
Abstract
Caspase and inhibitor of apoptosis (IAP) expression was examined in rats subjected to moderate traumatic brain injury (TBI) using a parasagittal fluid-percussion brain insult (1.7 to 2.2 atm). Within 1 hour after injury, caspase-8 and −9, two initiators of apoptosis, were predominantly expressed in superficial cortical areas adjacent to the impact site and in the thalamus. Caspase-3, an effector caspase, was evident at 6 hours throughout the traumatized cerebral cortex and hippocampus. Moreover, the authors observed that XIAP, cIAP-1, and cIAP-2, members of the IAP family, were constitutively expressed in the brain. Colocalization of XIAP-immunolabled cells with cell-specific markers indicated that XIAP is expressed within neurons and a subpopulation of oligodendrocytes. Immunoblots of brain extracts revealed that the processed forms of caspase-8, −9, and −3 are present as early as 1 hour after trauma. The appearance of activated caspases corresponded with the detection of cleavage of XIAP into fragments after injury and a concomitant increase in the levels of cIAP-1 and cIAP-2 in the traumatized hemispheres. The current data are consistent with the hypotheses that caspases in both the extrinsic and intrinsic apoptotic pathways are activated after moderate TBI and that IAPs may have a protective role within the brain with alterations in levels and cleavage of IAPs that contribute to cell death in this setting.
Traumatic brain injury (TBI) results in the death of cortical, hippocampal, and thalamic neurons (Dixon et al., 1987; Cortez et al., 1989; Adams, 1992; Dietrich et al., 1994; Graham and Gennarelli, 1997), and apoptotic mechanisms have been reported to participate in the pathogenesis (Ng et al., 1997; Yakovlev et al., 1997; Conti et al., 1998; Newcomb et al., 1999; Clark et al., 1999, 2000). One basic challenge is to define the temporal and regional pattern of apoptotic cell change after TBI, and to correlate this localization with principal effectors of the apoptotic pathways. It is clear that apoptosis is regulated by the caspase family of cysteine proteases that are classified into three groups according to their putative functions, substrate preference, and sequence homologies—apoptotic initiators or apical caspases, apoptotic effectors or downstream caspases, and inflammatory mediators (Thornberry and Lazebnik, 1998). The apical or initiator caspases such as caspase-8, −9, and −10 function at upstream positions in the apoptotic pathways primarily to cleave effector caspases. The effector caspases such as caspase-3, −6, and −7, function as the ‘executioners’ of the apoptotic program and mediate morphologic and biochemical parameters of apoptosis. Inflammatory mediators such as caspase-1, −4, −5, and −11 function in interleukin-1β (IL-1β) and IL-18 activation (Ghayur et al., 1997).
To avoid inappropriate cell death and disease, apoptotic mechanisms must be tightly controlled. Recent experimental evidence has revealed that apoptosis can be inhibited and controlled at several distinct points in the apoptotic pathways (Wehrli et al., 2000; Goyal, 2001). One group of naturally occurring inhibitors includes the family of proteins known as inhibitors of apoptosis (IAPs). Several mammalian homologues (XIAP, cIAP-1, cIAP-2, NAIP, Apollon, Survivin, pIAP) have been identified, most of which have been demonstrated to inhibit cell death (Deveraux and Reed, 1999). Some IAPs have been reported to directly bind and inhibit certain caspases, including caspase-3, −7, and −9 (Roy et al., 1997; Deveraux et al., 1998, 1999). Other IAPs (cIAP-1 and cIAP-2) bind to TNF-receptor associated factors (TRAFs) 1 and 2 (Rothe et al., 1995; Shu et al., 1996), whereas survivin has been implicated in the cell cycle (Ambrosini et al., 1997; Li et al., 1998). Thus, IAPs can inhibit both the intrinsic and extrinsic apoptotic pathways.
All IAPs contain one to three baculovirus IAP repeat (BIR) domains, which are characterized by a highly conserved ∼70 amino acid domain (Duckett et al., 1996). Mammalian XIAP contains three BIR domains. The BIR-2 domain serves as a regulatory element for caspase binding and potently inhibits caspase-3 and −7 (Deveraux and Reed, 1999; Chai et al., 2001; Huang et al., 2001; Riedl et al., 2001), whereas the BIR-3 domain inhibits caspase-9 (Deveraux et al., 1999). During apoptosis induced by the TNF family member Fas, XIAP is cleaved, separating the BIR-1-2 domains from the BIR-3 ring domain. Moreover, XIAP is cleaved into two fragments after moderate spinal cord injury (Keane et al., 2001). The significance of XIAP cleavage during apoptosis is unclear, but may be one mechanism for lowering the threshold of caspase activity necessary for inducing apoptosis (Deveraux and Reed, 1999).
Within the nervous system, IAPs may function to protect some specific types of compromised neurons. Virus-mediated overexpression of NAIP (Xu et al., 1997) or XIAP (Xu et al., 1999) can prevent ischemic neuronal loss in the hippocampus, and the CA1 neurons protected in this manner appeared to function normally (Xu et al., 1999). Animals treated with kainic acid, which induces delayed neuronal death particularly in the CA1 region of the hippocampus, show increased levels of XIAP in the CA3 region (Korhonen et al., 2001). In contrast, in cell culture experiments, transfection of primary cerebellar granule neurons with adenovirus encoding NAIP, XIAP, c-IAP-1, or c-IAP-2 delayed, but did not prevent apoptosis induced by K+ depolarization and serum deprivation (Simons et al., 1999). Necrotic cell death induced by L-glutamate treatment was unimpaired by this group of IAPs, suggesting that some IAP family proteins may not be sufficient to protect some types of neurons from insults associated with ischemia (Deveraux and Reed, 1999).
In this report, the authors investigated regulation of expression of initiator caspase-8 and −9, effector caspase-3, and IAP family members XIAP, c-IAP-1, and c-IAP-2 after TBI. The current results demonstrate that the cell death after TBI involves activation of multiple caspases in both the extrinsic and intrinsic apoptotic pathways. Associated with caspase expression is the up-regulation of c-IAP-1 and c-IAP-2 and cleavage of XIAP that also contributes to the apoptosis that occurs after moderate TBI.
