
Abstract
Metabotropic glutamate (mGlu) receptors have been considered as potential targets for neuroprotective drugs, but the lack of specific drugs has limited the development of neuroprotective strategies in experimental models of acute or chronic central nervous system (CNS) disorders. The advent of potent and centrally available subtype-selective ligands has overcome this limitation, leading to an extensive investigation of the role of mGlu receptor subtypes in neurodegeneration during the last 2 years. Examples of these drugs are the noncompetitive mGlu1 receptor antagonists, CPCCOEt and BAY-36-7620; the noncompetitive mGlu5 receptor antagonists, 2-methyl-6-(phenylethynyl)pyridine, SIB-1893, and SIB-1757; and the potent mGlu2/3 receptor agonists, LY354740 and LY379268. Pharmacologic blockade of mGlu1 or mGlu5 receptors or pharmacologic activation of mGlu2/3 or mGlu4/7/8 receptors produces neuroprotection in a variety of in vitro or in vivo models. MGlu1 receptor antagonists are promising drugs for the treatment of brain ischemia or for the prophylaxis of neuronal damage induced by synaptic hyperactivity. MGlu5 receptor antagonists may limit neuronal damage induced by a hyperactivity of N-methyl-d-aspartate (NMDA) receptors, because mGlu5 and NMDA receptors are physically and functionally connected in neuronal membranes. A series of observations suggest a potential application of mGlu5 receptor antagonists in chronic neurodegenerative disorders, such as amyotrophic lateral sclerosis and Alzheimer disease. MGlu2/3 receptor agonists inhibit glutamate release, but also promote the synthesis and release of neurotrophic factors in astrocytes. These drugs may therefore have a broad application as neuroprotective agents in a variety of CNS disorders. Finally, mGlu4/7/8 receptor agonists potently inhibit glutamate release and have a potential application in seizure disorders. The advantage of all these drugs with respect to NMDA or AMPA receptor agonists derives from the evidence that mGlu receptors do not “mediate,” but rather “modulate” excitatory synaptic transmission. Therefore, it can be expected that mGlu receptor ligands are devoid of the undesirable effects resulting from the inhibition of excitatory synaptic transmission, such as sedation or an impairment of learning and memory.
Neurodegeneration is a common outcome of a variety of acute or chronic central nervous system (CNS) disorders, such as stroke, temporal lobe epilepsy, Parkinson disease, Alzheimer disease, Huntington disease, and the AIDS-dementia complex. In most of these disorders, the current therapy is symptomatic and does not prevent or arrest the ongoing neurodegeneration. Like many other cells, neurons die by necrosis or apoptosis, and both phenotypes of death are commonly found in acute disorders such as stroke. Apoptotic death can develop as a direct consequence of the causative agent—such as the lack of oxygen and glucose in stroke or the presence of ß-amyloid fibrils in Alzheimer disease)—but also can be secondary to the loss of neurotrophic inputs normally released from neurons that primarily die. The coexistence of different processes of neuronal death must be outlined because the same therapeutic intervention that protects against necrotic death can aggravate apoptosis by trophic deprivation and vice versa (Lee et al., 1999). The intracellular events underlying the execution phase of cell death have been extensively dissected and ultimately involve the activation of caspases. However, these processes are not cell-specific; therefore, they cannot be easily targeted by neuroprotective drugs. For example, a long-term neuroprotective therapy with a caspase inhibitor will impair the occurrence of “helpful” apoptosis in cells infected by viruses or harboring genomic damage. A better strategy is to intervene against extracellular insults that specifically contribute to neuronal death. An excessive activation of ionotropic glutamate (iGlu) receptors, particularly N-methyl-d-aspartate (NMDA) receptors, has been widely implicated in the pathophysiology of neuronal damage in most of acute and chronic neurodegenerative disorders (Choi, 1992; Doble, 1999). A number of NMDA or alpha-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonists have been tested as neuroprotectants in experimental animals and humans. However, in spite of the promising results obtained in animal models, all clinical trials with iGlu receptor antagonists in stroke have been unsuccessful because of the follwing: 1) a narrow therapeutic window, 2) the occurrence of undesirable side effects, and 3) lack of efficacy (Lee et al., 1999). The impairment of fast excitatory synaptic transmission caused by iGlu receptor antagonists is certainly responsible for side effects such as sedation, ataxia, and memory loss, and also might contribute to the lack of efficacy of these drugs. According to the hypothesis of a dual role of Ca2+ in cell survival (Johnson et al., 1992), neuronal death may be triggered not only by a Ca2+ overload, but also by the reduction of free cytosolic Ca2+ below a critical threshold. This threshold is set by trophic factors produced by neighboring neurons or glial cells. A selective blockade of iGlu receptors therefore may deprive susceptible neurons from the amount of Ca2+ that could compensate for the loss of neurotrophic agents. Therefore, it can be expected that iGlu receptor antagonists protect neurons that die from an excessive increase in cytosolic free Ca2+, but facilitate apoptotic death in neurons that no longer receive neurotrophic input. There are two strategies to overcome this limitation. First, one can try to limit the intracellular consequences of an excessive activation of iGlu receptors without impairing the “physiologic effects” of receptor activation. Examples are provided by the receptor abuse–dependent antagonists (RADA drugs), such as melatonin or the gangliosides GM1 and GT1b, which were proven to be protective against excitotoxic neuronal death (Manev et al., 1990a, 1990b, 1997; Costa et al., 1994). Second, neuroprotective drugs can target specific categories of membrane receptors that “modulate” rather than mediate excitatory synaptic transmission but are nonetheless implicated in the pathophysiology of neuronal damage. Metabotropic glutamate (mGlu) receptors meet these criteria (Nicoletti et al., 1996) and will be discussed in this review.
