
Abstract
Apoptosis plays an essential role in the cascade of CNS cell degeneration after traumatic brain injury. However, the underlying mechanisms are poorly understood. The authors examined the temporal profile and cell subtype distribution of the proapoptotic protein Bid from 6 hours to 7 days after cortical impact injury in the rat. Increased protein levels of tBid were seen in the cortex ipsilateral to the injury site from 6 hours to 3 days after trauma. Immunohistologic examinations revealed expression of tBid in neurons, astrocytes, and oligodendrocytes from 6 hours to 3 days after impact injury, and concurrent assessment of DNA damage using TUNEL identified tBid-immunopositive cells with apoptoticlike morphology in the traumatized cortex. Moreover, Bid cleavage and activation of caspase-8 and caspase-9 occurred at similar time points and in similar brain regions (i.e., cortical layers 2 to 5) after impact injury. In contrast, there was no evidence of caspase-8 or caspase-9 processing or Bid cleavage in the ipsilateral hippocampus, contralateral cortex, and hippocampus up to 7 days after the injury. The results provide the first evidence of Bid cleavage in the traumatized cortex after experimental traumatic brain injury in vivo, and demonstrate that tBid is expressed in neurons and glial cells. Further, findings indicate that cleavage of Bid may be associated with the activation of the initiator caspase-8 and caspase-9. Finally, these data support the hypothesis that cleavage of Bid contributes to the apoptotic degeneration of different CNS cells in the injured cortex.
Apoptosis is a feature of acute neurodegenerative diseases, including cerebral ischemia and traumatic brain injury (TBI) (Conti et al., 1998;Faden, 1996;McIntosh et al., 1998;Rink et al., 1995). Moreover, members of the caspase family of cysteine proteases have been implicated as important regulators of apoptosis following TBI. For example, caspase-3 is the major executioner caspase involved in neuronal and glial apoptosis after TBI in vivo (Beer et al., 2000b;Clark et al., 2000;Yakovlev et al., 1997) and in vitro (Pike et al., 2000).
Two major caspase-3–activating pathways have been identified. The extrinsic pathway involves cell surface receptors such as Fas. Binding of the Fas ligand to Fas allows the recruitment and activation of the initiator caspase-8 (Li et al., 1998), which in turn activates executioner caspase-3 and caspase-7 (Stennicke et al., 1998;van de Craen et al., 1999). Importantly, recent data have emphasized a contributory role for the extrinsic pathway in TBI-induced CNS apoptosis (Beer et al., 2000a; 2001;Keane et al., 2001).
The intrinsic apoptotic pathway is initiated by the release of cytochrome c to the cytosol. Cytochrome c then binds to the adaptor protein Apaf-1, which allows the recruitment and activation of the initiator caspase-9 in the presence dATP (Crompton, 2000;Green and Reed, 1998). Activated caspase-9 then results in caspase-3 processing (Slee et al., 1999). A role for caspase-9 activation in CNS apoptosis in TBI in vivo has been established recently (Yakovlev et al., 2001).
One proapoptotic member of the Bcl2 family, referred to as Bid, may play an important role in the sequence of events leading to caspase activation (Crompton, 2000). For example, recent in vitro studies have shown that Bid is a specific substrate of caspase-8 in the Fas apoptotic signaling pathway (Li et al., 1998). While inactive full-length (p22) Bid is present in the cytosol, tBid (p15; a truncated form of Bid) translocates to the mitochondria and induces conformational changes in Bax and Bak, triggering cytochrome c release into the cytosol with subsequent activation of caspase-9 (Grinberg et al., 2002, Krajewska et al., 2002;McDonnell et al., 1999). Thus, Bid represents an important mediator of cross-talk between the death receptor and the mitochondria pathways.
Although previous findings regarding the processing of caspase-8, caspase-9, and caspase-3 would suggest a contributory role of Bid-induced apoptosis of CNS cells after brain trauma, no studies to date have examined the temporal and spatial profile of Bid cleavage after TBI in vivo. To further investigate the processing of Bid after experimental TBI, rodents were subjected to a widely used model of experimental mechanical brain injury: lateral cortical impact injury (Dixon et al., 1991;Franz et al., 1999). Western blot analyses of cortical and hippocampal samples were used to determine the expression and cleavage of Bid after TBI. Immunohistochemical examinations were performed to investigate the cell subtype distribution of tBid, and terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-biotin nick end labeling (TUNEL) was used to assess whether tBid-immunopositive cells exhibit morphological features of apoptosis-related DNA damage. Finally, Western blotting and immunolabeling for tBid, caspase-8, and activated caspase-9 was performed to address whether these proteins are expressed at similar time points and in similar brain regions after experimental TBI in vivo.