MATERIALS AND METHODS
Animals and surgery
Male Sprague-Dawley rats (250 to 300 g, n = 60) were anesthetized with 3% halothane, 30% oxygen, and a balance of nitrous oxide. Tracheal intubation was performed, and the rats were placed in a stereotaxic frame with a 4.8 mm craniotomy made over the right parietal cortex (3.8 mm posterior to bregma and 2.5 mm lateral to bregma). A plastic injury tube was placed and anchored, and a fluid-percussion device was used to produce injury the next day (Dixon et al., 1987). Intubated and anesthetized rats (70% nitrous oxide, 1.5% halothane, 30% oxygen) were traumatized with moderate head injury (1.7 to 2.2 atm, n = 40). Femoral arterial and venous catheters (PE-50) were inserted for fluid administration, blood gas determination, and blood pressure monitoring. Brain temperature was monitored indirectly with a thermistor probe inserted into the left temporalis muscle (Jiang et al., 1991). In sham-operated, nontraumatized controls (n = 20), all surgical steps were conducted, including placement of plastic injury tube.
Antibodies
Immunohistochemical and immunoblotting procedures used antibodies from several sources to establish antibody specificity and confirm immunostaining and protein expression. Rabbit anti–caspase-3 (MF-393), anti–caspase-8 (MF-438), and anti–caspase-9 (MF-443) antibodies were kindly supplied by Dr. Donald W. Nicholson (Merck-Frosst, Montreal, Canada) and were used at 1:1000 dilution. Immunogens used to produce these antibodies were the large caspase subunit in each case and they recognized intact proenzymes, the fully matured large subunits, and any processing intermediates that might occur during maturation. Rabbit anti-cleaved caspase-3 (17 kDa, 1:1000) (cat. No. 9661) and anti-cleaved caspase-9 (37 kDa, 1:2000) (cat. No. 9501S) were purchased from Cell Signaling Technology (Beverly, MA, U.S.A.) and are specific for the processed forms of these enzymes. Rabbit anti–caspase-8 (p20, 1:500) (cat. No. SC-7890) was supplied by Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A., and recognized the p20 and proforms of caspase-8. Affinity purified rabbit anti-human/mouse cIAP-1 (cat. No. AF818, 1.0 μg/mL), anti-human/mouse cIAP-2 (cat. No AF817, 1.5 μg/mL) were purchased from R&D Systems (Minneapolis, MN, U.S.A.). Specificity of binding of antibodies obtained from R&D Systems was evaluated on cellular extract controls that were provided by the manufacturer. Monoclonal anti-hILP/XIAP (cat. No. H62120, 1:250) was purchased from Transduction Laboratores (Lexington, KY, U.S.A.) and rabbit anti-XIAP (cat. No. 2042) was purchased from Cell Signaling (Beverly, MA, U.S.A.). Mouse anti-β-tubulin (cat no. 556321, 1.0 μg/mL) was purchased from BD Transduction (San Diego, CA, U.S.A.).
Immunohistochemistry
At 1 and 6 hours, 1, 3, or 7 days, rats were anesthetized and perfused transcardially with isotonic saline for 5 minutes, followed by fixative containing a mixture of 40% formaldehyde, glacial acetic acid, and methanol (FAM, 1:1:8 by volume) for 20 minutes. After perfusion, brains were immersed in FAM at 4°C for 24 hours. Brains were blocked and embedded in paraffin for tissue sectioning. Serial cross-sections representing the traumatic epicenter were stained with anti–caspase-3, −8, −9, or rabbit polyclonal anti-XIAP antibodies followed by biotinylated horse anti-rabbit immunoglobulin (1:1000; Vector Elite ABC kit) and streptavidin-horseradish peroxidase followed by 3–3′-diaminobenzidine (DAB) until a brown reaction product was observed. Negative controls without primary antibody and controls using an irrelevant antibody of the same isotope were included in each experiment.
Immunofluorescence microscopy
Ten-micron sections were deparaffinized, and endogenous peroxidase activity was blocked by treatment with 6% hydrogen peroxide and methanol for 15 minutes. Tissue sections were rinsed with phosphate-buffered saline (PBS; pH 7.4) and steamed with 10 mmol/L citrate buffer, pH 6.0 for 20 minutes. Sections were incubated overnight at 4°C with rabbit polyclonal XIAP (1:200), or mouse monoclonal anti-neuronal nuclei (NeuN) (MAB 377, 1:400; Chemicon International, Temecula, CA, U.S.A.), or mouse anti-oligodendrocyte/myelin specific monoclonal antibody (MAB 328, 1:500; Chemicon International, Temecula, CA, U.S.A.), or mouse anti-glial fibrillary acidic protein (GFAP) monoclonal antibody (MAB 360, 1:8000; Chemicon International, Temecula, CA, U.S.A.). Primary antibody binding was detected with Alexa Fluor 488 or Alexa Fluor 584 (1:200, Molecular Probes; Eugene, OR, U.S.A.). Immunofluorescence was detected and processed with an Olympus IX 70 inverted fluorescence microscope equipped with an Optronics DEI-750 CCD camera (Olympus, Tokyo, Japan).
Immunoblotting
For detection of caspases and IAPs, a 2-mm2 section of cortex or hippocampus (n = 5) was homogenized in cell extraction buffer (100 mmol/L HEPES, pH 7.5, 1% Triton X-100, 10 mmol/L dithiothreitol, 1 mmol/L phenylmethylsulfonyl fluoride, 5 μg/mL leupeptin, 1 μg/mL pepstatin A, 1 mmol/L EDTA). Proteins were resolved on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene fluoride membranes, and placed in blocking buffer (PBS, 0.1% Tween-20, 0.4% I-block; Tropix) for 1 hour (Keane et al., 1997). Membranes then were incubated with anti-caspase (1:1500) or anti-XIAP monoclonal antibodies (1:1500; Transduction Laboratories, San Diego, CA, U.S.A.) or polyclonal anti-XIAP antibodies (1:2000; Apoptag) or anti-cIAP-1 or cIAP-2 (R&D Systems). Visualization of the signal was by enhanced chemiluminescence. To control for protein loading, the immunoblots were stripped with Restore, Western blotting stripping buffer (Pierce, Rockford, IL, U.S.A.), and probed for tubulin using monoclonal anti-β-tubulin (MAB 556321; 1.0 μg/mL, BD Transduction, San Diego, CA). Quantification of bands corresponding to changes in protein levels was made using scanned densitometric analysis and NIH Image Program 1.62f.
