
Abstract
The authors developed a novel positron emission tomography method to estimate changes in the synaptic level of dopamine ([DA]) induced by direct dopamine agonists (for example, apomorphine) in patients with Parkinson disease. The method is based on the typical asymmetry of the nigrostriatal lesion that often occurs in Parkinson disease. Using the between-side difference (ipsilateral (I) and contralateral (C) putamen to the more affected body side) of the inverse of the putamen [11C]raclopride binding potential (BP), the authors obtained
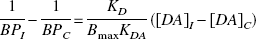
at baseline (that is, before apomorphine administration) and
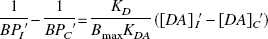
after apomorphine administration (assuming the concentration of apomorphine is equal in both putamina). The between-side difference in the estimated synaptic concentration of dopamine (diff[DA]) should remain constant unless apomorphine affects dopamine release differently between the two sides. The authors found that apomorphine given subcutaneously at doses of 0.03 and 0.06 mg/kg induced significant changes in their estimate of diff[DA] (P < 0.05). Such changes were more pronounced when only patients with a stable response to levodopa were considered (P < 0.01). These findings provide in vivo evidence that direct dopamine agonists can inhibit the release of endogenous dopamine. The authors propose that this effect is mainly mediated by the activation of presynaptic D2 /D3 dopamine receptors.
The progression of Parkinson disease (PD) is invariably accompanied by changes in the therapeutic response to levodopa (Marsden and Parkes, 1976) and to dopamine agonists such as apomorphine (Verhagen Metman et al., 1997). In the case of apomorphine, such changes include a shortened response duration, a steeper dose-response curve, and a narrower therapeutic window (Verhagen Metman et al., 1997).
The physiologic alterations underlying these changes have been the subject of some studies. One hypothesis is that they are primarily caused by postsynaptic alterations in receptor density or sensitivity (Bravi et al., 1994; Verhagen Metman et al., 1997; Zigmond et al., 1990). However, some recent studies suggest that changes in presynaptic autoregulatory function may accompany disease progression and that these changes may contribute to the development of altered therapeutic response (Ekesbo et al., 1999; Tedroff et al., 1996).
These studies were based on the idea that dopamine synthesis, storage, and release are “autoregulated” in such a way as to maintain the synaptic concentration of dopamine. There is substantial evidence for this autoregulation and for its mediation by dopamine receptors. For instance, in studies of the nigrostriatal system in various species, dopamine agonists have been shown to negatively regulate tyrosine hydroxylase activity (Cho et al., 1997), aromatic amino acid decarboxylase activity (Hadjiconstantinou et al., 1993), electrical firing (Bunney et al., 1973), and cerebrospinal fluid dopamine levels (Przedborski et al., 1995).
In a recent study, Ekesbo et al. (1999) found that although patients with early PD were able to down-regulate the uptake of L-[11C]-dopa in response to apomorphine challenge, this ability was lost in patients with more advanced disease. Based on these findings, they proposed that there might be a loss of autoreceptor function with the progression of PD.
Because the ultimate purpose of presynaptic autoregulatory function is to regulate the synaptic concentration of dopamine, a loss of this function accompanying disease progression should lead to a loss in the ability of dopamine agonists such as apomorphine to negatively regulate the concentration of dopamine in the synapse. To test this hypothesis, the authors designed a positron emission tomography (PET) study using [11C]raclopride to estimate apomorphine-induced changes in the synaptic concentration of dopamine and to explore their dependence on disease severity. This technique is based on the ability of endogenous dopamine (and direct dopamine agonists) to compete with [11C]raclopride for D2/D3 dopamine receptors; the use of this technique to measure changes in the synaptic concentration of dopamine in humans in vivo has been well established (Breier et al., 1997; Laruelle, 2000; Seeman et al., 1989; Stoessl and Ruth, 1999; Volkow et al., 1994, 1997).
Comparisons between individuals are likely to be confounded by individual differences in disease duration, treatment protocol, age, and apomorphine pharmacokinetics that may obscure subtle relations between apomorphine response and disease severity. Therefore, rather than comparing different individuals, the authors compared the response to apomorphine between the two putamina in patients with asymmetric, but bilaterally symptomatic, PD. In these patients, the putamen contralateral to the side of the body with the more severe symptoms (henceforth referred to as the contralateral putamen) is presumed to be more affected than that ipsilateral to the more severely affected body side (henceforth referred to as the ipsilateral putamen). Both putamina should be identical in all respects except for disease severity and pathophysiologic correlates (for example, dopamine synthesis capacity). Therefore, comparisons should not be confounded by factors such as those outlined above.