STRUCTURAL AND FUNCTIONAL FEATURES OF mGlu RECEPTORS
MGlu receptors belong to family 3 of G-protein–coupled receptors, which also include gamma aminobutyric acid type B receptor (GABA-B), Ca2+-sensing, and some olfactory, pheromone, and taste receptors. The extracellular domain of mGlu receptors harbors the agonist binding site and is structurally related to bacterial periplasmic amino acid binding proteins (O'Hara et al., 1993). X-ray crystallographic studies of the mGlu1 receptor support the original hypothesis (O'Hara et al., 1993) that glutamate binds to a cleft separating two globular lobes, switching the equilibrium toward the closed conformation of the two lobes. In addition, the receptor acts as a dimer in such a way that the closure of one of the two extracellular domains resulting from glutamate binding produces a drastic change in conformation that reduces the distance of the two C-terminus domains of the promoters (Kunishima et al., 2000). The second intracellular loop is the least conserved among the mGlu receptor subtypes and is critical for the selectivity of G-protein coupling, whereas the third intracellular loop is mostly involved in G protein activation and controls the coupling efficacy in cooperation with the first loop and the C-terminal tail (De Blasi et al., 2001). mGlu receptors form a family of 8 subtypes (mGlu1 to mGlu8), which are subdivided into 3 groups on the basis of structural homology, pharmacologic profile, and transduction pathways (Fig. 1). Group-I includes mGlu1 (splice variants: mGlu1a, -b, -c, -d, -e) and mGlu5 (splice variants: mGlu5a and -b) receptors, which are coupled to polyphosphoinositide (PI) hydrolysis (Tanabe et al., 1992; Minakami et al., 1994; Joly et al., 1995; Laurie et al., 1996; Flor et al., 1996; Schoepp et al., 1999). Activation of these receptors generates inositol-1,4,5-trisphoshate (InsP3) and diacylglycerol, which releases intracellular Ca2+ and activates protein kinase C (PKC), respectively (Fig. 1). However, in transfected cells, activation of mGlu1a receptors produces a single peaked intracellular Ca2+ response, whereas activation of mGlu5 receptors generates an oscillatory increase in intracellular Ca2+. The latter property relies on the presence of a threonine residue just distal to the seventh TM domain of the mGlu5 receptor, which is phosphorylated by PKC (Kawabata et al., 1996). Whether or not this difference is physiologically relevant is unclear. Native mGlu1 and mGlu5 receptors also can negatively modulate a variety of K+ channels and activate inward cationic currents (Pin and Duvoisin, 1995; Chuang et al., 2000), with the net effect of increasing neuronal excitability. MGlu5 receptors also are expressed in astrocytes and microglia (Biber et al., 1999). Group-II mGlu receptors include mGlu2 and mGlu3 (Flor et al., 1995a; Emile et al., 1996), which are coupled to Gi proteins in heterologous expression systems. Activation of these receptors reduces cyclic adenosine monophosphate formation (Fig. 1), but also can produce pleiotropic effects mediated by the beta-gamma subunits of the G protein, such as the activation of mitogen-activated protein kinase and the phosphatidylinositol-3-kinase (PI-3-K) pathways (Ferraguti et al., 1999). Both receptor subtypes are preferentially localized in the preterminal region of axons, where their activation attenuates glutamate release. mGlu3 receptors, however, are widely expressed on astrocytes, where their functional role is becoming increasingly important (see below). Group-III mGlu receptors include the subtypes mGlu4 (splice variants: mGlu4a and -b), mGlu6, mGlu7 (splice variants mGlu7a and -b), and mGlu8 (splices variants: mGlu8a and -b), which are all coupled to Gi proteins in transfected cells (Okamoto et al., 1994; Flor et al., 1995b; 1997; Duvoisin et al., 1995; Corti et al., 1997). mGlu4, and mGlu7 and mGlu8 receptors are all presynaptically localized, and their activation inhibits the release of glutamate or GABA. mGlu6 receptors are exclusively expressed by ON bipolar cells in the retina, and are physiologically involved in the amplification of visual inputs. In these cells, mGlu6 receptors are coupled to a cyclic guanosine monophosphate (cGMP) phosphodiesterase (Fig. 1) (Nakanishi, 1994). There is no evidence of mGlu receptor localization in oligodendrocytes.