RESULTS
Activation of upstream caspase-8 and −9 after traumatic brain injury
The temporal pattern of upstream caspase-8 and −9 after TBI was determined by immunohistochemical staining with anti–caspase-8 (Fig. 1) and anti–caspase9 (Fig. 2) antibodies. In sham-operated animals, no positive immunostaining was observed with the anti–caspase-8 antibody (Fig. 1A). By 6 hours after TBI, a few pyknotic cells in the subcortical area underlying the impact site demonstrated immunoreactivity for caspase-8 that was predominantly localized to the nucleus and cell processes (Fig. 1B). This pattern of immunoreactivity also was observed in cortical cells at 3 days after TBI (Fig. 1C), and by 7 days (Fig. 1D), the number and intensity of caspase-8–positive cells was dramatically reduced.
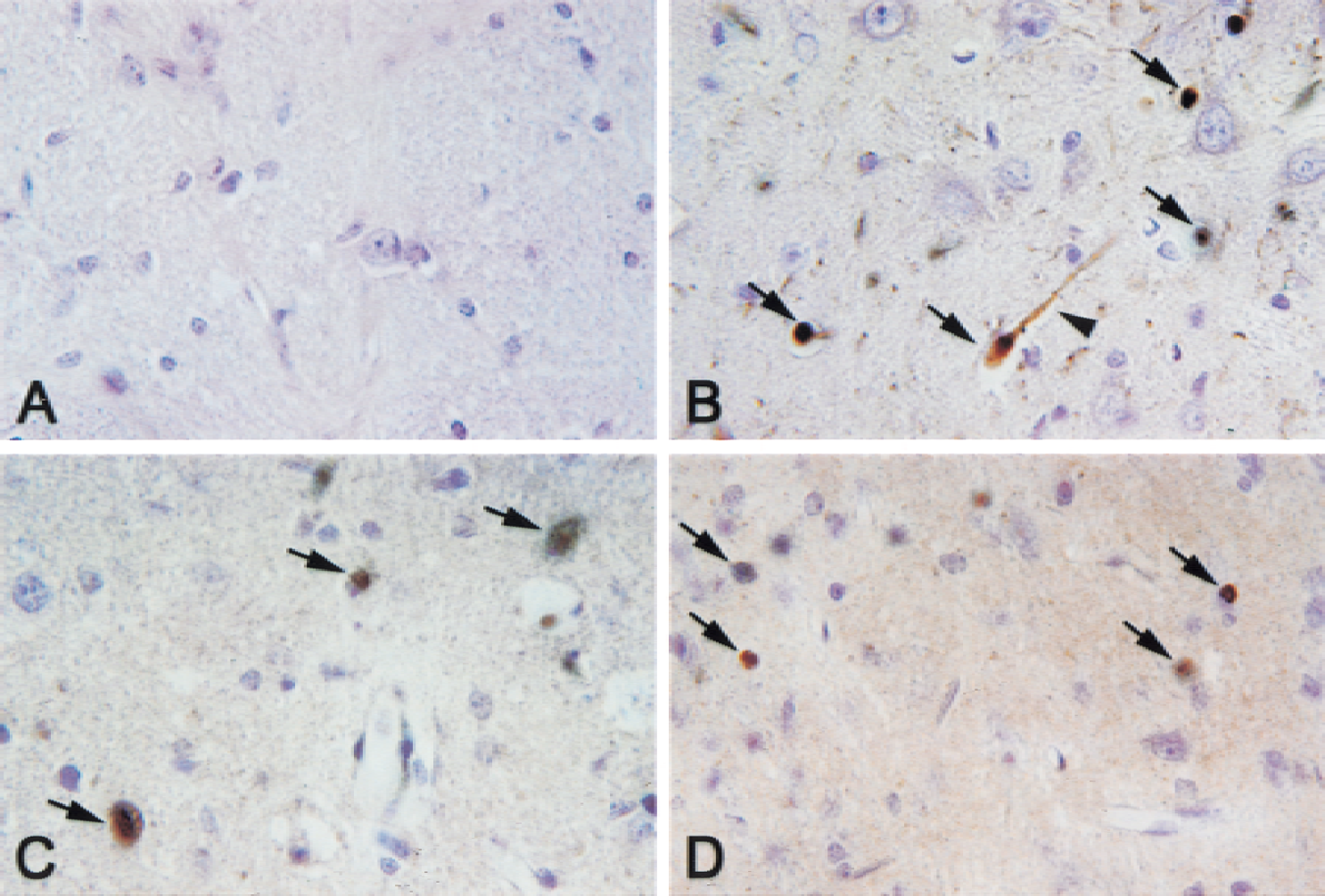
Immunohistochemistry of caspase-8 after traumatic brain injury (TBI). Caspase-8 is expressed predominantly in the nucleus (arrows) and cell processes (arrowhead) in superficial cortical areas underlying the impact site. Cerebral cortex in sham-operated control animals
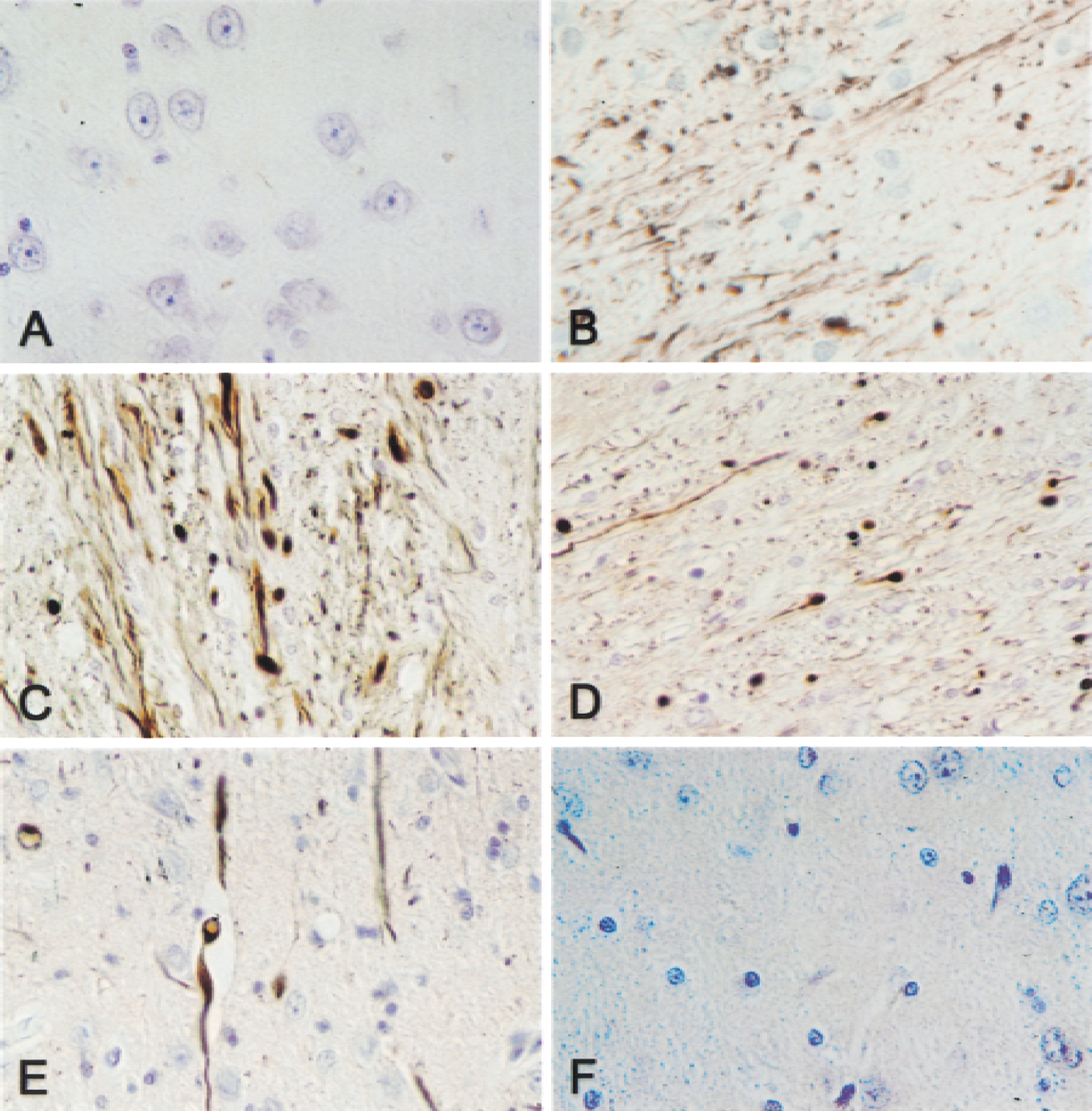
Immunohistochemistry of caspase-9
A quite different pattern of immunostaining was obtained with the anti–caspase-9 antibody (Fig. 2). No immunostaining for caspase-9 was observed in brains of sham control animals (Fig. 2A), whereas positive immunoreactivity for caspase-9 was detected predominantly in the thalamus (Fig. 2B) and subcortical regions in the traumatized hemisphere as early as 1 hour after TBI. By 1 day after TBI, a marked increase in caspase-9 immunoreactivity