking anti-TLR2 antibody (clone T2.5) after 45 minutes of cerebral ischemia and compared with control antibody (isotype) treated wild-type mice. Whereas T2.5-treated mice had no reduction in infarct volumes at 48 hours after reperfusion, they did have decreased numbers of CD11b-positive inflammatory cells and decreased neuronal death compared with isotype-treated control mice. Comparison of the isotype antibody treatment to control (saline) treatment showed no effects on infarct volumes or neuronal survival. However, mice treated with the control isotype antibody had increased numbers of CD11b-positive inflammatory cells compared with saline-treated animals. Thus, antibody treatment itself (i.e., control isotype antibody, but potentially of any antibody) may have adverse effects and limit therapeutic benefit of anti-TLR2-antibody therapy. We conclude that TLR2 mediates leukocyte and microglial infiltration and neuronal death, which can be attenuated by TLR2 inhibition. The TLR2 inhibition
Introduction
Ischemic brain injury after focal cerebral ischemia results from a complex pattern of pathophysiological events including exitotoxicity, periinfarct depolarizations, inflammation, and apoptosis (Dirnagl et al, 1999; Kariko et al, 2004; Hossmann, 2006; McColl et al, 2009). The contribution of the inflammatory mechanisms to the postischemic neuronal damage is well known (Dirnagl, 1999; Hossmann, 2006; Trendelenburg, 2008). Toll-like receptors (TLRs) were recently shown to have an important role for the postischemic inflammatory response after transient focal cerebral ischemia (Babcock et al, 2006; Cao et al, 2007; Caso et al, 2008; Ziegler et al, 2007).
The TLRs represent a family of pattern-recognition transmembrane receptors, which recognize a large number of conserved structural motifs, called pathogen-associated molecular patterns. Although TLRs have a crucial role in the innate immune response to invading pathogens and infection, recent findings also suggest an important role in neurodegenerative disorders: TLRs are thought to be activated by endogenous ‘danger signals’ released from injured or necrotic cells and contribute to the initiation of the inflammatory response as well as apoptotic cell death (Asea et al, 2002; Kariko et al, 2004; Aliprantis et al, 2000; Park et al, 2004; O'Neill et al, 2009; Kielian, 2006; Matzinger, 2002; Trendelenburg, 2008; Yao et al, 2009).
It was shown that activation of TLR signaling mediates ischemic preconditioning (Pradillo et al, 2009; Marsh et al, 2009; Stevens and Stenzel-Poore, 2006; Stevens et al, 2008; Kariko et al, 2004). Moreover, TLR2 and TLR4 were recently shown to contribute to ischemic brain damage in mice, potentially by activating proapoptotic pathways and the release of proinflammatory cytokines (Caso et al, 2008; Cao et al, 2007; Marsh et al, 2009; Kilic et al, 2008; Kariko et al, 2004; McColl et al, 2009; Tang et al, 2007; Ziegler et al, 2007). Furthermore, an association of TLR4 gene polymorphisms with ischemic stroke was found in human patients (Lin et al, 2005). The TLR2 is mainly expressed in microglia, but also in selected neurons (Babcock et al, 2006; Ziegler et al, 2007; Tang et al, 2007; Lalancette-Hébert et al, 2009). The TLR2 is activated by endogenous ‘danger signals,’ such as HMGB1, apolipoprotein(apo)CIII, or heat shock proteins HSP70 and GP96 (Asea et al, 2002; Vabulas et al, 2002; Kawakami et al, 2008; Curtin et al, 2009) and was shown to contribute to neuroinflammation and neuronal death (Tang et al, 2007; Hoffmann et al, 2007).