MATERIALS AND METHODS
Rat model of traumatic brain injury
A controlled cortical impact device was used to induce a moderate level of TBI as described previously (Beer et al., 2001;Dixon et al., 1991;Franz et al., 1999). Briefly, adult male Sprague-Dawley rats (250 to 350 g) were intubated and anesthetized with 2% halothane in a 2:1 mixture of N2O:O2. Core body temperature was monitored continuously by a rectal thermistor probe and maintained at 36.5°C to 37.5°C. Animals were mounted in a stereotaxic frame on the injury device in a prone position, and were secured by ear and incisor bars. The head was held in a horizontal plane with respect to the interaural lines. A midline incision was made, the soft tissues were reflected, and two 7-mm craniotomies were made between lambda and bregma and centered 5 mm laterally on either side of the central suture. The dura was kept intact over the cortex. Injury was induced by impacting the right cortex (ipsilateral cortex) with a 6-mm-diameter tip at a rate of 4 m/sec. The injury device was set to produce a tissue deformation of 2 mm. Velocity was measured directly by the linear velocity displacement transducer (Model 500 HR; Shaevitz, Detroit, MI, U.S.A.), which produces an analog signal that was recorded by a PC-based data acquisition system for analysis of time/displacement parameters of the impactor. After cortical impact, animals were extubated and immediately assessed for recovery of reflexes (Dixon et al., 1991). Sham-injured animals underwent identical surgical procedures but did not receive an impact injury. Naive animals were not exposed to any injury-related surgical procedures. A total of 56 animals were used in this study (naive, n = 8; sham-injured rats, n = 8; injured rats, n = 40). All animal studies carefully conformed to the guidelines outlined in the Guide for the Care and Use of Laboratory Animals from the Austrian Department of Health and Science, and were approved by the University of Innsbruck Medical School Animal Welfare Committee. Importantly, all efforts were made to minimize animal suffering and to reduce the number of animals used.
Sample preparation, sodium dodecyl sulfate-polyacrylamide gel electrophoresis, immunoblotting, and quantification
All animals (n = 28) received a lethal dose of intraperitoneal phenobarbital (20 mg/kg; Tyrol Pharma, Austria) and were killed by decapitation 6 hours or 1, 2, 3, or 7 days after TBI (n = 4 for each time point after injury; n = 2 for sham-injured and naive animals). Sham-injured animals were killed 6 hours and 2 days after sham surgery, respectively. Both cortices and hippocampi (ipsilateral and contralateral to the injury site) were removed. Excision of both cortices beneath the craniotomies extended approximately 4 mm laterally, approximately 7 mm rostrocaudally, and to a depth extending to the white matter. All samples were immediately frozen in liquid N2. The microdissected tissue was homogenized at 4°C in ice-cold homogenization buffer containing 20 mmol/L piperazine-N,N′-bis (2-ethanesulfonic acid) (pH 7.1), 2 mmol/L ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid, 1 mmol/L ethylenediamine tetraacetic acid, 1 mmol/L dithiothreitol, 0.3 mmol/L phenylmethylsulfonylfluoride, and 0.1 mmol/L leupeptin. Chelators and protease inhibitors (Sigma, St. Louis, MO, U.S.A.) were added to prevent endogenous in vitro activation of proteases and subsequent artifactual degradation of Bid and caspases during tissue processing.
Protein concentrations were determined by bicinchoninic acid microprotein assays (Sigma) with albumin standards. Protein-balanced samples were prepared for polyacrylamide gel electrophoresis. Sixty micrograms of protein per lane was routinely resolved on 16% Tris/glycine gels (Invitrogen, Groningen, The Netherlands). After separation, proteins were immediately transferred to nitrocellulose membranes using Western blotting as described previously (Beer et al., 2001). Coomassie blue (Bio-Rad, Hercules, CA, U.S.A.) and Ponceau red (Sigma) staining were routinely performed to confirm successful transfer of protein and to insure that equal amounts of protein were loaded in each lane. Five-percent nonfat milk in phosphate-buffered saline (PBS) with 0.05% Tween 20 was used to reduce nonspecific binding. Immunoblots were probed with a mouse monoclonal antibody (diluted 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) reacting with the p20 subunit and precursor of caspase-8 (Beer et al., 2001), a rabbit polyclonal antiserum (diluted 1:2500) against the large subunit (p35) of caspase-9 (The Burnham Institute, La Jolla, CA, U.S.A.;Krajewski et al., 1999), a rabbit polyclonal antibody (diluted 1:2500; Pharmingen, San Diego, CA, U.S.A.) directed against full-length Bid, or a polyclonal anti-Bid antiserum (diluted 1:2500; The Burnham Institute) recognizing cleaved tBid (Krajewska et al., 2002). Specificity of the anti-Bid antisera was confirmed by preabsorption with recombinant full-length or truncated Bid (obtained by incubation of recombinant Bid with recombinant active caspase-8), respectively. After incubation with primary antibodies overnight at 4°C, nitrocellulose membranes (Amersham Pharmacia Biotech, Uppsala, Sweden) were incubated with secondary antibodies linked to horseradish peroxidase (Amersham Pharmacia Biotech) for 1 hour at ambient temperature. Enhanced chemiluminescence reagents (Amersham Pharmacia Biotech) were used to visualize the immunolabeling on X-ray film. In each blot, the constitutively expressed protein α-tubulin (Sigma) was used as an internal standard to further indicate that sample processing was performed correctly.
