
Abstract
XIAP is a member of the inhibitor of apoptosis (IAP) gene family that, in addition to suppressing cell death by inhibition and polyubiquitination of caspases, is involved in an increasing number of signaling cascades. Moreover, the function and regulation of XIAP in the central nervous system (CNS) is poorly understood. In this study, the authors investigated the cell-type expression, the subcellular distribution, ubiquitination of XIAP, and levels of Smac/DIABLO in the normal adult rat brain and in brains subjected to moderate traumatic brain injury (TBI). In the normal brain, XIAP was predominantly expressed in the perinuclear region of neurons. Traumatized brains showed dramatic alterations in cellular and regional expression of XIAP early after injury. Stereologic analyses of the number of XIAP-positive cells within the hippocampus of both hemispheres showed a biphasic response. Immunoprecipitation and immunoblots of extracts derived from different brain regions demonstrated that a single ubiquitin modifies XIAP. Normal cortex contained significantly higher levels of monoubiquitinated XIAP than hippocampus. TBI induced alterations in levels of monoubiquitinated XIAP that correlated with changes in XIAP distribution and immunoreactivity, suggesting that monoubiquitination of XIAP may be a regulator of XIAP location or activity. Similar levels of Smac/DIABLO were present in lysates of normal and traumatized brains. These data demonstrate for the first time a region-specific regulation of XIAP monoubiquitination in the normal adult rat brain, and after TBI, that may be a key event in the regulation of XIAP function contributing to the pathogenesis following injury.
Apoptosis of neurons and glia contributes to the overall pathology of clinical and experimental brain trauma. Apoptotic (Yakovlev et al., 1997;Conti et al., 1998;Newcomb et al., 1999;Clark et al., 1999, 2000;Keane et al., 2001), and antiapoptotic (Keane et al., 2001) signaling pathways are activated after traumatic brain injury (TBI), and it is generally accepted that a shift in the balance between pro- and antiapoptotic protein factors toward the expression of proteins that promote cell death may be one mechanism underlying apoptotic cell death. The key effectors of apoptosis are the cysteine proteases in the caspase family, which systematically dismantle and eliminate cells. The members of the inhibitor of apoptosis (IAP) gene family suppress cell death through the dual action of binding to caspases via their BIR domains (Deveraux and Reed, 1999) and the ubiquitination of caspases via their RING finger (Huang et al., 2000;Suzuki et al., 2001); thus they act to raise the apoptotic threshold.
Of the eight known IAP family members, XIAP is probably the best characterized. Several recent studies with XIAP have shown that its biologic properties are not restricted to caspase inhibition but extend to the induction of cell cycle arrest (Levkau et al., 2001) and involvement in the signaling that is mediated by members of the transforming growth factor-β (TGF-β)/bone morphogenetic (BMP) superfamily (Salvesen and Duckett, 2002;Lotocki and Keane, 2002). XIAP has been reported to bind to the type-I BMP receptor (Yamaguchi et al., 1999) and the TGF-β receptor (Birkey-Reffey et al., 2001). Moreover, XIAP has been shown to augment TGF-β and BMP responses. With respect to TGF-β signaling, XIAP has been reported to activate nuclear factor of κB (NF-κB), c-Jun amino-terminal kinase (JNK), and SMAD-dependent transcription (Birkey-Reffey et al., 2001). However, it is not clear whether activation of these signaling pathways by XIAP confers prosurvival or proapoptotic effects. Despite intense investigation of the mechanism by which XIAP binds to caspases, an understanding of the caspase-independent function of XIAP in signaling functions has not been reconciled.
XIAP has been shown to induce the ubiquitination of caspase-3 (Suzuki et al., 2001). Protein ubiquitination involves the sequential action of ubiquitin activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin protein ligase (E3) (Hershko and Ciechanover, 1998). IAPs contain a COOH-terminal RING domain with E3 ligase activity (Yang et al., 2000) and have been shown to polyubiquitinate (Yang et al., 2000;Suzuki et al., 2001) and monoubiquitinate target proteins (Huang et al., 2000). Polyubiquitination (the sequential formation of oligomeric ubiquitin chains) directs target protein for degradation by the 26S proteasome, whereas the function of monoubiquitination (the addition of a single ubiquitin moiety) is less clear. Instead of sending proteins to their death in the proteasome, monoubiquitination has been shown to regulate diverse cellular processes that include the location and activity of cellular proteins (Hicke, 2001).
XIAP also may protect cells from damage by polyubiquitination of the proapoptotic molecule, Smac, targeting it for proteosomal degradation (MacFarlane et al., 2002). Thus some IAPs may serve as Smac-binding proteins, therefore effectively acting as apoptotic sinks (Salvesen and Duckett, 2002;Lotocki and Keane, 2002). This action of IAPs gives rise to the possibility that certain IAPs exert their antiapoptotic properties by sequestration of Smac or other IAP binding proteins. In Drosophila, IAP and Reaper-like proteins appear to mutually control each other's abundance through ubiquitin-mediated degradation (Olson et al., 2003). Not only do the Reaper-like proteins stimulate the ubiquination and degradation of IAPs, but the IAPs also stimulate the ubiquination and degradation of the Reaper-like proteins. This so-called two-way street interaction between IAPs and the Reaper-like proteins appears to be a significant factor in determining their biologic activities and antiapoptotic mode of action (Olson et al., 2003). Whether similar protein interactions occur in mammalian cells has not been reported.
In the authors' previous work, they reported that the death of CNS cells after TBI involves alterations in levels and cleavage of IAP proteins (Keane et al., 2001). To gain a better understanding of the physiologic functions of XIAP in the normal and traumatized brain, the present authors determined the cell-type expression, the subcellular distribution, the ubiquitination of XIAP, and the levels of Smac/DIABLO in the normal adult rat brain and in brains subjected to moderate TBI. The data demonstrate that XIAP is expressed predominantly in neurons in the normal brain and is monoubiquitinated in a region-specific fashion. TBI induced a dramatic alteration in the subcellular expression pattern of XIAP throughout the brain that correlated with levels of XIAP monoubiquitination. These studies suggest that monoubiquitination of XIAP may serve as a regulator of XIAP location and activity and that TBI induces alterations in XIAP subcellular distribution and monoubiquitination that may contribute to pathomechanisms after injury.