There is growing interest in pharmacological modulation of TLR signaling due to the TLR-mediated effects in stroke and various other diseases. However, whereas specific TLR4 inhibitors were already tested in human patients, there is only limited information with regard to the use of TLR2 inhibitors
This study was designed to address the question whether the application of anti-TLR2 antibody after transient focal ischemia
Materials and methods
Animals
Adult 10–12 weeks old male TLR2−/– mice (Takeuchi et al, 1999) were generously provided by Dr S Akira (Department of Host Defense, Osaka University, Osaka, Japan). The TLR2−/– mice had been backcrossed to the original C57Bl/6J background for > 10 generations. Thus, male C57Bl/6J mice (Charles River, Sulzfeld, Germany) were used as wild-type (control) mice, as well as for TLR2-inhibition studies. During surgery and ischemia, body temperature was measured in all animals and maintained between 37.0°C and 37.5°C, with a heating pad. All animal handling and surgery were performed in accordance with the
Induction of Focal Cerebral Ischemia
Middle cerebral artery occlusion (MCAO) was induced by inserting a silicone-coated filament (Xantopren M Mucosa and Activator NF Optosil Xantopren, Heraeus Kulzer, Wehrheim, Germany) via the internal carotid artery as described by Hara et al (1996). For sham control, the operation was performed without inserting the filament. Mice were anesthetized with 2.5% isofluran for induction of surgery and maintained 1.5% isofluran in 70% N2O and 30% O2 during surgery via a face mask. Anesthesia did not exceed 10 minutes. The animals were reanesthetized for 1 minute after induction of MCAO (occlusion time: 60 minutes in the experiments with the TLR2-knockout mice, 45 minutes in the experiments with the
In Vivo Application of T2.5 and TLR2 Isotype Control Antibody
C57Bl6 wild-type mice were treated with TLR2-blocking antibody T2.5 or isotype control antibody, respectively, after 45 minutes MCAO. In all, 45 minutes MCAO was chosen to compensate for increased infarct sizes observed in the
Assessment of Infarct Volume
At 48 hours after the induction of ischemia, mice were deeply anesthetized and killed. The brains were removed rapidly from the skull and snap frozen in 2-methylbutane on dry ice. Brains were sectioned (12 μm) on a microtome, dried, and stained with hematoxylin (Merck, Darmstadt, Germany). The sections were digitized, the area of infarction (‘direct infarct volume’) was quantified on a PC using Sigma Scan Pro Software (Jandel Scientific, San Rafael, CA, USA; Sigma, St Louis, MO, USA) and infarct volumes were calculated. A correction for edema was applied by calculating the ‘indirect’ infarct volume as the volume of the contralateral hemisphere minus the noninfarcted volume of the ipsilateral hemisphere. The difference between ‘direct' and ‘indirect’ infarct volumes represents brain swelling.
Statistical Analysis
For comparison of infarct volumes, inflammatory cell count, and neuronal survival between the treated groups analysis of variance (
Immunohistochemistry
Staining was performed on 12 μm coronal cryosections at interaural positions 6.6, 5.3, 3.9, 1.9, and 0 mm from mice after 48 hours of reperfusion. The cryosections were thawmounted onto glass slides. Sections were used to determine stroke volume (see above). Slides were air dried for 30 minutes and fixed for 5 minutes in −20°C methanol and acetone (1:1). For counting leukocyte and activated microglia, sections were blocked using a solution containing 3% normal goat serum and 0.3% Triton X-100 (Sigma) in PBS for 60 minutes. The slides were incubated over night at 4°C with rat anti-CD11b antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) at a dilution of 1:200. Neuron staining was performed according to the manufacture's instructions using M.O.M. Vector Kit (Vector, Burlingame, CA, USA) as well as mouse anti-NeuN (Chemicon, Temecula, CA, USA) at a dilution of 1:200. For detection of primary antibody Cy3-conjugated goat anti-mouse and goat anti-rat were used for secondary antibody (Molecular Probes, Leiden, The Netherlands). Omission of the primary antibody was used to ensure specificity of the secondary antibodies.