Immunohistochemistry
Animals from all treatment groups (n = 28) received a lethal injection of intraperitoneal phenobarbital (20 mg/kg) before perfusion. Rats were transcardially perfused through the left ventricle (120 mL 0.9% saline and 200 mL 4% paraformaldehyde) at 6 hours or 1, 2, 3, or 7 days after TBI (n = 4 for each time point after injury; n = 4 for sham and naive animals). The brains were removed, grossly sectioned coronally at 2-mm intervals, and routinely embedded in paraffin. Sections were then cut at 3 to 4 microns on a rotary microtome, mounted on aminoalkylsilated glass slides, and processed for immunohistochemical staining as described previously (Beer et al., 2000a,2000b; 2001). Consecutive sections from the primary injury zone were probed with anticaspase-8 (rabbit polyclonal antiserum, The Burnham Institute) (Beer et al., 2001), anticaspase-9 p35 (rabbit polyclonal antiserum, The Burnham Institute) (Krajewski et al., 1999) and anti-Bid (rabbit polyclonal antiserum, The Burnham Institute) (Krajewska et al., 2002). Antibodies were diluted 1:5000 in 10% fetal calf serum in PBS and permitted to bind overnight at 4°C. Biotinylated goat antirabbit secondary antibody (Vector Laboratories, Burlingame, CA, U.S.A.) was then applied at a dilution of 1:200 in 3% rat serum in PBS for 1 hour at ambient temperature followed by avidin-peroxidase (Sigma), diluted 1:100 in PBS, also for 1 hour at ambient temperature. The reaction was visualized by treatment with 0.05% 3,3-diaminobenzidine tetrahydrochloride solution in PBS containing 0.05% H2O2. The color reaction was stopped with several washes of PBS. Immunostaining results were confirmed by preimmune serum from the same animals and by preabsorption of the polyclonal antibodies with the relevant recombinant protein. In addition, sections without primary antibodies were similarly processed to control for binding of the secondary antibody. On control sections, no specific immunoreactivity was detected.
For double immunostaining using brightfield chromagens, sections were pretreated with the rabbit polyclonal antiserum against Bid as described previously. Sections were then incubated with a mouse monoclonal anti-NeuN (neuron-specific nuclear protein) antibody (Wolf et al., 1996) (Chemicon, Temecula, CA, U.S.A.) for neuronal staining. For staining of astrocytes and oligodendrocytes, a mouse monoclonal anti—glial fibrillary acid protein (GFAP) antibody (Debus et al., 1983) (Roche Molecular Biochemicals, Mannheim, Germany) or an anti-CNPase (2′,3′-cyclic-nucleotide-3′-phosphodiesterase) monoclonal antibody (Sprinkle, 1989) (Sternberger Monoclonals Inc., Lutherville, MD, U.S.A.) was used. All antibodies were diluted 1:500 in 10% fetal calf serum in PBS and were allowed to bind overnight at 4°C. Sections were rinsed and incubated with a biotinylated horse antimouse antibody (Vector Laboratories) at a dilution of 1:200 for 1 hour at ambient temperature, followed by incubation with an alkaline phosphatase avidin-biotin substrate and then reaction with blue chromagen (Vector Blue; Vector Laboratories). The color reaction was stopped with several washes of PBS. Sections were dehydrated with graded ethanol, cleared in a xylene substitute (Histoclear; National Diagnostics, Atlanta, GA, U.S.A.), mounted in Permount (Fisher Scientific, Nepean, Ontario, Canada), and coverslipped. Control experiments were performed in which the primary antibodies were omitted. No staining was observed under these conditions.