MATERIALS AND METHODS
Fluid percussion brain injury
The University of Miami Animal Care and Use Committee approved all procedures. Male Sprague-Dawley rats weighing 250 to 350 g (n = 63; 9 per group) were surgically prepared under halothane anesthesia and subjected to moderate (1.7–2.1 atm) levels of TBI, as described previously (Keane et al., 2001). After TBI, all rats were returned to their cages and allowed to recover from the surgical procedures. Sham-operated animals underwent all surgical procedures but were not traumatized. Naïve rats were anesthetized and perfused.
Perfusion fixation
Animals were anesthetized with 2% halothane and perfused with 300 ml of 4% paraformaldehyde solution, and the brains were removed and placed in 4% paraformaldehyde at 4°C for 20 hours. Brains were transferred to 20% sucrose in 0.1 M phosphate-buffer solution and kept in sucrose solution at 4°C until they were ready to be sectioned.
Immunohistochemistry
To determine the precise coexpression and cellular distribution of IAPs, immunostained sections of normal adult brains were examined with a Zeiss laser scanning confocal microscope (Zeiss, Inc., Thornwood, NY, U.S.A.). Adult male rats were perfused with 4% paraformaldehyde solution, as described in the previous section, and processed for cryostat sectioning (Leica SM 2000R Sliding Microtome, Leica Microsystems, Chantilly, VA, U.S.A.). Sections (35 μm) were blocked by treatment with appropriate preimmune serum. Tissue sections were rinsed with 0.1 M phosphate-buffered saline (PBS; pH 7.4). Sections were incubated overnight at 4°C with the primary antibodies: rabbit anti-XIAP (1:200; Cell Signaling, Beverly, MA, U.S.A.), mouse anti-neuronal nuclei (NeuN) (1:1000; Chemicon International, Temecula, CA, U.S.A.), mouse anti-neurofilament (1:1000; Chemicon International, Temecula, CA, U.S.A.), mouse anti-oligodendrocyte specific protein (1:1000; Chemicon International, Temecula, CA, U.S.A.), mouse anti-rat CD11b (1:1000; Accurate Chemical, Westbury, NY, U.S.A.), mouse anti-GFAP (1:1000; BD Biosciences, San Diego, CA, U.S.A.). Primary antibody binding was detected either with Alexa Fluor secondary antibody conjugates (1:200; Molecular Probes, Eugene, OR, U.S.A.) (confocal analysis) or with biotinylated secondary antibodies (1:200; Vector Laboratories Inc., Burlingame, CA, U.S.A.) (diaminobenzidine [DAB] staining). Controls lacking the primary antibody were run in parallel. Sections were coverslipped with Vectashield mounting medium (Vector Laboratories Inc., Burlingame, CA, U.S.A.) for confocal analysis and with Permount (Fisher Scientific, Pittsburgh, PA, U.S.A.) for DAB stained sections.
Stereology
Serial cryostat sections (35 μm) of the rat brain at the level of injury were divided into four groups; each group contained 10 sections representing the traumatized area. For estimation of the number of XIAP-immunopositive cells, DAB was used as the chromogen. Both the right (traumatized) and left (control) regions of hippocampus, CA1, CA3, CA4, and dentate gyrus (DG) in sham, naïve, and animals at 1, 6, and 24 hours, 3 and 7 days after TBI, were analyzed using an Axiophot (Zeiss, Inc., Thornwood, NY, U.S.A.) research microscope, furnished with fully motorized 3D LEP stage, Optronix cooled video camera, and MicroBrightfield Inc. Stereo-Investigator software package. To perform cell number estimation in the structure volume, the optical fractionator method and optical disector probe were used. Dimension of the optical disector was designed based upon the cell distribution on the section, and optical fractionator grid size was determined based upon the results of the preliminary count of the naïve brain sample to allow 120 to 300 counts per region of the hippocampus. All regions were analyzed separately. Immunoreactive cells were those that had degrees of cell body stain greater than controls lacking primary antibody. Total cell counts were analyzed using two-way ANOVA followed by Tukey-HSD post hoc comparison.
Coimmunoprecipitation
Sections of cerebral cortex and hippocampus (2 mm2) were homogenized in T-Per Protein Extraction Reagent (Pierce, Rockford, IL, U.S.A.) containing 10 mM DTT, 1 mM PMSF, 5 μg/ml leupeptin, 1 μg/ml pepstatin A, and 1 mM EDTA on ice for 1 hour. Equal amounts of protein in lysates were immunoprecipitated with anti-ubiquitin monoclonal antibodies (Chemicon International, Temecula, CA, U.S.A.) using the Catch and Release Immunoprecipitation System (Upstate Cell Signaling Solutions #17-319, Charlottesville, VA, U.S.A.) according to the manufacturer's instructions. The anti-ubiquitin monoclonal antibody recognizes both monoubiquitinated and polyubiquitinated proteins (Oberdorf et al., 1999). Eluates were mixed with 2X Laemelli sample buffer and used for immunoblot analysis.
Immunoblot analysis
Immunoprecipitates or equal amounts of protein in lysates were resolved on 8.5% SDS-PAGE, transferred to polyvinylidene fluoride (PVDF) membranes, and placed in blocking buffer (0.1% Tween-20, 0.4% I-block in PBS, Applied Systems, Foster City, CA, U.S.A.) for 1 hour (Keane et al., 1997). One hundred microliters of cerebrospinal fluid (CSF) samples (n = 3 per group) were obtained by puncturing the cisterna magna with a 27-gauge needle connected to PE50 tubing, and equal amounts of protein were resolved on SDS-PAGE. Recombinant XIAP (R&D Systems, Minneapolis, MN, U.S.A.) was run as the control. Membranes were incubated with anti-XIAP (1:250; BD Biosciences, San Diego, CA, U.S.A.) or anti-Smac (2 μg/ml; Zymed Laboratories, South San Francisco, CA, U.S.A.). β-tubulin was used as internal standard and control for protein loading. Visualization of the signal was made by enhanced chemiluminescence.