MATERIALS AND METHODS
Patient details
Five patients (4 male, 1 female) with clinically definite PD (as defined by Calne et al., 1992) and clear asymmetry of parkinsonian signs were studied. All subjects gave written informed consent. The study was approved by the University of British Columbia Ethics Committee. Demographics and clinical details are summarized in Table 1. All patients were taking levodopa with a decarboxylase inhibitor at the time of the study. In addition to levodopa, 2 patients also were taking tolcapone (400 and 600 mg/day). There were no other antiparkinsonian medications, but 1 patient was also taking a psychoactive drug for depression (sertraline, 100 mg/day). Quantitative measurements based on the Modified Columbia Scale (MCS) (Duvoisin, 1971) during the “off” state (after 12 to 18 hours without medications) were performed on the morning of the scans.
Demographics and clinical characteristics of patients
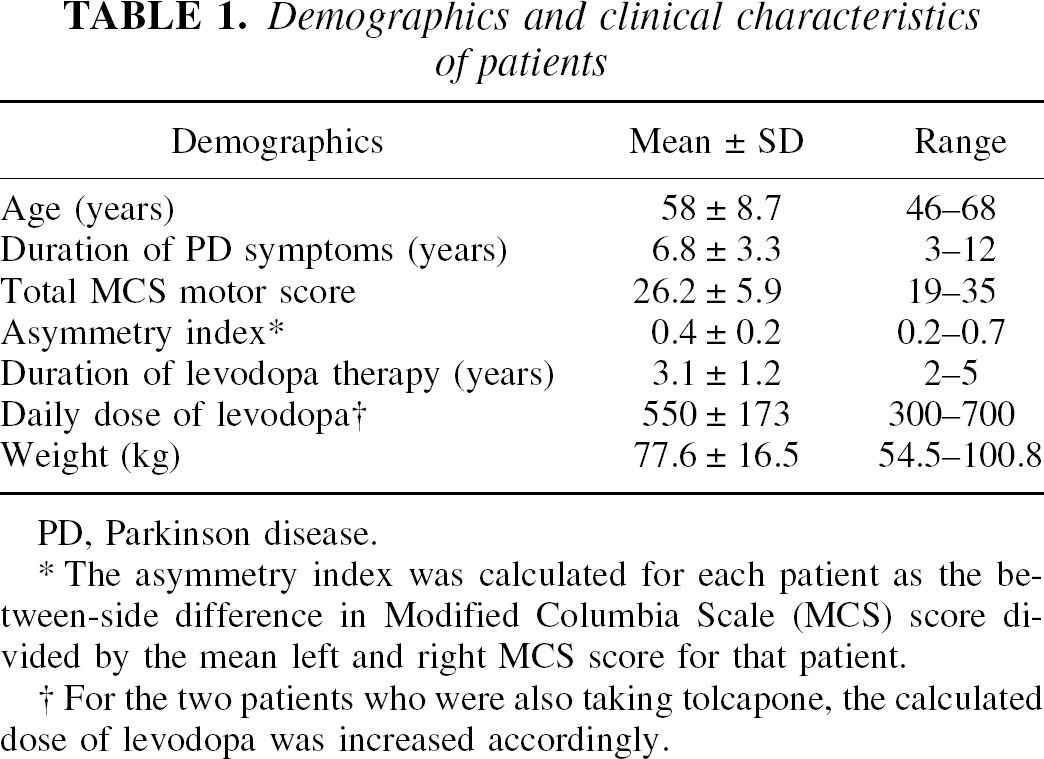
PD, Parkinson disease.
The asymmetry index was calculated for each patient as the between-side difference in Modified Columbia Scale (MCS) score divided by the mean left and right MCS score for that patient.
For the two patients who were also taking tolcapone, the calculated dose of levodopa was increased accordingly.