Cell Counting
A measure of 12 μm coronal cryosections were immunostained with anti-NeuN and anti-Cd11b antibodies. Labeled cells were counted in the whole affected ischemic hemisphere at interaural position 3.9 using a microscope and Stereo Investigator 7 Software (MicroBrightField Bioscience, San Diego, CA, USA) to reduce bias caused by different assignments of specific brain structures or different infarct localizations despite loosing regional information in contrast to cell number measurements in specific areas (Collins et al, 2010; Harhausen et al, 2010; Ziegler et al, 2009). The neuronal survival was determined by the ratio, which was calculated by the number of NeuN-positive cells in the ischemic hemisphere divided by the number of NeuN-positive cells in the nonischemic/contralateral hemisphere.
In Vitro Assays
Murine macrophage RAW 264.7 cells were grown in high-glucose DMEM (Biochrom KG, Berlin, Germany) supplemented with 10% FCS (Biochrom KG) and seeded at a density of 25,000 cells/cm2. After 2 days, cells were preincubated with 50 μg/mL anti-TLR2 antibody T2.5 (eBioscience Inc., San Diego, CA, USA) for 30 minutes and then stimulated with 0.1 μg/mL pam3Cys-SKKK (EMC Microcollections GmbH, Tübingen, Germany). Six hours after stimulation, 90 μL of supernatants were harvested for tumor necrosis factor bioassay and after 24 hours, the nitrite concentration was measured by Griess reaction. Tumor necrosis factor-α Bioassay and Griess reaction was performed as described previously (Freyer et al, 1999).