was seen in the thalamus ipsilateral to the injury site (Fig. 2C) that was localized to cell processes. Interestingly, a similar pattern of immunoreactivity in the contralateral thalamus also was observed with the anti–caspase-9 antibody (Fig. 2D), although the number of fibers and the intensity of staining were reduced. By 3 days, caspase-9 staining was confined to cells in the cortex and thalamus ipsilateral to the site of injury (Fig. 2E), and demonstrated diffuse staining throughout the cell. Little or no observable positive immunostaining with the anti-caspase-9 antibody was detected in brain by 7 days after TBI (Fig. 2F).
Cellular expression of downstream caspase-3
Figure 3 demonstrates the pattern of caspase-3 expression in brain after TBI. No observable positive immunoreactivity was present in sham control animals (Fig. 3A). By 6 hours after TBI, nuclear staining was seen in small shrunken cells in the cortex (Fig. 3B), whereas by 1 day after TBI, small cells in the cortex demonstrated a similar pattern of immunoreactivity, although more intense (Fig. 3C). Caspase-3 immunopositive neurons were detected in the hippocampus by 3 days postinjury, a time at which the authors could clearly identify apoptotic bodies, a morphologic hallmark of apoptosis (Fig. 3D). Caspase-3 immunoreactivity and apoptotic bodies also were found within the thalamus at 3 days after TBI (Fig. 3E, arrows). By 7 days, caspase-3 appeared in shrunken, fragmented cells within the thalamus (Fig. 3F). Thus, distinct patterns of upstream and downstream caspases can be found in the brain after moderate TBI.