Positron emission tomography protocol
All patients underwent three consecutive [11C]raclopride PET scans on the same day according to the following protocol: the first scan was performed 12 to 18 hours after withdrawal of all antiparkinsonian medications, the second scan was performed 2.5 hours after the start of the first scan, and the third scan was performed 2.5 hours after the start of the second scan. At the beginning of scan 2, patients were administered 0.03 mg/kg apomorphine subcutaneously; at the beginning of scan 3, patients were administered 0.06 mg/kg apomorphine subcutaneously. These doses were chosen because they span the range of therapeutic doses for a wide variety of patients (Verhagen Metman et al., 1997). All patients were pretreated with 60 mg/day of oral domperidone for 2 days before the scans. In addition, 45 minutes before both scans 2 and 3, the patients were given 20 mg oral domperidone. This drug is a peripherally selective D2 antagonist used to combat the peripheral effects of apomorphine. The time between scans was to allow for the decay of [11C]raclopride and the clearance of apomorphine (de la Fuente-Fernández et al., 2001).
All PET scans were performed in three-dimensional mode using an ECAT 953B/31 tomograph (Siemens Canada/CTI Knoxville, TN, U.S.A.). Attenuation correction was achieved by performing a 15-minute transmission scan using68 Ge rods. For the assessment of D2 binding, 16 sequential frames were obtained over 60 minutes, starting at the time of intravenous injection of 5 mCi [11C]raclopride (specific activity, mean ± SEM = 4438.7 ± 681.4 Ci/mmol at time of injection). The three-dimensional emission data were reconstructed using previously described algorithms with corrections for scatter and detector normalization (Oakes et al., 1998; Sossi et al., 1998). An integrated image with 31 planes (each 3.37-mm-thick) was made from the emission data (from 30 to 60 minutes) for each subject. On the summed image obtained from the 5 axial planes in which the striatum was best visualized, one circular region of interest (ROI) of 61.2 mm2 was positioned by inspection on each caudate nucleus and adjusted to maximize the average ROI activity. Three circular ROIs of 61.2 mm2 were placed without overlap along the axis of each putamen (for rostral, intermediate, and caudal putamen) and were similarly adjusted. The background activity was averaged from a single elliptical ROI (2017 mm2) drawn over the cerebellum on the sum of 2 contiguous axial planes. The binding potential (BP = Bmax /KDapp) was determined using a graphical approach and tissue input function as described by Logan et al. (1996). The reproducibility of [11C]raclopride PET coupled with Logan analysis has been shown previously to be high (Volkow et al., 1993).
Data analysis
Assumptions.
The current study focuses on changes in the putamen because this part of the striatum is most involved in motor control; asymmetric PD patients were identified based on their clinical status. The analysis described below makes use of three assumptions that merit discussion. Namely, it supposes (1) that the interaction between endogenous dopamine and [11C]raclopride is purely competitive and homogeneous, (2) that the D2 receptor density in the contralateral putamen is greater than or equal to that in the ipsilateral putamen, and (3) that apomorphine is distributed evenly between both putamina. The terms used below are defined in Table 2. Implicit in the first assumption is that KD and KDA remain unaltered. Hence, identical constants are assumed for ipsilateral and contralateral putamen. The same applies for KAPO.
Definition of terms

For all the terms defined above, the superscript' in the text indicates a value pertaining to a scan in the presence of apomorphine. The subscripts I and C refer to values in the putamen ipsilateral and contralateral, respectively, to the more affected side of the body.
Several studies have examined the nature of the interaction between dopamine and raclopride at D2 receptors. Some investigators have reported a noncompetitive interaction between dopamine and raclopride in vitro, which has been explained by referring to dopamine-induced interconversion between two affinity states of the D2 receptor (Hall et al., 1990; Seeman et al., 1989, 1990). However, the one study to examine this question in primates in vivo (a situation much more relevant to the current study) demonstrated a purely competitive interaction at a homogenous population of receptors (Ginovart et al., 1997). The discrepancy may be explained by noting that the percentage of D2 receptors in the high-affinity state is anywhere from 28% (Wreggett and Seeman, 1984) to 55% (DeLean et al., 1982) in vitro, whereas approximately 90% of D2 receptors are in the high-affinity state in vivo (Richfield et al., 1986), thus forming a nearly homogenous population.
Regarding the relative magnitudes of Bmax(C) and Bmax(I), there is substantial evidence to support the assertion that Bmax(C) is roughly equal to or is greater than Bmax(I). Several groups have presented imaging studies on patients with PD supporting either that Bmax(C) = Bmax(I) (Ichise et al., 1999) or that Bmax(C) > Bmax(I) (Antonini et al., 1994; Rinne et al., 1993). Because these studies based their conclusions solely on comparisons of differences in BP between ipsilateral and contralateral putamen, they did not differentiate whether differences in BP were caused by differences in Bmax or in receptor occupancy by dopamine (that is, differences in KDapp). However, these studies are consistent with animal studies in which Bmax has been explicitly measured. These show either that Bmax(C) = Bmax(I) (Hume et al., 1995) or that Bmax(C) > Bmax(I) (Creese and Snyder, 1979). The authors are unaware of any studies demonstrating that Bmax(C) < Bmax(I). Furthermore, PET studies using both [11C]N-methylspiperone and [11C]raclopride suggest that increases in putamen D2 binding in patients with early PD reflect increased receptor density rather than differences in occupancy (Kaasinen et al., 2000).