Results
TLR2 Deficiency Reduces Direct Infarct Volume, Neuroinflammation, and Preserves Neuronal Survival in the Ischemic Hemisphere
As it is known that TLR2-deficient mice had reduced infarct volumes at 48 hours of reperfusion compared with wild-type mice (Ziegler et al, 2007), we now tested whether TLR2-deficient mice also have reduced inflammatory cell accumulation, as measured by the CD11b-positive cell counts (CD11b stains inflammatory cells such as macrophages/monocytes and activated microglia) in the infarcted brain at 48 hours reperfusion after 1 hour MCAO compared with wild-type control mice. As shown in Figure 1A and reported before (Ziegler et al, 2007), TLR2-deficient mice had significant reduced (direct) infarct volume compared with wild-type mice. Moreover, there were less CD11b-positive cells in the ischemic hemisphere in TLR2-deficient mice (mean value ± s.d. 614 ± 140) compared with wild-type control mice (mean ± s.d. 918 ± 112;
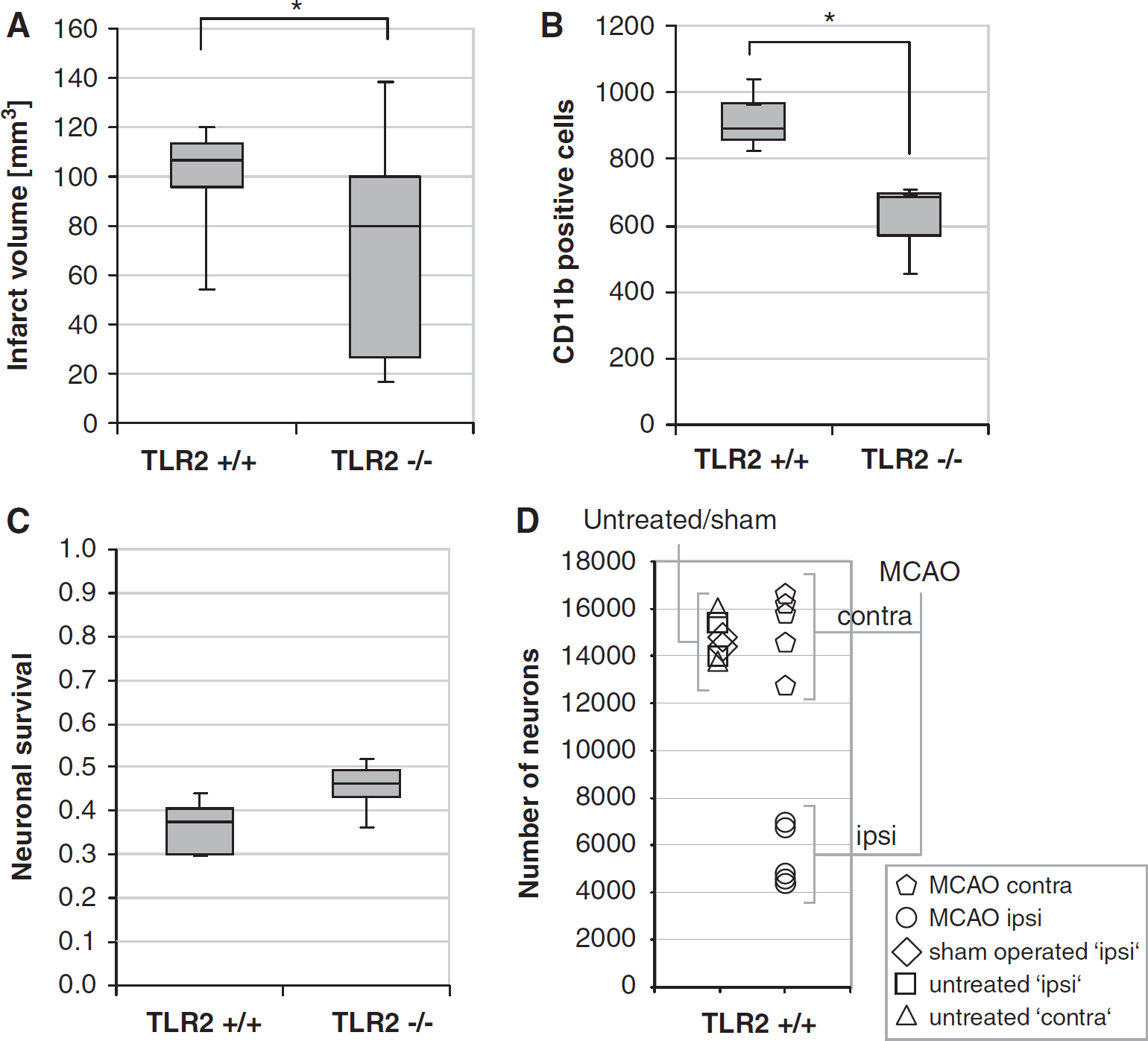
The TLR2 deficiency is associated with attenuated infarct volume, inflammatory cell accumulation, and with improved neuronal survival. (