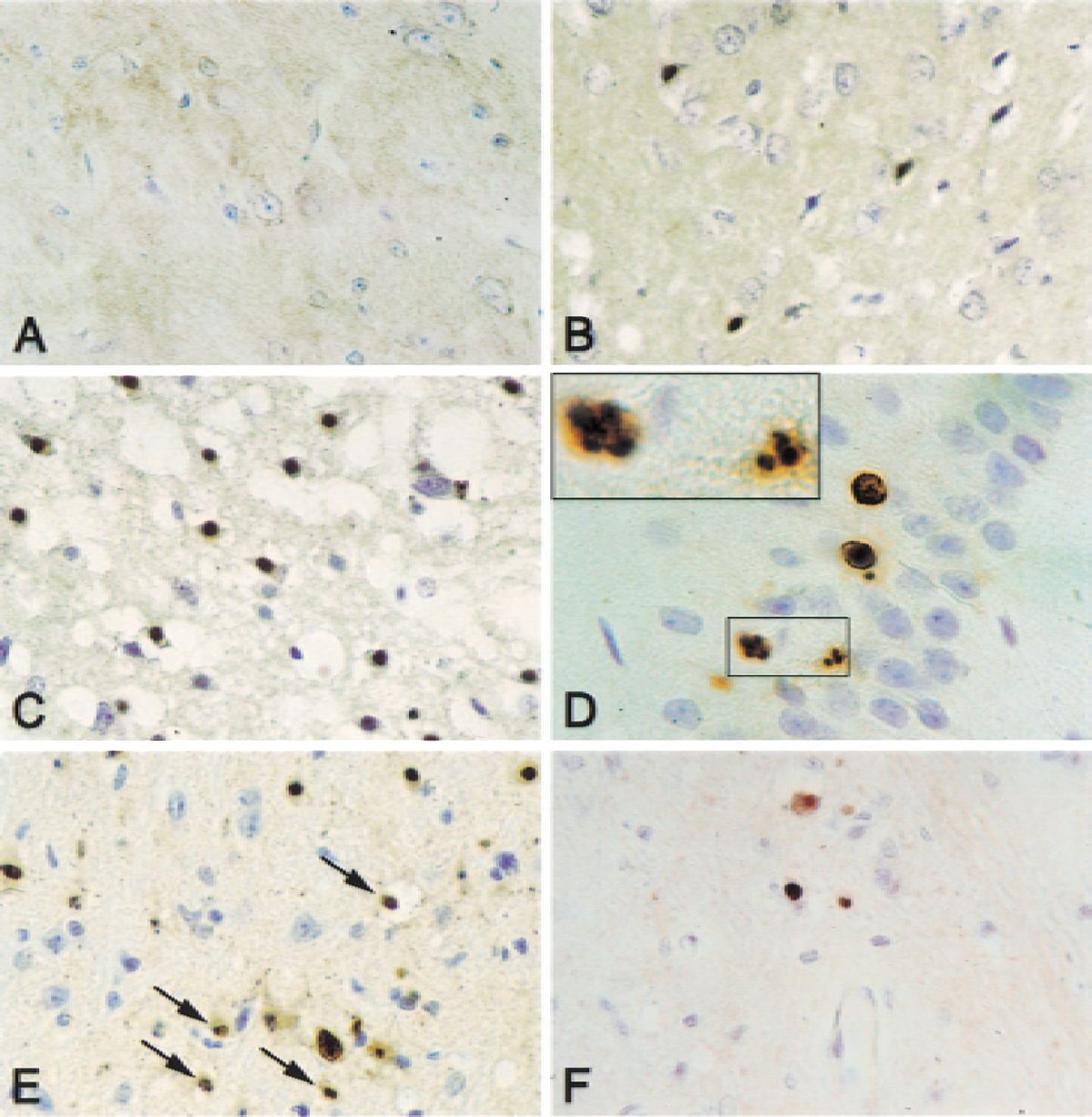
Immunohistochemistry of active caspase-3 after traumatic brain injury (TBI). Caspase-3–positive cells were detected in the cerebral cortex, hippocampus, and thalamus after TBI. Cortex in sham-operated control animals
XIAP expression after traumatic brain injury
To examine the regulation of XIAP protein expression in traumatized brain, the temporal pattern of changes in XIAP immunostaining was assessed. Figure 4 shows positive XIAP immunoreactivity in uninjured sham controls (Fig. 4A) that was confined to neurons and localized predominantly in the nucleus, although some cortical neurons showed weak cytoplasmic staining as well. By 6 hours after TBI, there was an increase in the expression of XIAP that was seen in both the nuclear and cytoplasmic compartments (Fig. 4B). By 3 days, XIAP was strongly expressed in neurons of the cortex (Fig. 4C) and hippocampus (Fig. 4E) in the nucleus and cytoplasmic processes. Moreover, strong XIAP immunostaining also was found in cells lining or associated with blood vessels (Fig. 4D) and in hippocampal neurons in the contralateral hemisphere (Fig. 4F). By 7 days, intense XIAP immunoreactivity was present in neurons of the cortex (Fig. 4G) and in cells positioned around blood vessels (Fig. 4H).
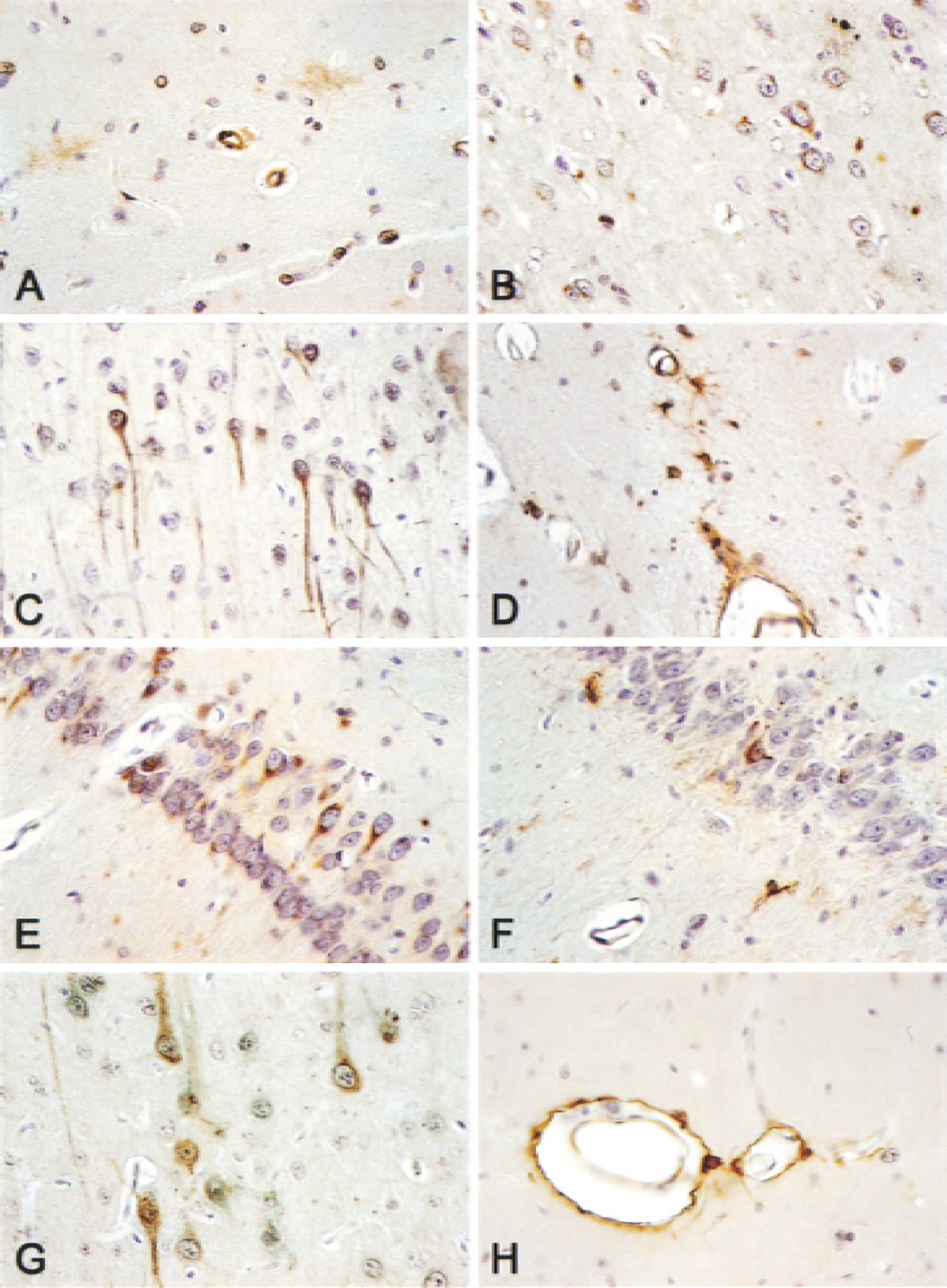
Immunohistochemistry of XIAP after traumatic brain injury (TBI). XIAP is expressed in cerebral cortex, hippocampus, and thalamus. Cortex in sham-operated control animals
Cellular distribution of XIAP in the brain