Regarding the distribution of apomorphine between both putamina, there is little reason to suppose that the concentration of apomorphine should be greater in one putamen than the other. The only remotely plausible situation in which this would occur is if there were differences in perfusion between the ipsilateral and contralateral putamen (Wolfson et al., 1985). Using [99m TC]-HM-PAO single photon emission computed tomography to monitor striatal perfusion in patients with asymmetric PD, Pizzolato et al. (1993) demonstrated that this is not the case. Using PET, Perlmutter and Raichle (1985) also reported no significant local blood flow changes in the putamen of hemiparkinsonian patients. Most importantly, even if there were some asymmetry in putamen blood flow, it should affect apomorphine and [11C]raclopride delivery equally. Consequently, the effect of blood flow asymmetry, if any, already would be present at baseline for [11C]raclopride delivery, and the degree of competition between apomorphine and [11C]raclopride for D2 /D3 dopamine receptors would tend to remain the same in both sides of the putamen.
Analysis.
It can be shown that the BP as determined by the Logan analysis is related to Bmax and KD as follows (Logan et al., 1996):
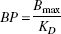
In the presence of dopamine, KD in Eq. 1 is replaced by an “apparent” dissociation rate constant defined by Eq. 2 (Bylund and Yamamura, 1990):
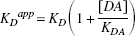
Substituting KDapp from Eq. 2 for KD in Eq. 1 and taking the inverse results in
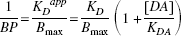
In the presence of apomorphine, KDapp takes the form
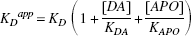
so
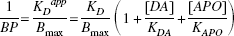
Equations 3 and 5 indicate that the inverse of the BP could be used as an estimate of the synaptic concentration of dopamine if other factors potentially affecting the binding (age, treatment, and so on) could be maintained constant. Thus, by comparing the inverse of the BP between ipsilateral and contralateral putamen and under different conditions, one should be able to draw inferences about the corresponding synaptic concentrations of dopamine. This can be seen most clearly when Bmax(C) = Bmax(I), although, as demonstrated in the Appendix, similar conclusions can be drawn when Bmax(C) > Bmax(I).
From Eq. 3, when Bmax(I) = Bmax(C) = Bmax and apomorphine is absent,
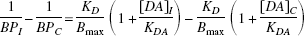
Rearranging results in
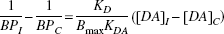
From Eq. 5, in the presence of apomorphine,
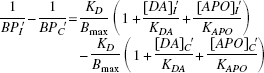
Assuming that the concentrations of apomorphine in both putamina are equal, the
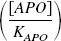
terms in Eq. 8 cancel out and Eq. 8 can be rewritten as
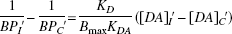
To see how Eqs. 7 and 9 can be used to compare the apomorphine-induced drop in synaptic dopamine in the ipsilateral and contralateral putamen, consider the case where this drop is the same on both sides:
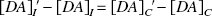
Rearranging results in
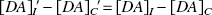
Substituting this into Eqs. 7 and 9 leads directly to the conclusion that
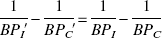
From the above consideration, it can be seen that changes in the difference between 1/BPI and 1/BPC because of the presence or absence of apomorphine must reflect some differential response to apomorphine between the two putamina. Thus, comparisons of the [11C]raclopride BP between the ipsilateral and contralateral putamen with and without apomorphine allow for the investigation of the relation between disease severity and the ability to modulate synaptic dopamine levels in response to apomorphine challenge. The authors compared
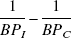
at baseline and after the two injections of apomorphine by repeated measures analysis of variance. Correlations between baseline
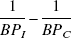
and apomorphine-induced changes were calculated using both parametric and nonparametric methods. The authors also applied the method proposed by Altman (1991) to avoid the possibility of spurious correlations.