RESULTS
XIAP expression in the normal adult rat brain
Figure 1 shows confocal images of the cell type expression and regional distribution of XIAP in the normal adult rat brain. Sections were stained for XIAP (red) and the neuronal nuclear marker, NeuN (green). XIAP immunoreactivity colocalized with NeuN in all brain regions examined including cortex (Rows 1 and 3) and hippocampus (Row 2), indicating that XIAP is expressed in neurons in the normal adult rat brain. Intense XIAP immunoreactivity was localized in the perinuclear region, and punctate staining was present in the cell cytoplasm (Row 3). Weak XIAP staining was detected in some neuronal processes. To determine whether XIAP was expressed in glial cells, sections were double labeled with antibodies against XIAP and antibodies against glial specific proteins: GFAP (astrocytes), OX42 (microglia), or oligodendrocyte specific protein (oligo) (Fig. 2). In contrast with neurons, XIAP immunoreactivity was not detected in oligodendrocytes (Row 1), astrocytes (Row 2), or microglia (Row 3). However, in a previous study (Keane et al., 2001), a very small number of oligodendrocytes lining the ventricles were XIAP-immunoreactive. These studies demonstrate that XIAP is predominantly expressed in neurons of the normal adult rat brain.
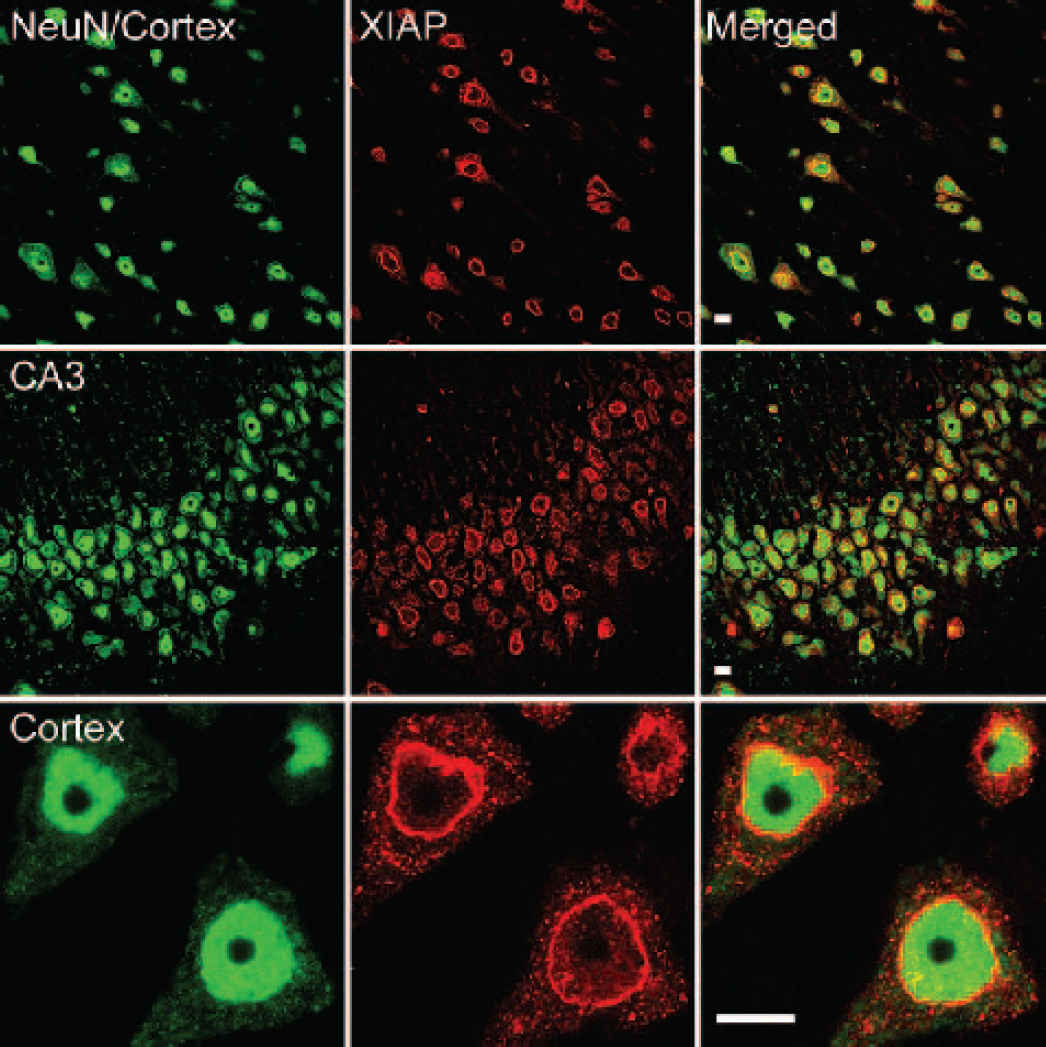
XIAP expression in cortical and hippocampal neurons of normal adult rat brain. Confocal analyses with anti-XIAP antibody (Column 2, red) and neuronal nuclear marker, NeuN (Column 1, green), and merged images (Column 3, yellow/orange). XIAP colocalizes in neurons in the cortex (Rows 1 and 3) and hippocampus (Row 2). Row 3 images show XIAP is primarily expressed in the perinuclear compartment with punctate staining in the cell body. Bars = 10 μm. NeuN, anti-neuronal nuclei.