To determine cellular distribution of XIAP, the authors characterized colocalization of XIAP-immunolabeled cells with cell-specific markers (Fig. 5). The localization of XIAP (red) in neurons (green) was confirmed by costaining with anti-NeuN, and appeared to be confined to the nuclear compartment. The authors next determined whether XIAP (green) was localized to oligodendrocytes (red) within the brain. Anti-oligodendrocyte and central nervous system (CNS) myelin colocalized (yellow) with anti-XIAP in a subpopulation of oligodendrocytes associated with the lining of the ventricles. However, attempts to colocalize XIAP in oligodendroglia from other brain regions were not successful. Finally, the absence of XIAP (red) in astrocytes (green) was demonstrated by costaining with anti-GFAP. Astrocytes within the cortex and hippocampus did not colocalize the two markers, indicating that XIAP is not expressed within this glial cell population in these brain areas. Together, these observations suggest that XIAP is differentially expressed by neurons and a subpopulation of oligodendrocytes, but not by astrocytes within the cerebral cortex and hippocampus.
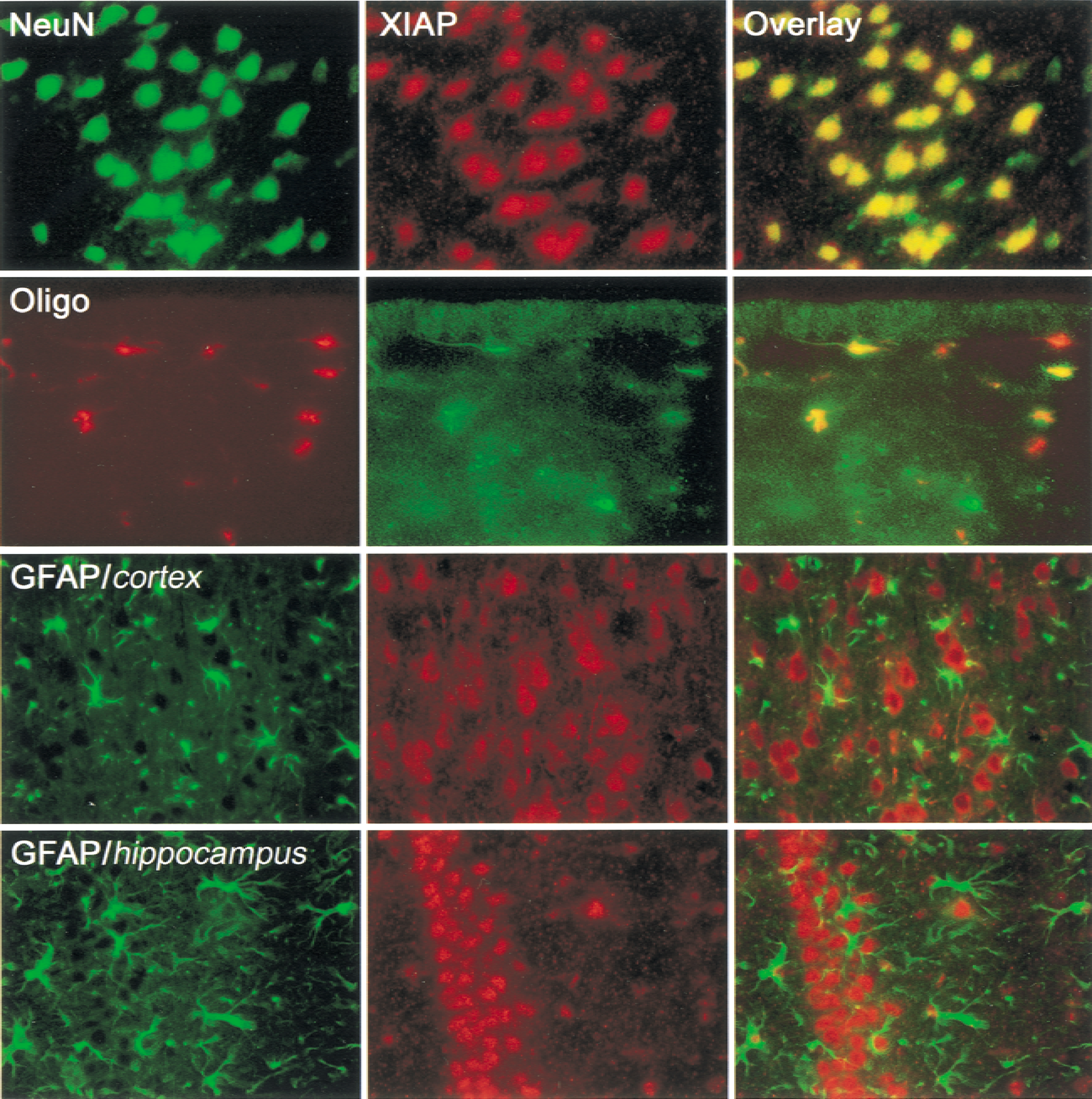
XIAP is colocalized in neurons and a subpopulation of oligodendrocytes, but not astrocytes in the cerebral cortex or hippocampus. Neuronal nuclei (NeuN) marker (green), XIAP (red), and overlaid images (yellow). Oligodendrocyte marker (red), XIAP (green), and overlaid images (yellow). Astrocyte marker, glial fibrillary acidic protein (GFAP; green), XIAP (red), overlaid images show no colocalization in cortex or hippocampus. Magnification ×600.