RESULTS
All patients reported improvement in their parkinsonism after the apomorphine. However, the authors did not attempt to quantify this objectively because (1) this could result in movement during scanning that would seriously degrade the reliability of the approach, and (2) voluntary motor activation could result in release of endogenous dopamine, leading to further confound.
[11C]raclopride BP values for putamen ipsilateral and contralateral to the more affected body side are detailed in Table 3. The between-side difference of the inverse of [11C]raclopride BP decreased (significantly) by 25% after apomorphine injection (repeated measures analysis of variance, F = 4.85, df = 2, 8, P < 0.05) (Figs. 1 and 2). Although both doses of apomorphine gave similar results (Fig. 2), the highest dose led to a slightly higher effect in four of the five patients (Fig. 1). There was only one patient (Patient 4 in Table 3 and Fig. 1) with no apparent apomorphine-induced changes on 0.03 mg/kg of apomorphine, and a small paradoxic effect on 0.06 mg/kg of apomorphine (Fig. 1). This patient was the only one with levodopa-related motor fluctuations and dyskinesias at the time of the study (the others were stable responders), despite the fact that this patient had the lowest motor score (MCS = 19). This is also the patient who was on sertraline. The exclusion of this patient led to a highly significant effect of apomorphine (repeated measures analysis of variance, F = 23.78, df = 2, 6, P = 0.0014).
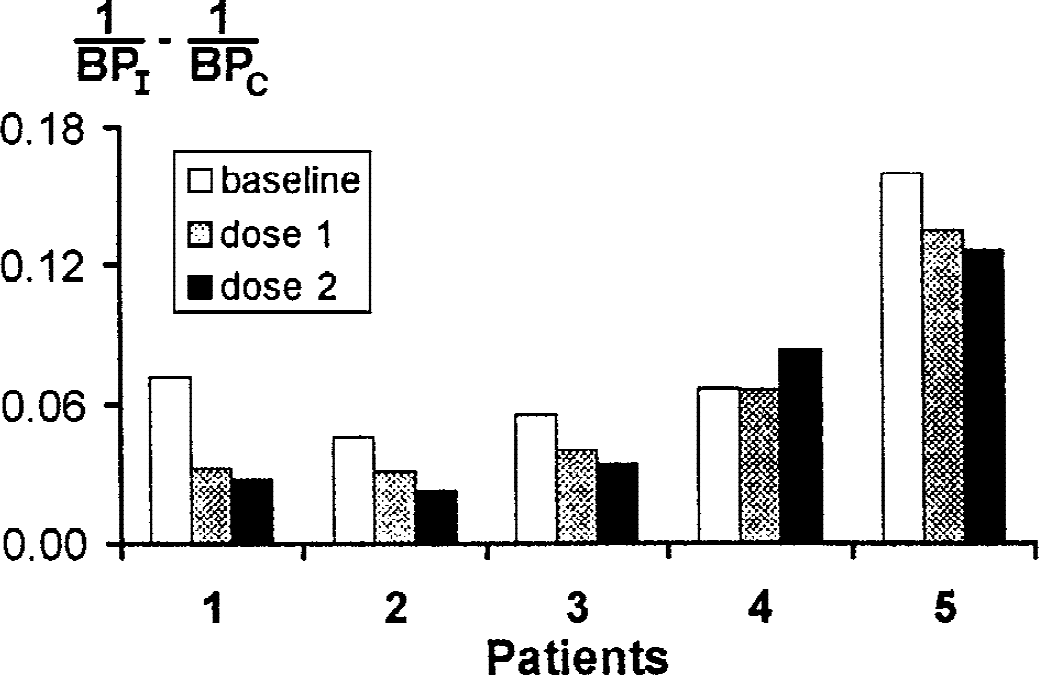
Individual values for the difference in the inverse of the [11C]raclopride binding potential (BP) between the putamen ipsilateral (I) and contralateral (C) to the more affected body side of patients with Parkinson disease, both before (baseline) and after apomorphine (dose 1 = 0.03 mg/kg; dose 2 = 0.06 mg/kg) given subcutaneously. Patient 4 had levodopa-related motor fluctuations and dyskinesias.