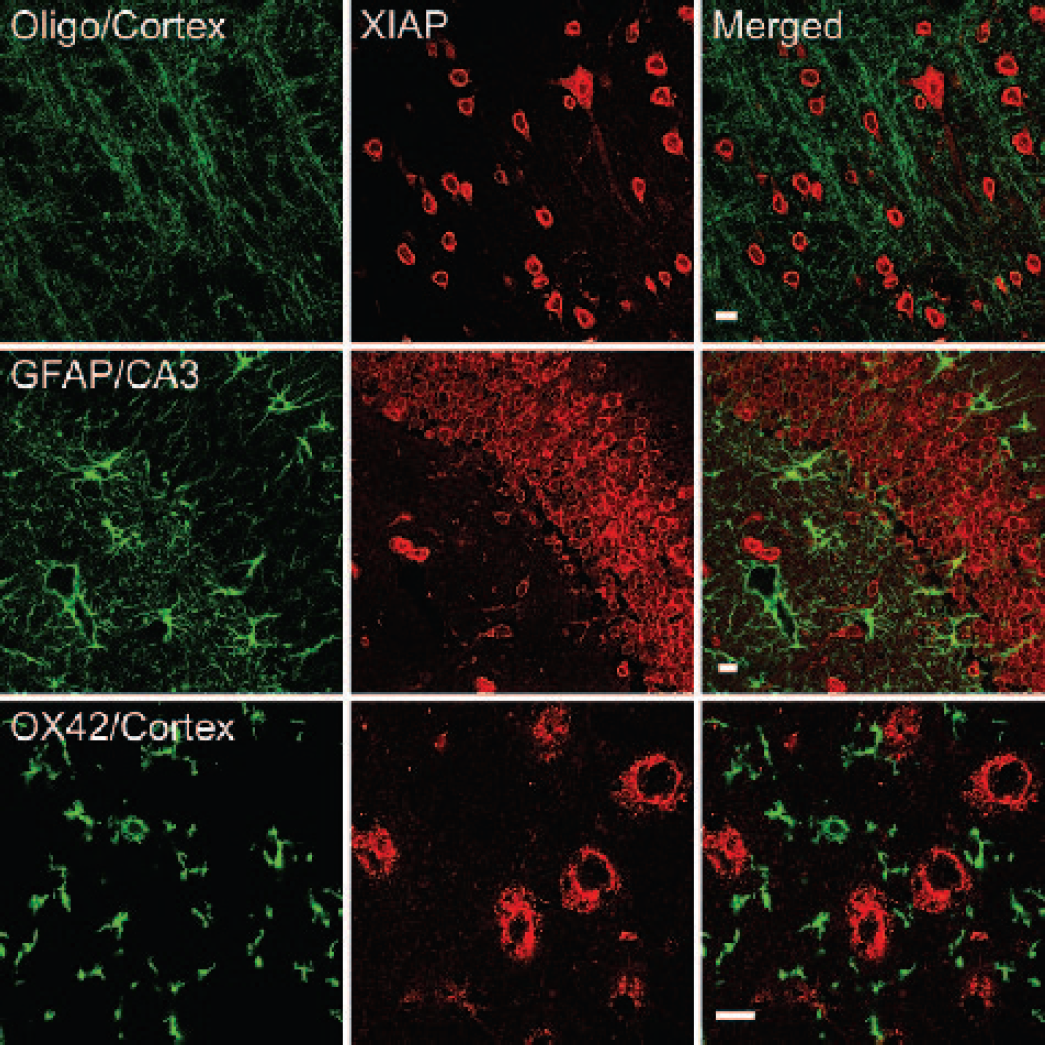
XIAP does not colocalize in glial cells. Confocal images of sections stained with antibodies specific for oligodendrocyte (oligo), astrocytes (GFAP) and microglia markers (OX42); Glial markers (Column 1, green); XIAP (Column 2, red). Note lack of yellow/orange in merged images (Column 3). Bars = 10 μm.
XIAP expression in the rat brain after TBI
To examine whether TBI induced changes in the pattern of expression of XIAP, rats were subjected to moderate TBI, and brains were examined by confocal microscopy (Fig. 3). Early after injury, there was a dramatic change in the pattern of XIAP and NeuN staining. Neurons in the injury epicenter exhibited an irregular triangular morphology and alteration of NeuN nuclear staining (Rows 1 and 2), hallmarks of necrotic cell death (Zipfel et al., 2000). XIAP immunoreactivity in the perinuclear region was dramatically reduced, but an increase in XIAP immunoreactivity was seen in the extracellular space that exhibited a punctate appearance (Rows 1 and 5). A significant proportion of neurons in the injured cortex exhibited hallmarks of apoptotic cell death that included nuclear margination and fragmentation (Rows 3 and 4, arrows). These apoptotic neurons (Fig. 3, Row 3 inset, arrowhead) exhibited a different pattern of XIAP immunoreactivity from necrotic neurons (Rows 1 and 2). Perinuclear XIAP staining was not present, but only punctate staining in the cytoplasmic compartment was observed. Cortical neurons located ipsilaterally but away from the lesion, and neurons in the contralateral hemisphere showed a NeuN staining pattern (Row 5) similar to normal neurons (Fig. 1, Row 3). Of importance is the fact that the intensity and pattern of XIAP expression in these neurons and neurons in the hippocampus was strikingly altered with a decrease in perinuclear localization and an increase in punctate extracellular staining (Fig. 3), indicating that alterations in the neuronal subcellular distribution of XIAP occurs in both hemispheres in response to TBI. Additionally, a proportion of hippocampal neurons (Row 4, arrows) exhibited nuclear changes indicative of apoptosis. Oligodendrocytes, astrocytes, and microglia failed to stain with anti-XIAP antibodies after TBI as in the normal adult rat brain (data not shown). These results demonstrate that TBI induces dramatic alterations in the XIAP expression pattern in neurons and that different XIAP expression patterns occur in normal, apoptotic, and necrotic neurons. Because punctate XIAP staining was seen in the extracellular space after TBI, the present authors investigated the possibility that XIAP was present in cerebrospinal fluid (CSF). CSF samples from naive, sham, and traumatized rats failed to reveal detectable levels of XIAP by immunoblot analysis using anti-XIAP antibodies (data not shown).