Immunoblot analysis of caspases and IAPs after traumatic brain injury
Immunoblot analysis was performed to determine the time course of caspase activation after TBI and to establish antibody specificity (Fig. 6). The cleaved p20 subunit of caspase-8 (Fig. 6A), the cleaved p37 subunit of caspase-9 (Fig. 6B), and the cleaved p17 subunit of caspase-3 (Fig. 6C) are generated within 1 hour after TBI in the injured cortical area (R), whereas these processed caspase subunits were less prominent in lysates of the contralateral control hemisphere (L). Increased levels of the processed forms of caspase-8, −9, and −3 were detected in extracts up to 3 days after injury and decreased thereafter (Fig. 6A to 6C). These data support the idea that principal players of the intrinsic and extrinsic apoptotic pathways are activated after TBI.
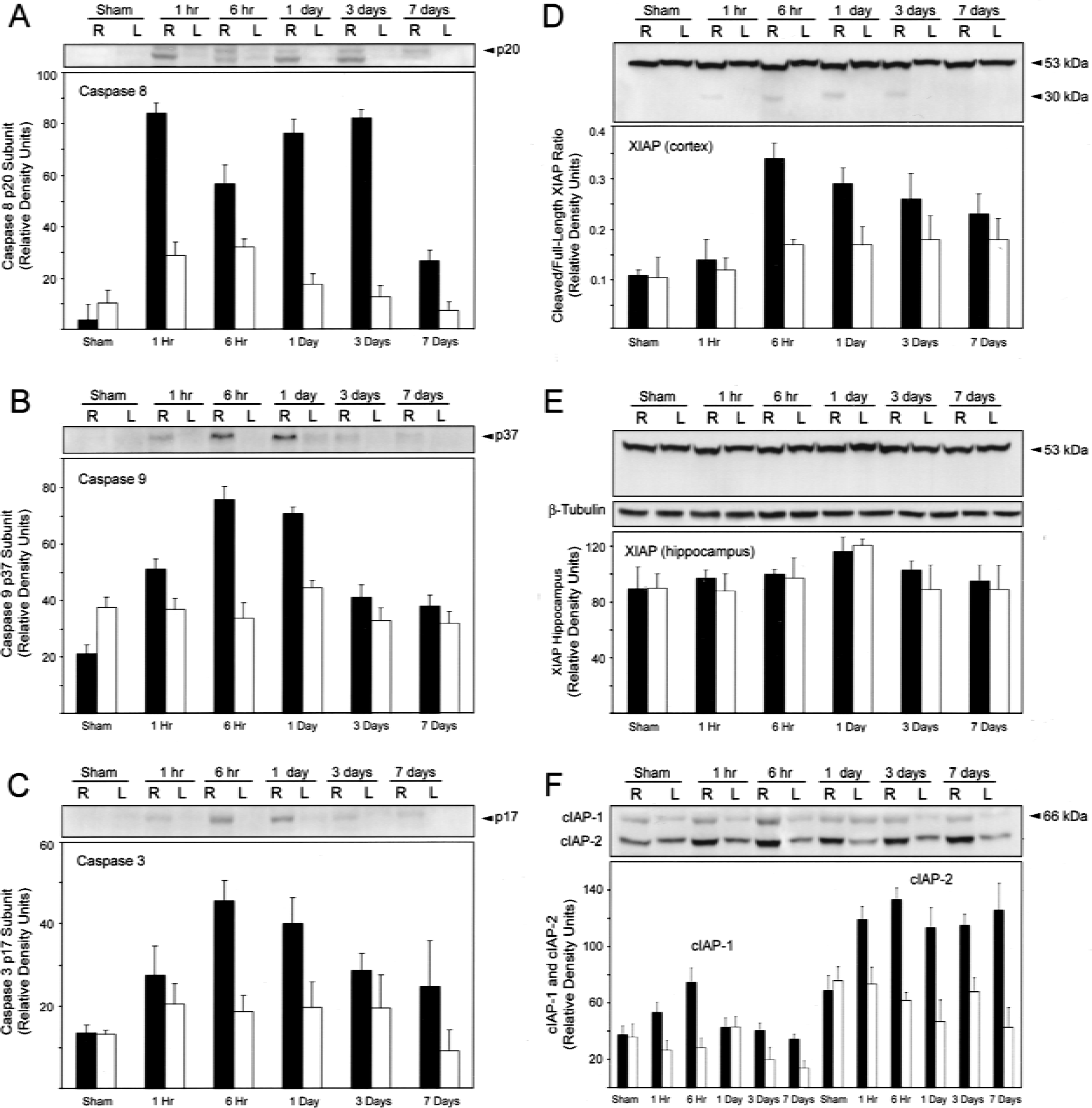
Time course of activation of caspase-8
Associated with the appearance of processed caspase-8, −9, and −3 is the cleavage of XIAP within the cortex (Fig. 6D). Within 1 hour after TBI, the full-length 53-kDa protein has been cleaved to generate a 30-kDa fragment that reacts with both the monoclonal and polyclonal anti-XIAP antibodies. Increased levels of the XIAP cleaved fragment were detected in injured cortical extracts (R) between 1 hour and 3 days after TBI; but by 7 days, the cleavage fragment was not detected. Within the hippocampus, a small increase in the level of XIAP was detected after TBI, but the authors did not detect cleavage of XIAP into smaller fragments by immunoblotting procedures (Fig. 6E).