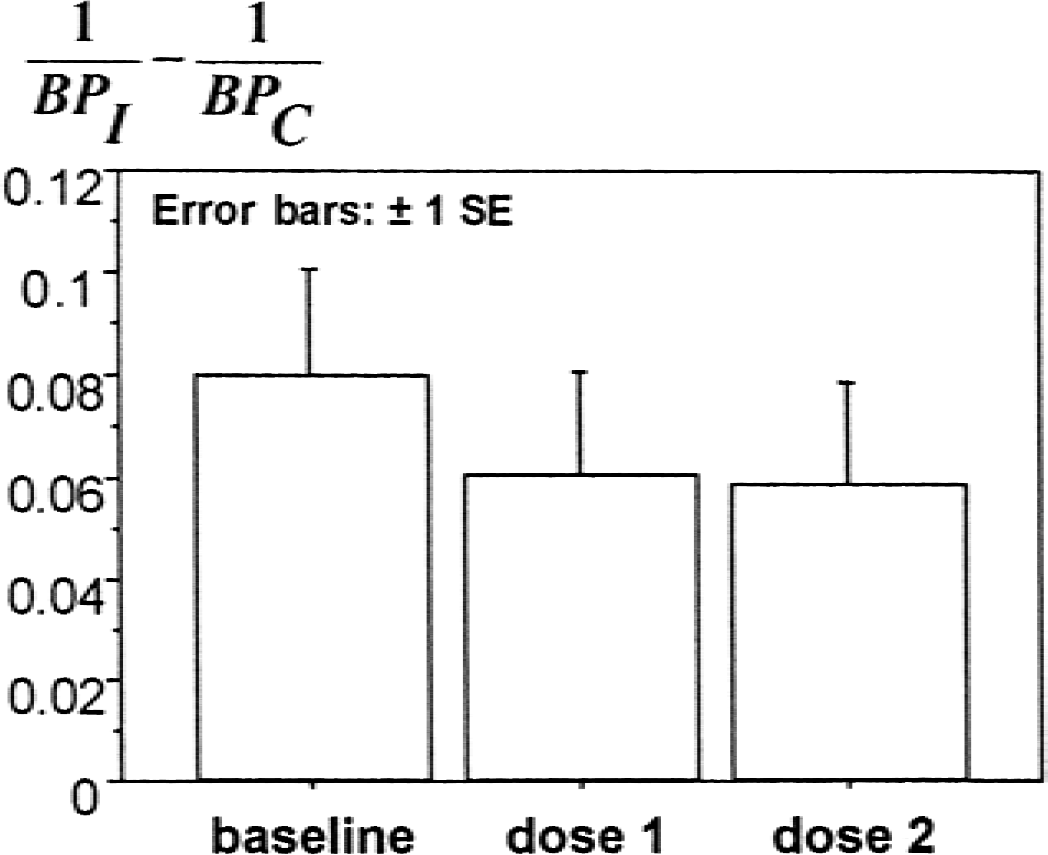
Chart from the repeated measures analysis of variance of the between-side differences in the inverse of the putamen [11C]raclopride binding potential (BP) before (baseline) and after apomorphine (dose 1 = 0.03 mg/kg; dose 2 = 0.06 mg/kg). This difference (calculated for each patient, n = 5) decreases after apomorphine injection (P < 0.05), which suggests that [DA]I′ − [DA]C′ < [DA]I − [DA]C. This is best explained by a preferential reduction of [DA]I′ (that is, apomorphine-induced presynaptic inhibition of endogenous dopamine release affecting preferentially the putamen ipsilateral to the more affected body side). I and C represent putamen ipsilateral and contralateral, respectively, to the more affected body side of patients with Parkinson disease.
Putamen [11C]raclopride binding potential values
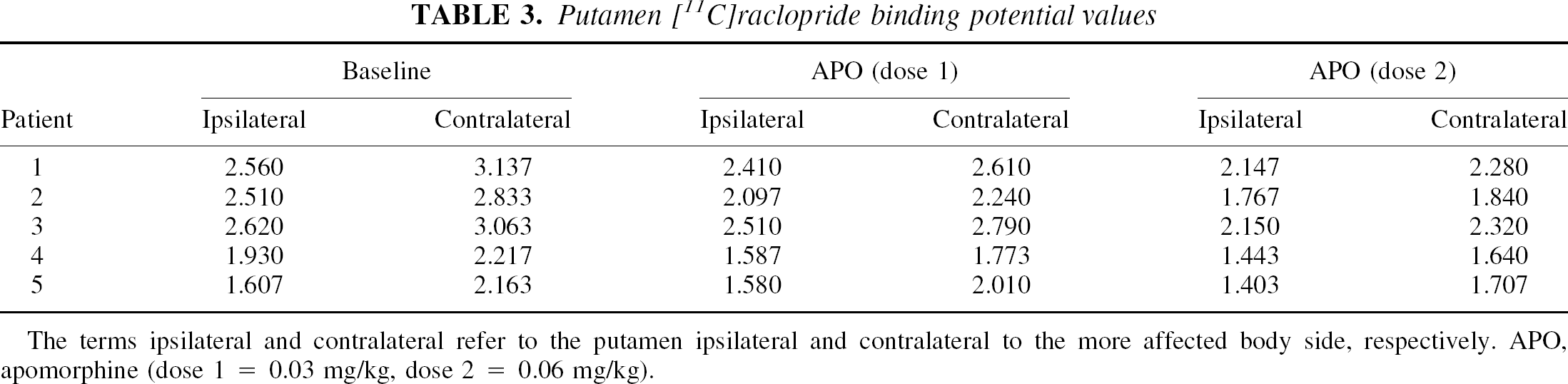
The terms ipsilateral and contralateral refer to the putamen ipsilateral and contralateral to the more affected body side, respectively. APO, apomorphine (dose 1 = 0.03 mg/kg, dose 2 = 0.06 mg/kg).