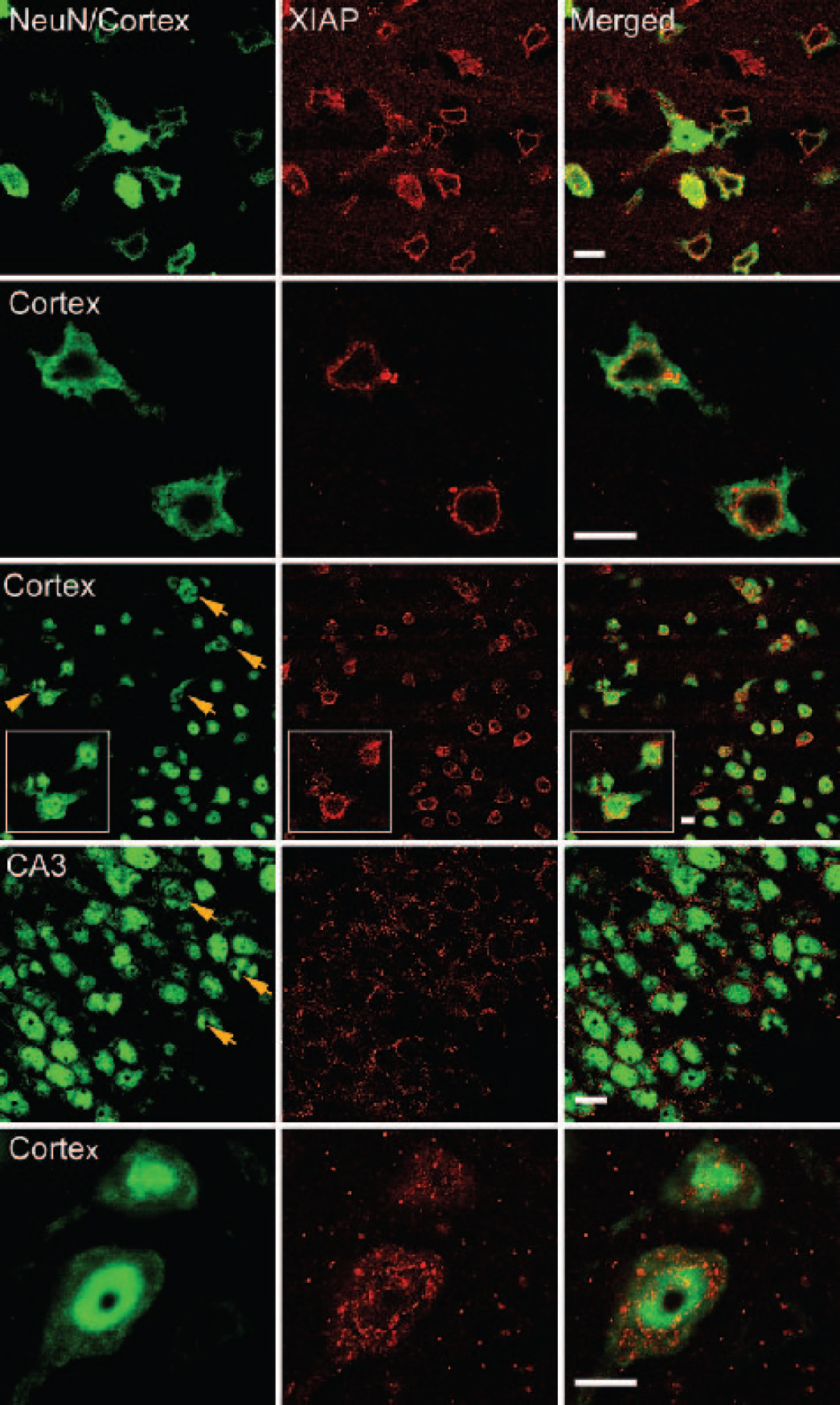
TBI induces a dramatic change in the pattern of expression of XIAP in necrotic (Rows 1, 2), apoptotic (Rows 3 and 4) and healthy neurons (Row 5). Confocal images of brain sections stained with anti-NeuN (Column 1, green), anti-XIAP (Column 2, red), and merged images (Column 3, yellow/orange). By 6 hours after injury, neurons in the lesion epicenter showed a necrotic morphology and a strong decrease in NeuN and perinuclear XIAP immunoreactivity (Rows 1 and 2). A significant number of cortical (Row 3) and hippocampal (Row 4) neurons ipsilateral to the injury demonstrated apoptotic bodies (arrows) and nuclear margination (Row 3 inset, arrowhead). Apoptotic neurons showed a different NeuN and XIAP staining pattern from necrotic neurons. Neurons in the contralateral hemisphere (Row 5) exhibited a healthy morphology and NeuN staining pattern, but showed reorganization of XIAP in the perinuclear compartment and an increase in punctate staining in the cytoplasm and extracellular space. Bars = 10 μm. NeuN, anti-neuronal nuclei.
Stereologic analyses of XIAP immunoreactive cells in hippocampus
To quantify changes in XIAP expression in the hippocampus after TBI, sections were stained with anti-XIAP using DAB as the chromogen. TBI induced a biphasic response in XIAP immunoreactivity in the hippocampus (Fig. 4) in which the intensity of staining was dramatically reduced early after injury, increased by 24, and then gradually decreased by 3 days after TBI. To quantify the changes in the number of immunopositive cells in the hippocampus after TBI, DAB stained sections were counted using stereologic analyses (Fig. 5). There were no significant differences between naive and sham-operated animals; therefore, TBI groups were compared with the sham group. Within 1 hour after TBI, there was a significant (P < 0.001) decrease in the number of XIAP immunoreactive cells in all regions of the hippocampus. This decrease continued by 6 hours and was also observed in immunofluorescently labeled sections analyzed by confocal microscopy (Fig. 3). However, by 24 hours, the number of XIAP positive cells had significantly (P < 0.001) increased. The increase in the number of XIAP-positive cells supports our previous report that TBI produces an increase in XIAP levels in hippocampal lysates at 24 hours postinjury (Keane et al., 2001). By 3 and 7 days, the number of XIAP positive cells had again declined. Thus it appears that moderate TBI induces a biphasic XIAP response in both the ipsilateral and contralateral areas of the hippocampus.