In parallel to the decreased cell accumulation of CD11b-positive cells, there was an attenuated loss of neuronal cells in the ischemic hemispheres of TLR2-deficient mice compared with that of wild-type mice. It was measured as the relative survival of neurons (neuronal survival ratio: 0.46 ± 0.06 versus 0.36 ± 0.06; given as mean ± s.d.;
TLR2 Antibody Blocks TLR2-Mediated Signaling In Vitro
To evaluate whether the monoclonal anti-TLR2 antibody clone T2.5 (Meng et al, 2004) blocks TLR2 activation
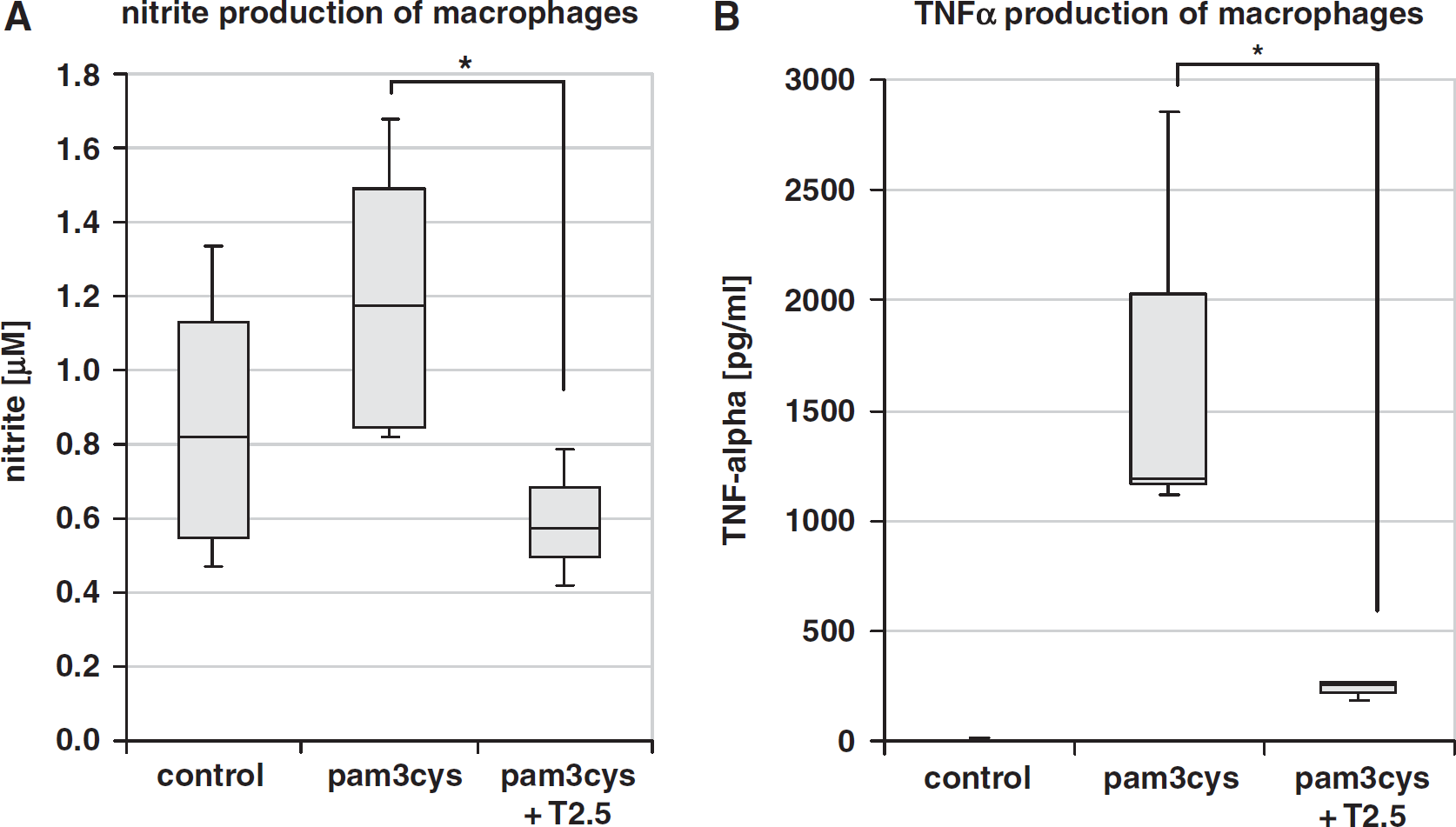
The TLR2-mediated inflammatory response can be blocked by anti-TLR2 antibody
TLR2-Blocking Antibody and Influence on the Infarct Volume
To evaluate a potential therapeutical effect of TLR2 inhibition, 0.05 μg TLR2-blocking antibody T2.5 was applied via flexible tube into the internal carotid artery of C57Bl/6 wild-type mice directly at the begin of reperfusion after induction of cerebral ischemia. Infarct volumes of wild-type mice treated with anti-TLR2 antibody were compared with those of wild-type mice treated with the same amount of isotype control antibody, which does not block TLR2 signaling. As shown in Figure 3A, there is no significant reduction of infarct volume in T2.5-treated mice compared with isotype-treated mice (mean ± s.d. 88 ± 25 mm3 versus 109 ± 34 mm3). For type II error considerations, see Materials and methods (Statistics).