To determine whether other IAP family members may compensate for cleaved XIAP, immunoblot analysis with anti–cIAP-1 and anti–cIAP-2 was performed on cortical extracts (Fig. 6F). Increased levels of both cIAP-1 and cIAP-2 were present in extracts of injured cortices as early as 1 hour after TBI. Although the level of cIAP-2 remained elevated until at least 7 days, the level of cIAP-1 decreased after 6 hours after TBI. These data suggest that other IAP family members may compensate for the lowered antiapoptotic activity resulting from XIAP cleavage after TBI.
DISCUSSION
In this article, the authors show activation of multiple caspases and IAPs and cleavage of XIAP in the cortex after moderate TBI that contribute to apoptosis in this setting. Activation of initiator caspase-8 and −9 was restricted to distinct brain structures and was first detected as early as 1 hour after injury. Moreover, activation of the effector caspase-3 was detected as early as 1 hour after injury within cells of the cerebral cortex, and at later stages in the hippocampus. These findings are consistent with and extend earlier findings describing the presence of apoptotic cells in brain trauma (Rink et al., 1995; Colicos and Dash, 1996; Yakovlev et al., 1997; Fox et al., 1998; Kaya et al., 1999; Newcomb et al., 1999; Clark et al., 1999, 2000).
The current findings indicate that principal players in both the intrinsic and extrinsic arms of the apoptotic program are activated early after TBI, but the factors regulating expression and activation of these components of the apoptotic pathways remain unclear. In this respect, TNF-α is elevated in serum and cerebrospinal fluid of humans after TBI (Goodman et al., 1990; Ross et al., 1994). Tumor necrosis factor-α levels also are elevated after injury in clinically relevant models of brain trauma, such as lateral fluid-percussion (Taupin et al., 1993; Yakovlev and Faden, 1995; Fan et al., 1996) and closed head impact (Shohami et al., 1994). In these models, TNF-α increases occur from 1 to 4 hours after injury, suggesting that TNF-α may serve as a putative death signal generated after TBI and may activate the extrinsic apoptotic program. However, molecular and structural studies of TNF signaling after TBI are required before definitive conclusions can be drawn.
Cleavage of XIAP is associated with activation of caspase expression, suggesting that caspases may regulate XIAP. Apoptotic T lymphocytes induced by the death receptor and drug-mediated pathways contain at least one 29-kDa XIAP fragment that remains associated with the active subunits of caspase-3 and −7 (Johnson et al., 2000). Moreover, recently, the authors have shown that cell death of spinal cord cells after moderate spinal cord injury is associated with cleavage of XIAP (Keane et al., 2001). These observations suggest that XIAP may act as a suicide inactivator of caspases and undergo cleavage to allow the suicide program to ensue when sufficient levels of caspase activation are reached. Whether XIAP functions in a similar fashion in the CNS after TBI is unclear, but the current findings suggest that dysregulation of normal control mechanisms of XIAP expression contributes to apoptosis induced by TBI.
Associated with cleavage of XIAP is an increase in the levels of cIAP-1 and cIAP-2 in extracts of injured cortices as early as 1 hour after TBI. These data suggest that cIAP-1 and cIAP-2 may provide functional compensation for loss of XIAP during the induction of apoptosis after TBI. In support of this idea, XIAP-deficient mice consistently demonstrate increased levels of cIAP-1 and cIAP-2 protein (Harlin et al., 2001), indicating that there exists a compensatory mechanism that leads to up-regulation of other IAP family members when XIAP expression is lost or reduced. In addition, the authors have observed increased levels of cIAPs associated with XIAP cleavage in the spinal cord after injury (Keane et al., 2001), which further supports this interpretation.
The finding that XIAP is differentially expressed within subpopulations of neural cells within the brain suggests cell lineage-specific expression of IAPs. XIAP expression was evident in neurons within both the nuclear and cytoplasmic compartments after TBI. Similarly, a subpopulation of oligodendrocytes appeared to differentially express XIAP in the brain. XIAP-positive oligodendrocytes were positioned lining the ventricles. However, oligodendrocytes examined in other brain regions and astrocytes did not show detectable levels of XIAP, suggesting that XIAP may function physiologically to ensure cell survival in some neurons and glia. It is tempting to speculate that XIAP functions in neural cells to ensure that small amount of adventitial caspase activation do not amplify out of control, resulting in inappropriate cell death. This mechanism may guarantee that appropriate triggering of the apoptotic pathway only occurs when critical thresholds of caspase activation are surpassed.
In addition to apoptotic and necrotic mechanisms initiated after TBI, increasing evidence suggests that inflammatory mediators contribute to CNS cell death. In a cryogenic injury (rat) model, Knerlich and colleagues (1999) reported up-regulation of IL-1β and inducible nitric oxide synthase mRNA and immunoreactivity beginning at 6 hours and peaking at 24 hours in the penumbra region. In contrast, utilization of in vivo microdialysis and ELISA after traumatic injury (probe insertion) found pronounced elevation of IL-1β protein levels within 60 minutes, with peak elevation at 48 hours (Fassbender et al., 2000). With respect to TBI, transgenic knockout mice for caspase-1 exhibited significantly lower lesion volume and attenuation of IL-1β levels compared with wild-type littermates following a similar degree of cortical impact (Fink et al., 1999). Similar results were observed in wild-type mice injected with caspase-1 inhibitors (Fink et al., 1999), suggesting that IL-1β is a significant proinflammatory mediator of secondary injury after insult. In support of this idea, the authors have observed that actin in cortical cells is cleaved after TBI. Because actin is a substrate for caspase-1 during the apoptotic process (Kayalar et al., 1996), it is possible that inflammatory mechanisms mediated by caspase-1 could contribute to the demise of cells after TBI. Taken together, the current findings suggest that caspases in both the intrinsic and extrinsic apoptotic pathways, and possibly inflammatory mediators, are activated after moderate TBI. Moreover, dysregulation of normal control mechanisms that control IAP protein levels could contribute to cell death induced by CNS injury. Understanding alterations in levels and expression of IAPs and interactions with other proteins in the apoptotic pathways could lead to therapeutic strategies preventing cell death after TBI.
Footnotes
Acknowledgments:
The authors thank Dr. Donald W. Nicholson, Merck-Frosst, Montreal, Canada, for antibodies.