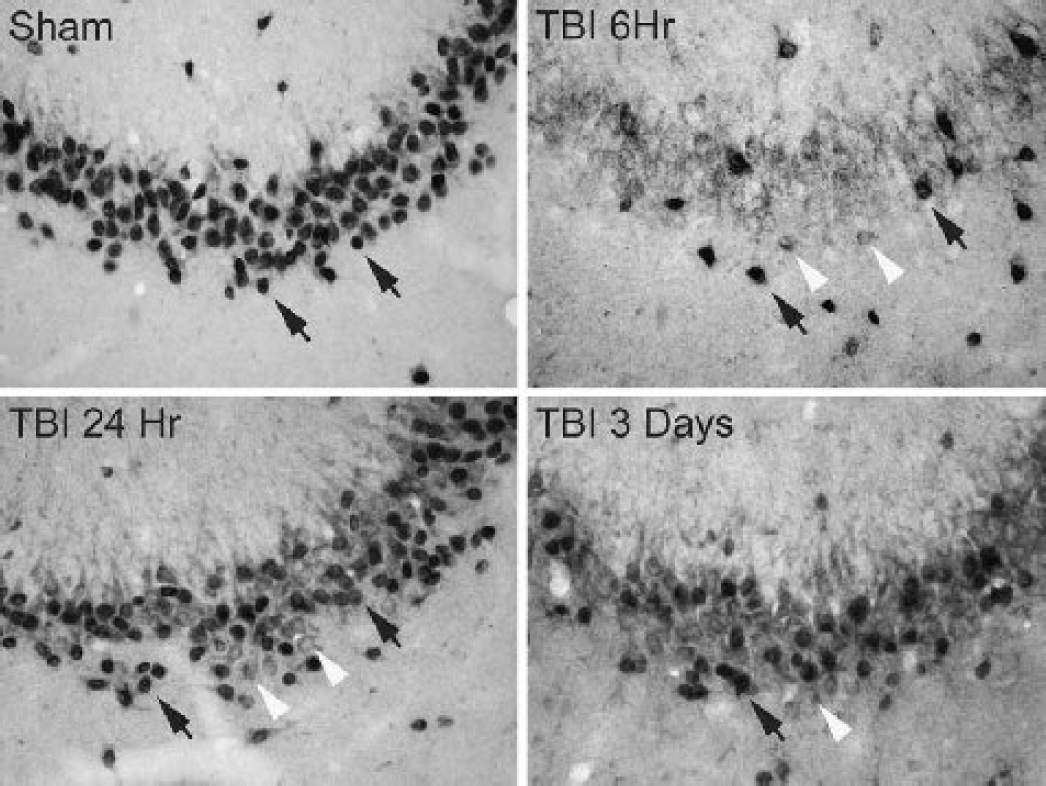
Temporal expression of XIAP in CA3 hippocampal neurons of sham and traumatized rats, 6 hours, 24 hours, and 3 days after TBI. Sections were immunostained with anti-XIAP and DAB as the chromogen. Black arrows indicate cells that were scored as XIAP-positive and white arrowheads indicate cells not counted. Magnification ×400. TBI, traumatic brain injury; DAB, diaminobenzidine.
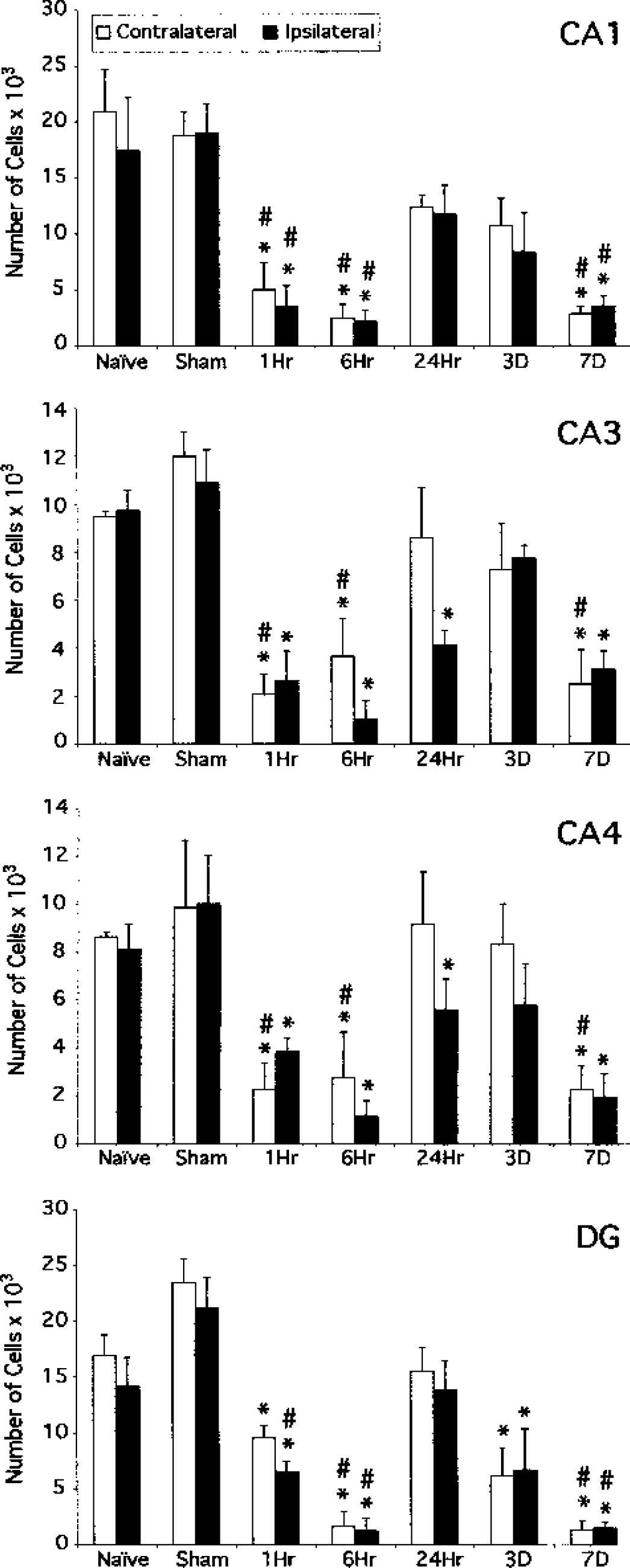
Stereologic analyses of XIAP temporal expression in the hippocampus (n = 6 per group) demonstrate a biphasic response. Black bars represent injury side (ipsilateral); white bars represent contralateral to injury side. The hippocampus was subdivided into CA1, CA3, CA4, and DG, and the number of XIAP-positive cells in each region was counted. XIAP immunoreactivity of all four regions decreased significantly in both sides of the hippocampus 1 and 6 hours after injury. By 24 hours, the levels of XIAP had increased significantly but did not reach those of sham controls. By 3 and 7 days postinjury, the number of XIAP positive cells again declined. For statistical analyses, the contralateral side was compared with the contralateral side, and the ipsilateral was compared with the ipsilateral side. There were no statistical differences between sham and naïve groups. ∗ P < 0.001vs. sham; #P < 0.001 vs. 24-hour group. DG, dentate gyrus.
Ubiquitination of XIAP in the normal rat brain and after TBI
To determine whether the ubiquitination was involved with XIAP regulation in the normal brain and after TBI, cortical and hippocampal lysates were immunoprecipitated with anti-ubiquitin and then immunoblotted with anti-XIAP antibodies. Recombinant XIAP was run as control (Fig. 6A). Anti-XIAP