The apomorphine-induced decrease in the between-side difference of the inverse of [11C]raclopride BP was observed in all subregions of the putamen (that is, rostral, intermediate, and caudal putamen), though with different intensity. Interestingly, the effect was lower in the most affected part of the putamen (caudal putamen, 19%) as compared with other less affected rostral subregions (34%). There was no correlation between baseline values and apomorphine-induced changes (for example, for the first dose of apomorphine, Pearson's correlation coefficient, r = 0.13, P = 0.83; Spearman's rank correlation coefficient, r = 0.20, P = 0.69) (Fig. 1).
DISCUSSION
Here, the authors developed a novel method to estimate relative changes in the synaptic levels of dopamine. The method allowed them to find a significant difference between ipsilateral and contralateral putamen with regard to apomorphine-induced changes in synaptic dopamine levels, which provides in vivo evidence for presynaptic inhibition of dopamine release. It was observed that apomorphine induced a significantly greater drop in synaptic dopamine in the ipsilateral putamen as compared with the contralateral putamen. Because the contralateral putamen is identical to the ipsilateral putamen except for disease severity and its physiologic correlates, the authors interpret this to mean that an increased severity of disease is accompanied by a decreased ability to regulate synaptic dopamine concentrations. This interpretation is consistent with the finding of Ekesbo et al. (1999) that apomorphine-induced regulation of L-[11C]dopa uptake is impaired in advanced PD. It is also consistent with a study by Santiago et al. (1991) demonstrating reduced autoreceptor mediated regulation of dopamine levels in MPP+ -treated rats. The argument could be made that the apomorphine-induced drop in synaptic dopamine levels is greater in the less affected ipsilateral putamen as compared with the contralateral putamen simply because of differences in baseline dopamine release (that is, greater release in the ipsilateral putamen). If this were the sole explanation for the current findings, however, one would expect that the effect of apomorphine would correlate with (1/BPI − 1/BPC) at baseline, and this was not the case.
Although a significant difference between ipsilateral and contralateral putamen in response to 0.03 mg/kg apomorphine was noted, no further difference was evident upon increasing the dose to 0.06 mg/kg. This finding suggests that a dose of 0.03 mg/kg is enough to saturate the presynaptic dopamine receptors responsible for mediating apomorphine-induced inhibition of dopamine synthesis and release. This would be consistent with a study by Skirboll et al. (1979) demonstrating that the apomorphine-induced inhibition of the electrophysiologic activity of substantia nigra pars compacta neurones in rats reached 100% of maximal effect at doses as low as 0.03 mg/kg apomorphine. In other words, presynaptic dopamine receptors have greater sensitivity than postsynaptic receptors.
The data in this study do not highlight any one particular site for the physiologic change underlying the loss of autoregulatory function. There are several sites at which apomorphine (and presumably other dopamine agonists and dopamine itself) could act to regulate dopamine synthesis and release. Possible sites include somatodendritic autoreceptors, presynaptic terminal autoreceptors, and postsynaptic receptors that may subsequently feed back through polyneural loops to influence dopaminergic cell function (Roth, 1984; Zigmond, 1994). Because the changes accompanying disease progression seem to be primarily presynaptic, it can be speculated that some loss of presynaptic (either terminal or somatodendritic) autoreceptors or some biochemical modification of their function may underlie the loss in autoregulatory function. The authors' observations support the notion that the mere loss of presynaptic autoreceptors might not completely explain the autoregulatory dysfunction observed in PD. Thus, the apomorphine effect decreased from 34% in rostral putamen to 19% in caudal putamen.