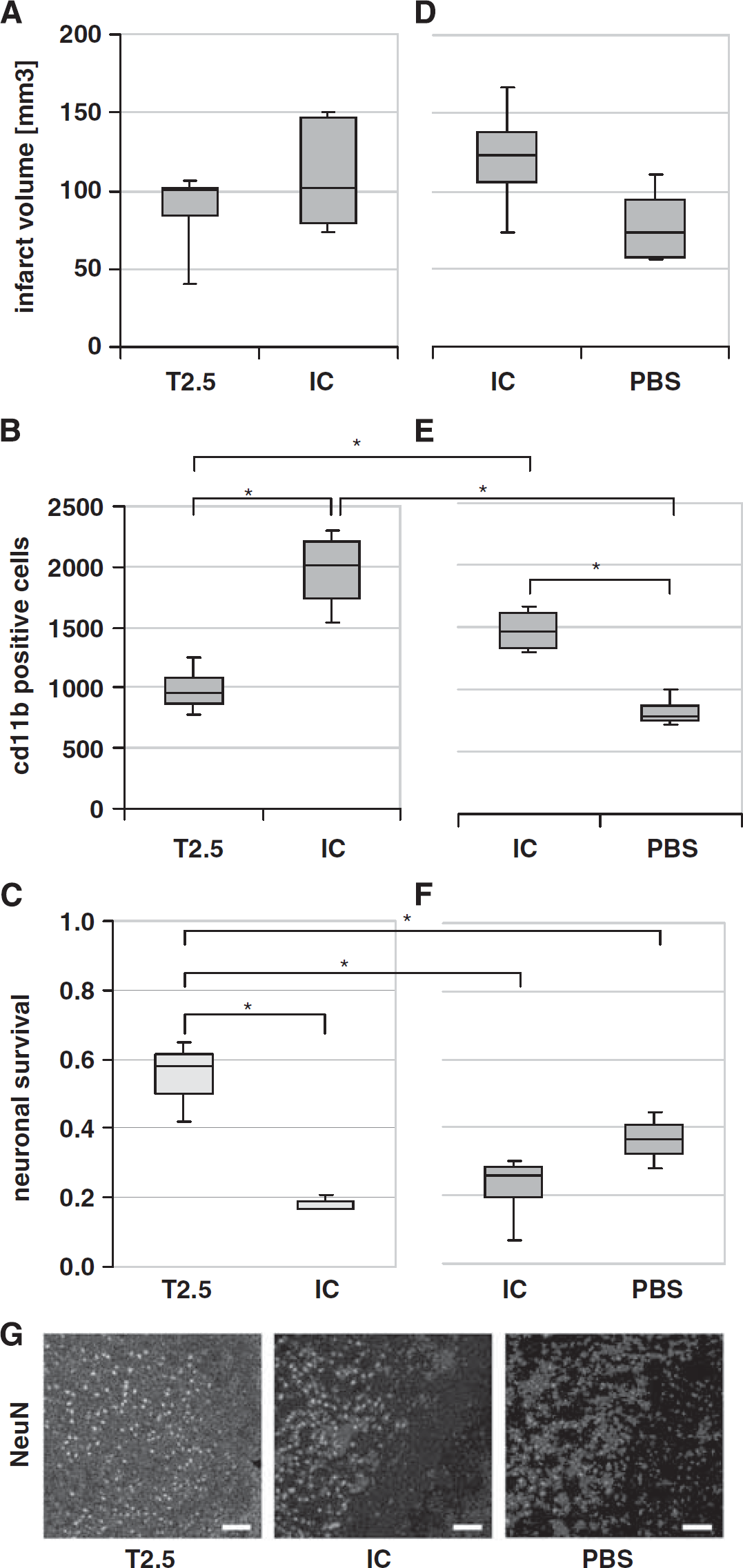
(
TLR2 Inhibition Reduces Postischemic Inflammatory Response In Vivo
Further, we tested whether TLR2-blocking antibody applied
TLR2 Inhibition In Vivo Protects Against Postischemic Neuronal Death
To test whether inhibition of TLR2 signaling by intraaterial application of TLR2-blocking antibody T2.5 after induction of MCAO preserves neuronal survival in the ischemic brain hemisphere, NeuN-positive cells were counted at 48 hours of reperfusion after MCAO for 45 minutes in C57Bl/6 wild-type mice. The animals were treated with either 0.05 μg TLR2-blocking antibody T2.5, or with the same amount of isotype control antibody. There were significantly more NeuN-positive cells in the ischemic hemisphere of T2.5-treated mice compared with the ischemic hemisphere of isotype-treated mice (mean ± s.d. 8,677 ± 2,340 versus 2,831 ± 554;
Isotype Control Antibody Application and Its Influence on Postischemic Outcome in Wild-Type Mice
To test whether antibody application by itself influences postischemic tissue damage, infarct volumes at 48 hours of reperfusion after induction of MCAO for 45 minutes were compared between wild-type mice treated with isotype control antibody and mice treated with only PBS. Treatment with 0.05 μg isotype control antibody did not lead to a significant increase of infarct volumes (mean ± s.d. 121 ± 39 mm3 versus 79 ± 27 mm3;
Analysis of Brain Swelling (Edema) After Treatment
Brain swelling (which was calculated as the difference between ‘indirect’ and ‘direct’ infarct volumes) was compared at 48 hours after induction of MCAO in TLR-deficient, wild-type mice, as well as in T2.5-, isotype-, and PBS-treated wild-type mice (Figures 4A–4C). There was no significant difference observed between each group when using Mann–Whitney
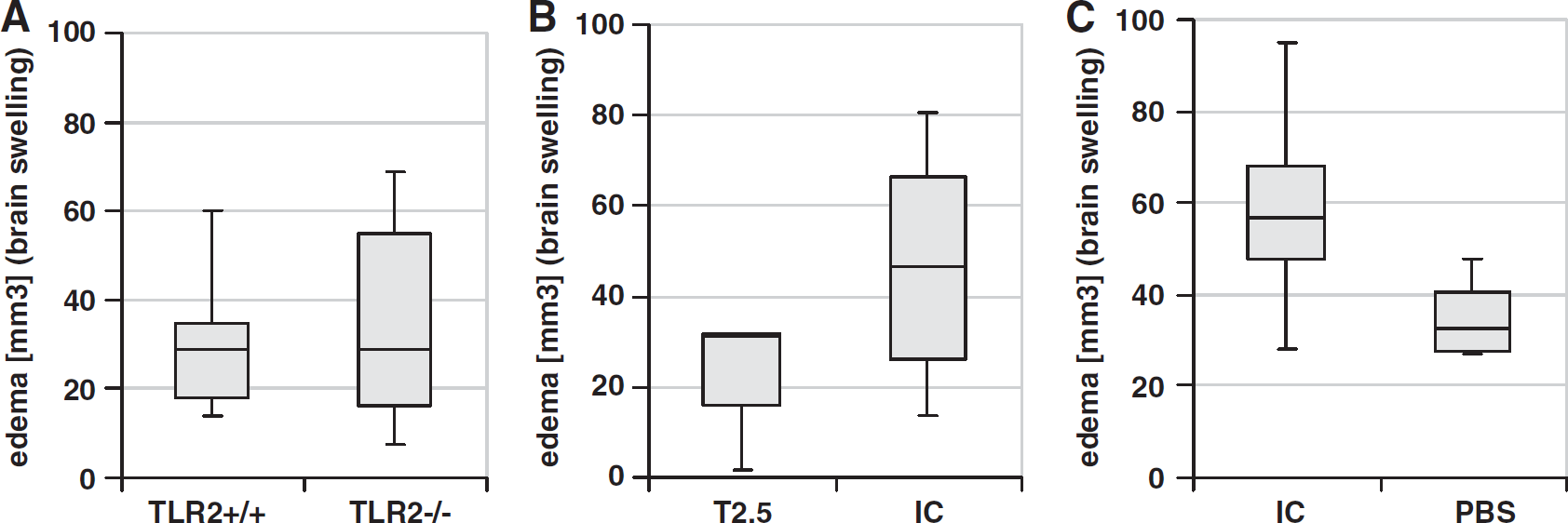
(
Discussion