For histochemical detection of DNA fragmentation on selected representative sections, TUNEL was performed as described by Gavrieli et al. (1992) with minor modifications (Beer et al., 2000a; 2001). The TUNEL reaction was visualized by treatment for 5 minutes with 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium substrate system (Dako Corporation, Carpinteria, CA, U.S.A.). For double-label experiments, sections were then processed for Bid immunohistochemistry as described earlier. Immunohistochemical staining was visualized by exposure to 3-amino-9-ethylcarbazole in N,N′-dimethylformamide (Sigma). Primary antibody, labeling mix, or secondary antibody was omitted in control sections. Sections were mounted using an aqueous mounting fluid (Dako Corporation) and examined under the light microscope.
Statistical analysis
Semiquantitative evaluation of immunoreactivity detected by Western blotting was performed using computer-assisted two-dimensional densitometric scanning on a Macintosh computer using the public domain NIH Image program (developed at the US National Institutes of Health and available at http://rsb.info.nih.gov/nih-image/). Relative band densities on Western blots (n = 1 per blot) were expressed as arbitrary densitometric units for each time point. This procedure was performed for the data from four independent experiments, for a total of four different animals per time point. Data acquired in arbitrary densitometric units were transformed to percentages of the densitometric levels observed for scans from sham animals on the same blot. Group differences were determined by analysis of variance and the Tukey post hoc honestly significant difference test. Values are mean ± SD of four independent experiments. Differences were considered significant when P < 0.05.
RESULTS
Western blotting
Cortical impact injury leads to cleavage of Bid in the ipsilateral cortex.
To determine whether Bid is cleaved after TBI, brain extracts from cortex and hippocampus ipsilateral and contralateral to the injury site were examined by Western blotting for the expression of full-length Bid (22 kd) and tBid (15 kd). In the ipsilateral cortex, cortical impact resulted in decreased protein levels of full-length Bid from 6 hours to 3 days after trauma (Fig. 1A), and the lowest immunoreactivity was observed at 1 day after injury (64% decrease relative to sham-injured animals). Concomitantly, tBid immunoreactivity increased within 6 hours after TBI, peaked at 1 day after the impact (793% increase relative to sham animals), and was still significantly increased 2 days and 3 days after impact injury (Fig. 1B). After 7 days, no statistically significant increase in tBid immunoreactivity was evident when compared with levels of sham-injured control animals. In the ipsilateral hippocampus, contralateral cortex, and hippocampus, TBI resulted in no apparent alteration in immunoreactivity of full-length and cleaved Bid (data not shown).
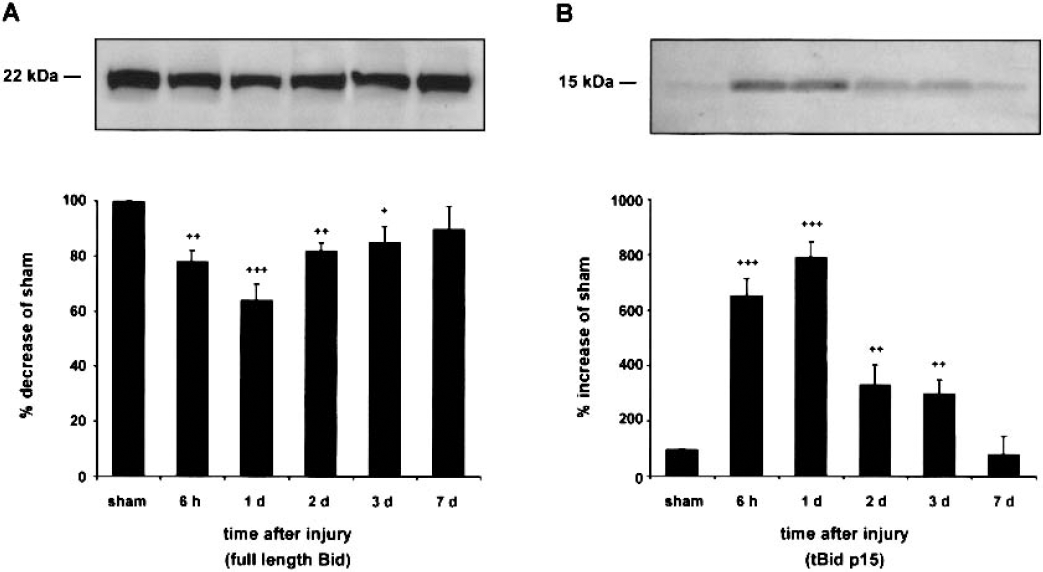
Time course of Bid protein expression
Proteolytic processing of caspase-8 and caspase-9 occurs in the injured cortex.