antibodies detected the recombinant XIAP protein with an apparent molecular weight of 57 kDa. However, samples immunoprecipitated with anti-ubiquitin contained slower migrating proteins, suggesting the regulated addition of a single 76-amino-acid (approximately 8 kDa) ubiquitin moiety. Cortical lysates of sham-operated animals contained increased levels of monoubiquitinated XIAP, indicating that the sham operation promotes monoubiquitination of XIAP in both hemispheres. By 1 hour after TBI, the levels of ubiquitinated XIAP in cortices increased and were maintained until 7 days after the injury. In contrast, hippocampal lysates of normal and sham-operated rats contained barely detectable levels of ubiquitinated XIAP. However, increased levels of ubiquitinated XIAP were present in hippocampal lysates by 24 hours and 3 days postinjury. By 7 days, the levels of ubiquitinated XIAP decreased. The increase in XIAP ubiquitination at 24 hours after TBI in the hippocampus correlates with the increased number of XIAP-positive hippocampal neurons (Fig. 5). These data demonstrate for the first time a region-specific regulation of XIAP ubiquitination in the normal and injured brain.
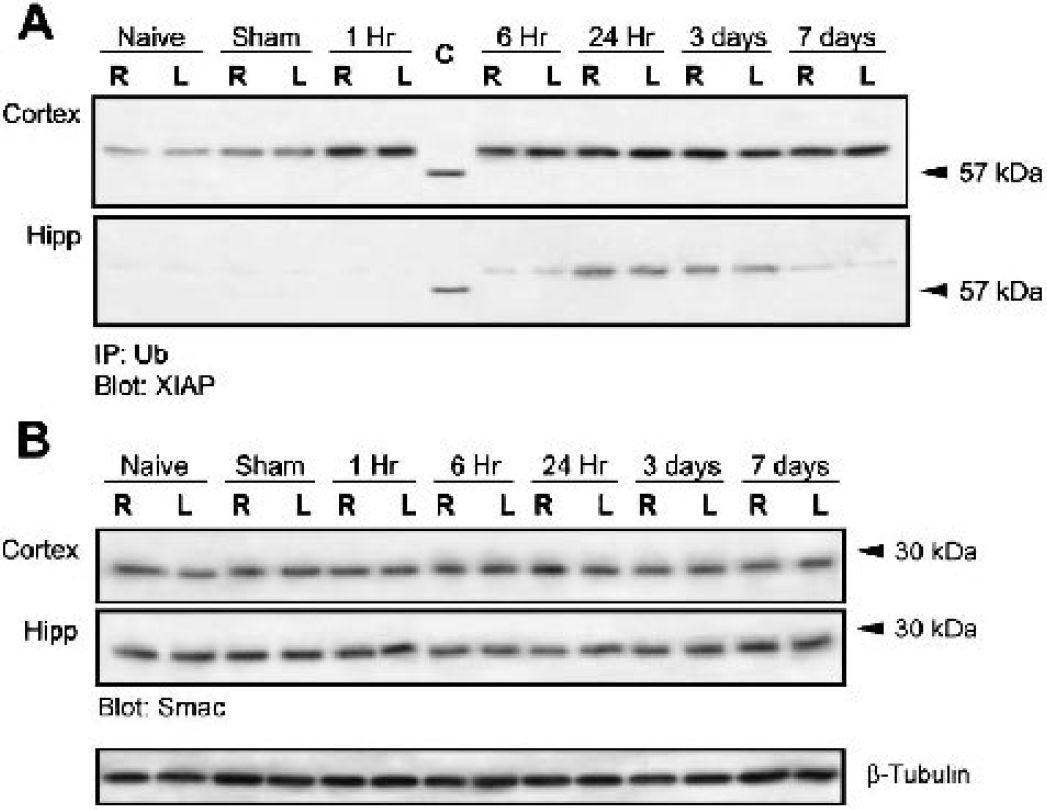
TBI promotes XIAP monoubiquitination
TBI does not induce alterations in the levels of Smac/DIABLO
Because XIAP has been shown to ubiquitinate Smac in vitro, targeting it for proteasomal degradation, the present authors determined whether TBI induced alterations in Smac/DIABLO expression that may contribute to proapoptotic mechanisms. As shown in Fig. 6B, lysates of normal and traumatized cortex and hippocampus showed similar levels of Smac/DIABLO, suggesting that Smac/DIABLO is not rapidly degraded by the proteasome after TBI.