Our data show a reduced postischemic neuronal death
Although there was only a tendency to reduced infarct volumes and brain swelling in mice treated with TLR2-blocking antibody, neuronal death as well as postischemic accumulation of inflammatory cells were significantly attenuated in T2.5-treated mice at 48 hours of reperfusion, which represents a time point at which infarct volumes were thought to be already consolidated and which was used for the analysis of TLR2-deficient mice in stroke (Ziegler et al, 2007). Our current findings are in good agreement with the recent data, which show a protection of TLR2-knockout mice against focal cerebral ischemia (Ziegler et al, 2007; Tang et al, 2007; Marsh et al, 2009). Moreover, the protective effect (as measured by the preservation of neurons as well as by the reduction of inflammatory cell accumulation in the ischemic hemisphere) observed by the application of the T2.5 antibody reaches a degree, which is comparable to that observed when TLR2-deficient mice were compared with wild-type mice (Figure 1).
However, it remains to be noted, that the isotype treatment itself has a detrimental effect compared with a pure PBS vehicle control, despite a significant benefit of the T2.5 treatment compared with control animals. Thus, the point of the right control (isotype versus vehicle) needs to be considered carefully. Whereas the application of the isotype was chosen in our study to detect TLR2-specific effects, the results of the experiments with PBS itself have demonstrated that an additional vehicle control is recommended in future studies using antibody treatment to control for unspecific or opposite effects of the antibodies.