Because Bid cleavage infers with the activation of caspase-8 and caspase-9, processing of these two caspases was examined by Western blot analysis. Using a monoclonal anticaspase-8 antibody that recognizes both procaspase-8 (p55) and its large subunit (p20), an increase of p55 and p20 caspase-8 immunoreactivity was observed in the ipsilateral cortex from 6 hours to 3 days after TBI (Fig. 2A). Immunoreactivity for caspase-8 p20 increased significantly within 6 hours, peaked at 1 day after the impact (860% increase as compared with sham animals), and was still significantly increased at 2 days and 3 days after impact injury. After 7 days, no significant increase was evident in the p55 and p20 fragments when compared with levels in sham-injured control animals. These results are consistent with a previous report on caspase-8 expression and proteolysis after experimental TBI (Beer et al., 2001). Further, immunoblots of cortical samples ipsilateral to the injury site showed an increase in the immunoreactivity for the large subunit (p35) of activated caspase-9 from 6 hours to 3 days after TBI. Densitometric measures of caspase-9 p35 protein levels from injured animals revealed a 325% increase relative to sham levels at 6 hours after TBI (Fig. 2B). The increase of caspase-9 p35 immunoreactivity reached a maximum level 2 days after impact injury (840% increase relative to sham-injured animals) and declined to a 520% increase relative to the sham group at 3 days after TBI. At 7 days after trauma, no statistically significant difference in caspase-9 p35 immunoreactivity was observed between cortical samples ipsilateral to the injury site and cortical samples from sham-injured animals.
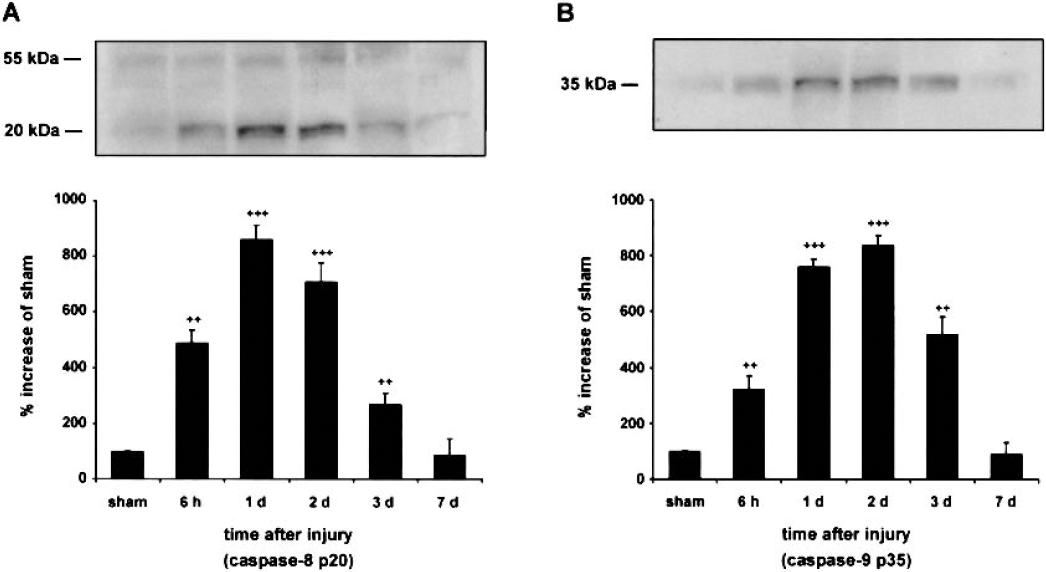
Time course of caspase-8 p20
No significant increases in caspase-8 and caspase-9 p35 immunoreactivity were seen between sham-injured and injured animals in cortical samples contralateral to the injury site and hippocampal samples ipsilateral and contralateral to the injury site 6 hours to 7 days after TBI (data not shown).
Immunohistochemistry
Immunohistochemical staining for tBid, caspase-8, and caspase-9, and cell subtype distribution of tBid after traumatic brain injury.
Ipsilateral and contralateral cortical and hippocampal tissues were examined rostrocaudally from +0.2 to −3.8 mm bregma. No immunoreactivity for tBid (Fig. 3A), caspase-8, and activated caspase-9 was present in the tissue from sham-injured or naive (data not shown) control rats. Positive immunoreactivity for tBid (Figs. 3B and 3E), caspase-8 (Fig. 3C), and processed caspase-9 (Fig. 3D) was found throughout the ipsilateral cortex at the primary injury zone (from −1.5 to −3.4 mm bregma, cortical layers 2 to 5) from 6 hours to 3 days after the trauma (time point = 1 day after trauma, −3.4 mm bregma). Immunoreactivity for tBid, caspase-8, and activated caspase-9 was absent in contralateral cortical samples and hippocampal samples ipsilateral and contralateral to the injury site at all time points investigated (data not shown).