Although any loss of homeostatic function is likely to be deleterious, in some ways, the loss of autoreceptor function in a patient with PD may be adaptive. For instance, a decrease in autoregulatory function, which normally suppresses dopamine synthesis and release, may result in increased synthesis and release of dopamine, an effect that potentially would be helpful in PD.
Decreased autoregulatory function may help to explain some of the observed changes in therapeutic response to apomorphine noted with PD progression (Verhagen Metman et al., 1997). For instance, a relative failure to down-regulate dopamine synthesis and release in response to apomorphine may contribute to the steeper dose-response curve noted in patients with more severe PD as compared with those with less severe PD. This is because in patients with relatively intact autoregulatory function, a decrease in synaptic dopamine would partially offset the stimulatory effect of apomorphine, whereas this would not occur in patients lacking autoregulatory function.
Although decreased autoregulatory function may have some benefits, it also may contribute to the increase in dopamine turnover (Abercrombie et al., 1990; Calne and Zigmond, 1991) that has been noted to accompany PD progression (Tedroff et al., 1996) and that the authors have observed to be associated with the development of motor fluctuations (de la Fuente-Fernández et al., 2000, 2001). Furthermore, it may contribute to the development of dyskinesias. To see how intact autoregulatory function may protect against the development of dyskinesias, or fluctuations, or both, consider the response of a patient with normal autoregulatory function to a pulse of levodopa. The levodopa would result in an initial increase in synaptic dopamine. This would act in turn on autoregulatory mechanisms to decrease the synthesis and release of dopamine. The result would be a decreased turnover of dopamine and a blunted spike in the synaptic concentration of dopamine in response to levodopa (and presumably a lower likelihood of dyskinesias). However, a patient lacking autoregulatory function would not be protected from spikes in synaptic dopamine concentration and turnover rate and thus may be more susceptible to the development of dyskinesias and motor fluctuations. In this context, a bilateral lack of autoregulatory function might explain why a dose of 0.03 mg/kg apomorphine failed to produce any substantial effect in the only patient (Patient 4) with levodopa-related motor complications included in the current study, even though there was a clear asymmetry of baseline raclopride binding, as well as clinical findings. However, this patient was also taking sertraline, which could have affected the results.
In demonstrating a decline in autoregulatory function with increased disease severity, this study supports the idea, proposed by Zigmond et al. (1990), that PD may be in part a disease of impaired neurotransmitter homeostasis and not merely the result of a loss of dopamine synthesis and storage capacity. As noted above, impaired autoregulatory function may account for some of the symptoms and complications associated with PD progression that are not fully explained by a simple loss of synthesis and storage capacity. Thus, it is conceivable that impairment of autoregulatory function is a key component of PD progression.
One technical aspect of the study deserves consideration. Although the Logan method used here is insensitive to global changes in cerebral blood flow, BP measurements could conceivably be affected by regional changes in cerebral blood flow. If this effect were asymmetric, one could incorrectly deduce that the changes in synaptic dopamine levels are asymmetric as an artefact of different tracer delivery between the two sides of putamen. This is most unlikely to explain the current findings, however, because, as noted above, putamen blood flow is not asymmetric in patients with asymmetric PD (Perlmutter and Raichle, 1985; but see Wolfson et al., 1985) and dopamine agonists do not demonstrably alter putamen blood flow (Perlmutter and Raichle, 1985). The authors' previous work also suggests that no significant local blood flow changes occur in the putamen after dopaminergic treatment (de la Fuente-Fernández et al., 2001). Furthermore, any hypothetical asymmetry in putamen blood flow, if present, should affect apomorphine and [11C]raclopride delivery equally, which would tend to maintain the degree of their competition for D2 /D3 dopamine receptors constant.
In conclusion, the new method reported here allowed the authors to provide evidence that an increased severity of PD is associated with a loss in the ability to regulate the synaptic concentration of dopamine. This phenomenon may contribute to the observed changes in therapeutic response with the progression of PD. Further study is needed to elucidate the pathophysiologic mechanisms underlying the loss of autoregulatory function and the role of this loss in PD symptomatology and progression.
Footnotes
Acknowledgments:
The authors are grateful to members of the UBC-TRIUMF PET team for assistance with the scans.