Interestingly, a very recent study using systemic application of another TLR2-blocking antibody (OP301), which was applied 5 minutes before reperfusion in an animal model of myocard ischemia, revealed a similar protection against ischemic heart injury (Arslan et al, 2010). However, great therapeutical enthusiasm about stroke treatment with blocking antibodies seems to be premature with regard to the effects observed with the isotype control antibody, which show that isotype control antibody leads to an increased inflammatory response compared with sham-treated (PBS-treated) animals (Figures 3D–3G).
Unfortunately, recent clinical studies regarding antiinflammatory stroke therapies using blocking antibodies have been discouraging so far. For example, the Enlimomab (anti-intracellular adhesion molecule-1 (ICAM-1)) antibody Acute Stroke Trail was halted following enrollment of 625 patients, because of its impairing effect on patient outcome after treatment compared with placebo-treated patients (Furuya et al, 2001). Whether this lack of therapeutical benefit may be caused by adverse effects of the IgG by itself requires further examination. Moreover, it would be interesting to see, whether a double TLR inhibition by the use of anti-TLR2 and anti-TLR4 antibodies, as used recently in a sepsis model by the group of Spiller et al (2008) would even improve the observed neuroprotective effect of single TLR2 inhibition. It also needs to be clarified, whether systemic, and not local parenchymal TLR2 inhibition is responsible for the neuroprotective effect observed, as it was shown recently in myocardial ischemia/reperfusion injury (Arslan et al, 2010).
Taken together, our data confirm high potential of TLR inhibition in stroke therapy. As several pharmacological TLR inhibitors are currently under development (O'Neill et al, 2009; Marsh et al, 2009; McColl et al, 2009), further studies using other TLR inhibitors in experimental stroke models are eagerly awaited.
Footnotes
The authors declare no conflict of interest.