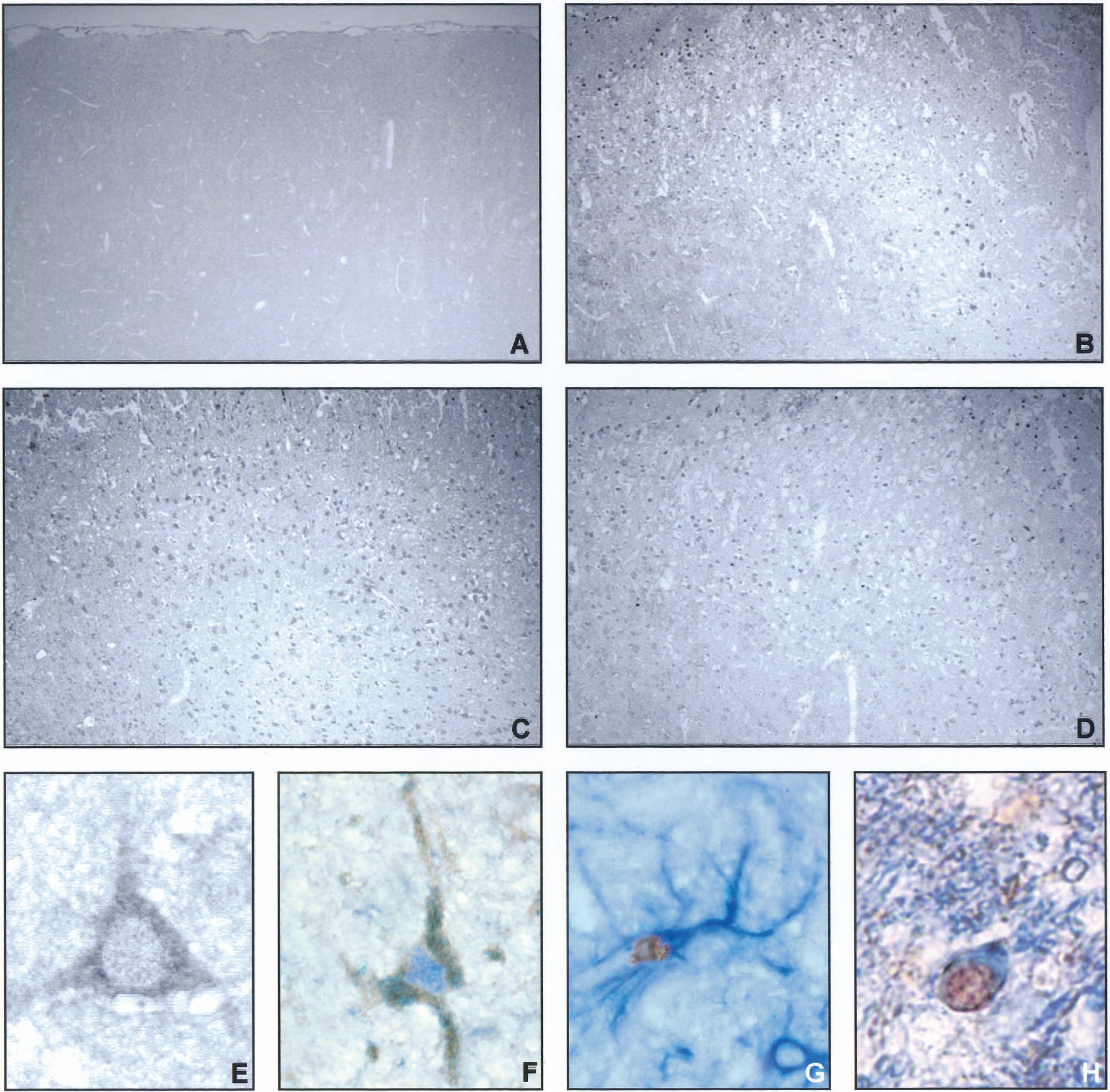
Immunohistochemical analysis of tBid, caspase-8 p20, caspase-9 p35, and cell-subtype distribution of tBid in the traumatized cortex (−3.4 mm bregma) 1 day after traumatic brain injury (TBI). Sham-injured controls showed no specific immunolabeling for tBid
To further investigate whether tBid is expressed in neurons or glial cells, we performed double-labeling experiments for tBid and the neuronal cell-specific marker NeuN, the astrocytic marker GFAP, and the oligodendroglial marker CNPase, respectively. These immunohistochemical analyses of tBid-positive cells from 6 hours to 3 days after TBI (Fig. 3F—H; time point = 1 day after trauma, −3.4 mm bregma) identified labeling with NeuN, GFAP, and CNPase, and demonstrated the expression of tBid in neurons (Fig. 3F), astrocytes (Fig. 3G), and oligodendrocytes (Fig. 3H).