DISCUSSION
These results demonstrate that XIAP is expressed in neurons of the CNS and is localized to the perinuclear region with punctate staining present around the neuronal soma. Weak XIAP reactivity was detected in some neuronal processes. Astrocytes, microglia, and the majority of oligodendrocytes did not show detectable levels of XIAP, suggesting that XIAP may function physiologically in neurons and some glia. These findings are consistent with and extend earlier reports describing the cellular expression of XIAP in neurons (Keane et al., 2001;Korhonen et al., 2001;Shibata et al., 2002) but are inconsistent with the report describing XIAP expression in astrocytes (Korhonen et al., 2001).
Moderate TBI rapidly induced a dramatic decrease in the intensity of perinuclear XIAP immunoreactivity in neurons and an increase in punctate staining in the cell soma and extracellular region, indicating that XIAP distributes to sites around the nucleus and extracellular space after a traumatic insult. This change in XIAP expression pattern occurred rapidly and was observed in all healthy neurons examined, indicating a CNS physiologic response to TBI. Different patterns of XIAP redistribution were observed in healthy, apoptotic, and necrotic neurons within and surrounding the lesion epicenter, suggesting that XIAP cellular distribution may be critical for cell survival. Although parasagittal fluid-percussion brain injury leads to histopathologic damage, primarily in the ipsilateral hemisphere, contralateral changes have been detected. For example, after moderate TBI, mild reductions in local cerebral metabolic rate of glucose have been observed (Dietrich et al., 1994). Likewise, hemodynamic depression is seen contralaterally as early as 30 minutes after fluid-percussion injury in rats (Dietrich et al., 1998). Also, mild but significant neuronal cell loss has been described in the contralateral hippocampus after fluid percussion brain injury (Lowenstein et al., 1992). Thus the observed changes in XIAP in the contralateral hemisphere may be a response to hemodynamic and metabolic stresses that occur remotely from the primary site of damage.
Stereologic analyses revealed a biphasic regulation of XIAP in the hippocampus after TBI in which the numbers of XIAP-positive neurons decreased sharply early after TBI but increased by 24 hours after the insult. Several other studies have demonstrated the involvement of XIAP in pathologic conditions of the brain (Korhonen et al., 2001;Shibata et al., 2002). A biphasic regulation of XIAP has been reported in the hippocampus following kainic acid-induced neuronal death (Korhonen et al., 2001), with XIAP expression increasing and then decreasing after lesion. Thus, it is possible that different injury paradigms may elicit different XIAP expression patterns in hippocampal neurons, but the physiologic basis for this response remains unclear.
The authors' observations suggest mechanisms that regulate XIAP location and function. In the normal rat brain and in brains subjected to moderate TBI, the authors demonstrate that a single ubiquitin modifies XIAP. Monoubiquitination has been shown to regulate endocytosis, viral budding, and protein location and activity (Hicke, 2001), but how monoubiquitination affects these diverse cellular responses remains unknown. Because alteration in monoubiquitination of XIAP was associated with alterations in XIAP expression patterns after TBI, this favors the possibility that XIAP monoubiquitination may be involved in regulation of XIAP subcellular distribution in neurons. It is tempting to hypothesize that monoubiquitination of XIAP may regulate XIAP function in signaling cascades, and work is underway to test this hypothesis.
During apoptosis, Smac, an IAP-binding protein, is released from mitochondria and potentiates apoptosis by relieving IAP inhibition of caspases. The RING finger domain of XIAP possesses E3 ligase activity and binds tightly to Smac. Both the association of XIAP with Smac and the RING finger domain of XIAP are essential for ubiquitination of Smac and proteasomal degradation (MacFarlane et al., 2002). Because XIAP targets proapoptotic molecules for proteasomal degradation, the present authors examined whether TBI induced a significant decrease in the levels of Smac/DIABLO. Similar levels of Smac/DIABLO were present in lysates of normal and traumatized brains, indicating that ubiquitination and degradation of Smac/DIABLO does not play an appreciable role in modulating apoptosis after TBI. However, the authors cannot rule out the possibility that degradation of small amounts of Smac/DIABLO undetected by the authors' assay are involved in regulation of apoptosis.
In a previous study, the present authors demonstrated that the cell death after TBI involves activation of multiple caspases in both the intrisinic and extrinsic apoptotic pathways (Keane et al., 2001). Associated with the appearance of processed caspases-8, −9, and −3 was the cleavage of the full-length 57-kD XIAP protein into a 30-kDa fragment as early as 1 hour after TBI. Cleavage of XIAP produces an N-terminal BIR1-2 fragment with reduced ability to inhibit caspases-3 and −7, therefore diminishing its ability to suppress apoptosis (Deveraux et al., 1999). Taken with the current findings, it appears that both cleavage and monoubiquitination of XIAP are involved with cell death after moderate TBI. Understanding the physiologic functions and modification of IAPs and interactions with binding proteins in signaling cascades could lead to therapeutic treatment paradigms targeting specific pathomechanisms after TBI.
Footnotes
Acknowledgments
The authors thank Dr. Helen M. Bramlett for assistance with statistics.