tBid-immunopositive cells exhibit nuclear apoptoticlike morphology.
To further verify an apoptotic component of posttraumatic cell death and to determine the role of tBid in trauma-induced apoptosis, sections immunopositive for tBid were processed for TUNEL to assess DNA damage. Double-labeling experiments (Fig. 4) demonstrated that a substantial proportion of TUNEL-positive cells with shrunken morphology, condensed nuclei, and chromatin margination (Fig. 4, arrows) also expressed tBid in layers 2 to 5 at the primary injury zone (time point = 1 day after trauma, bregma −3.4 mm). However, cells with tBid immunoreactivity or gross apoptoticlike morphology alone were also detected (Fig. 4).
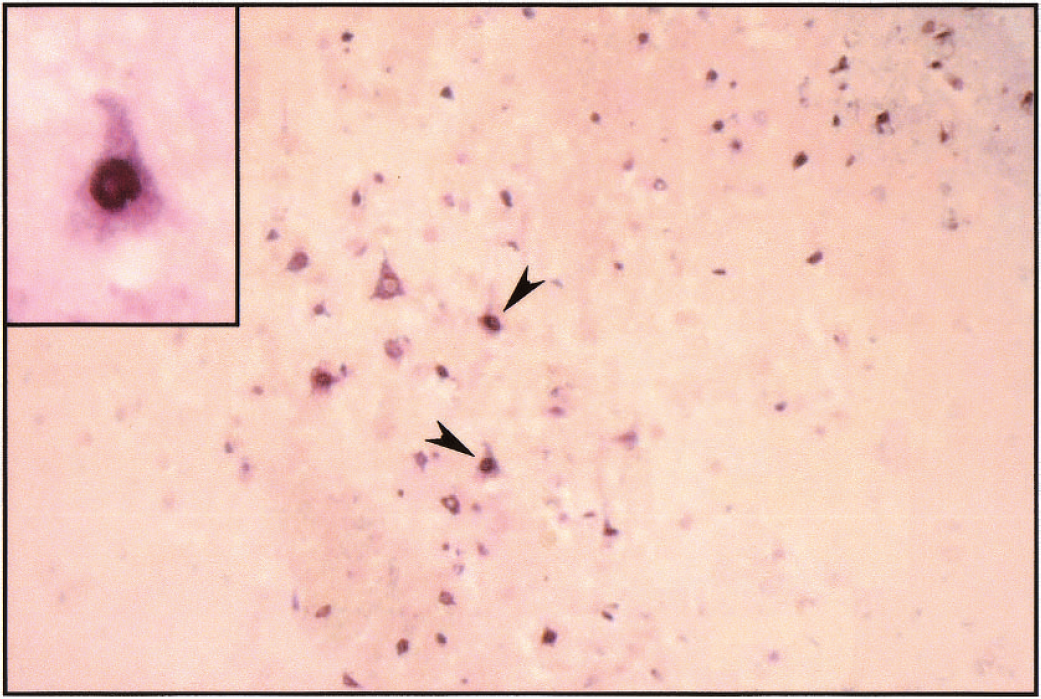
Appearance of tBid in TUNEL-positive cells in the traumatized cortex (−3.4 mm bregma) 1 day after traumatic brain injury. Combined immunohistochemistry for tBid (red color) and TUNEL (dark blue color) showed processing of Bid in cells with gross nuclear apoptoticlike morphology. The TUNEL-positive cells exhibited chromatin condensation and nuclear fragmentation (arrows). Magnification: 200×; insert, 1,000×.
DISCUSSION
Our results provide the first evidence for Bid expression and activation after experimental TBI. In the cortex ipsilateral to the injury site, increased immunoreactivity for activated tBid was detected from 6 hours to 3 days after TBI, and immunohistochemistry demonstrated expression of tBid in neurons, astrocytes, and oligodendrocytes at similar time points. In addition, our data revealed expression of tBid in CNS cells with apoptoticlike morphology in the traumatized cortex. Finally, our data suggest that caspase-8, caspase-9, and Bid are activated in similar brain regions of the injured cortex from 6 hours to 3 days after experimental brain trauma.
Only limited data exist regarding the putative role of Bid for apoptotic degeneration in chronic and acute neurologic disorders. For example, cleaved Bid has been implicated in apoptosis of neurons in Parkinson disease (Viswanath et al., 2001) and experimental epilepsy (Henshall et al., 2001). Further, tBid may also mediate apoptotic neuronal cell death after focal cerebral ischemia (Plesnila et al., 2001). In this regard, it is noteworthy that cortical impact injury may produce focal ischemia in the cortex ipsilateral to the injury site (Bryan et al., 1995). Thus, reduced cerebral blood flow may have contributed to Bid activation in our experiments.
Bid is a specific substrate of the initiator caspase-8 in the Fas apoptotic signaling pathway (Li et al., 1998). Importantly, we and others have documented increased expression of Fas and activation of caspase-8 after experimental brain trauma (Beer et al., 2000a; 2001;Keane et al., 2001). Our data now indicate that activated caspase-8 and tBid are expressed at similar time points and in similar cortical regions after cortical impact injury, which may suggest a contributory role of activated caspase-8 in the cleavage and activation of Bid. However, a current report also provided evidence that other proteases such as calpain may cleave Bid to an active fragment capable of mediating cytochrome c release in experimental myocardial ischemia (Chen et al., 2001). Therefore, future studies are needed to further investigate the potential role of other proteases in Bid processing after experimental brain injury, as calpain may play an important role in the degeneration of CNS cells after brain trauma (Kampfl et al., 1997;Saatman et al., 2000) and cerebral ischemia (Neumar et al., 1996) in vivo.
Our double-labeling experiments revealed the expression of tBid in neurons, astrocytes, and oligodendrocytes. Interestingly, expression of tBid has recently been shown in neurons and astrocytes after oxygen/glucose deprivation in vitro (Plesnila et al., 2001). Importantly, our results are the first to indicate that tBid is expressed in neurons and glial cells after up to 3 days after experimental brain injury in vivo, and that tBid is seen in CNS cells with apoptoticlike morphology. These results may suggest that even delayed treatment paradigms targeting Bid cleavage may prevent apoptotic cell death after acute brain injuries in vivo. In this regard, it is noteworthy that a recent report revealed that Bid-deficient neurons are more resistant to death after oxygen/glucose deprivation in vitro (Plesnila et al., 2001).
Current data suggest that tBid causes cytochrome c release from mitochondria, which results in the activation of caspase-9 and subsequently caspase-3 (Budihardjo et al., 1999). However, only limited data exist regarding the release of cytochrome c and the activation of caspase-9 after TBI in vivo (Buki et al 2000;Keane et al 2001;Yakovlev et al., 2001). Interestingly, the present study found that tBid and activated caspase-9 are expressed in similar brain regions at similar time points after cortical impact injury in the rat. These data may support the hypothesis that tBid contributes to the activation of caspase-9 in the traumatized cortex from 6 hours to 3 days after TBI. However, future studies are needed to further elucidate the exact role of tBid in caspase-9 activation after different acute CNS injuries in vitro and in vivo.
Our study failed to detect tBid, caspase-8, caspase-9, and apoptotic CNS morphology in the hippocampus ipsilateral to the injury site at the investigated time points. Previous reports clearly describe features of apoptotic neuronal degeneration in the hippocampus (Clark et al., 2000;Yakovlev et al., 1997). However, previous studies in our laboratory (Beer et al., 2000a, 2000b; 2001;Franz et al., 1999) have shown that cortical impact injury may not necessarily be associated with hippocampal neuronal degeneration. Probable reasons for discrepancies in the appearance of hippocampal damage may be subtle methodological differences in animal models of TBI. For example, differences in angulation and velocity of the impact devices could account for the absence of hippocampal degeneration.
In summary, our data suggest that activation of Bid occurs in the traumatized cortex after cortical impact injury in the rat, and that Bid cleavage is associated with caspase-8 and caspase-9 activation. As Bid is strategically located upstream of mitochondria and caspase-3 processing, it may represent an attractive therapeutic target for CNS diseases in which apoptotic cell death is prominent. However, this implicates the need for further investigations of the significance of Bid cleavage inhibition on functional outcome after TBI.
Footnotes
Acknowledgments
The authors thank Marianne Leisser (Brain Research Institute, University of Vienna, Vienna, Austria) and Kurt Hohenstein for their expert technical assistance.