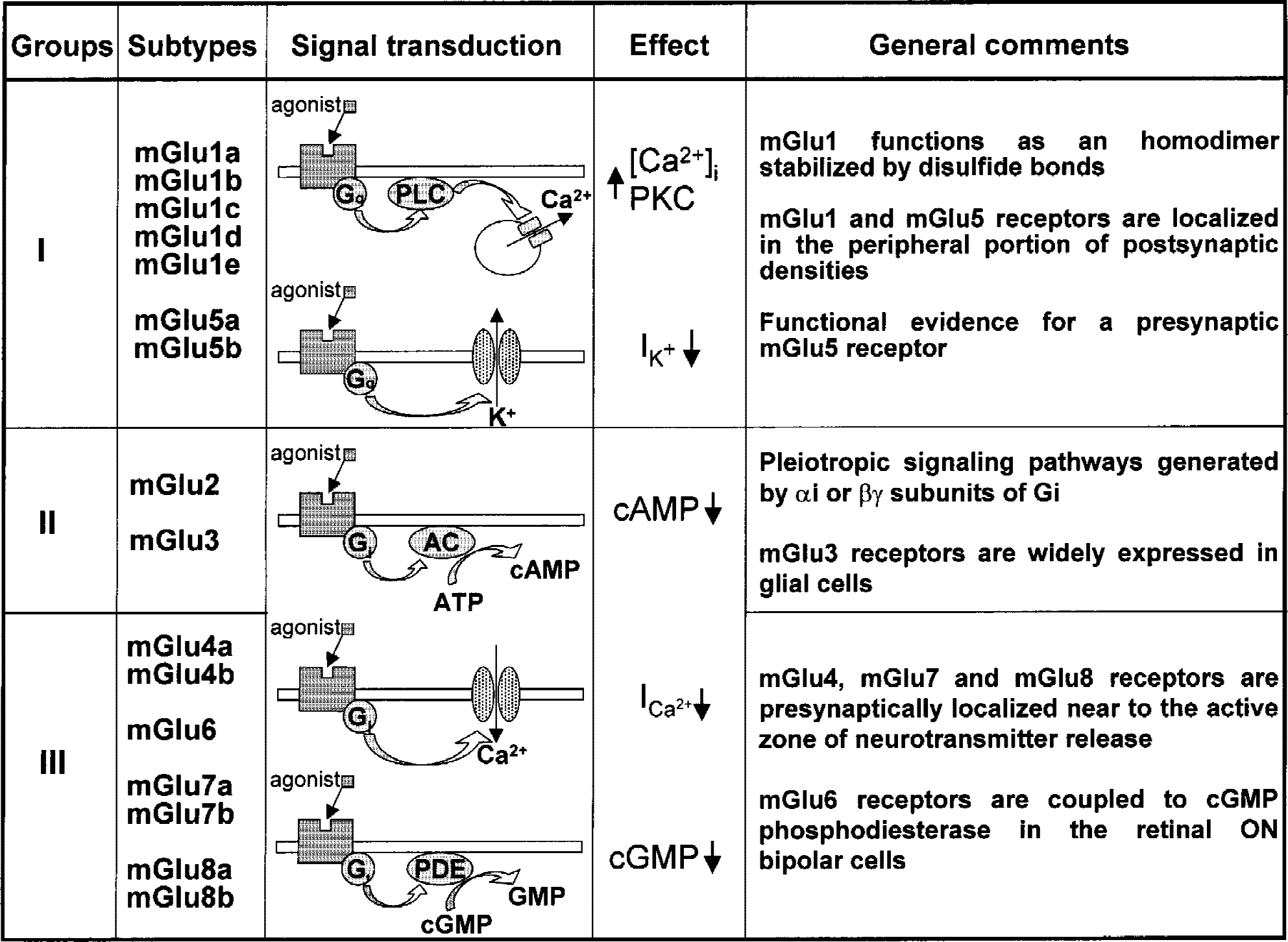
General features of metabotropic glutamate (mGlu) receptors. PLC, phospholipase C; PKC, protein kinase C; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; PDE, phosphodiesterase.
RECENT ADVANCES IN THE PHARMACOLOGY OF mGlu RECEPTORS
mGlu can be pharmacologically differentiated from iGlu with the agonist 1S,3R-1-amino-cyclopentane-1,3-dicarboxylic acid (1S,3R-ACPD), which, however, is not subtype-selective and activates both group-I and group-II mGlu receptors (Schoepp et al., 1999) (Table 1). Group-I mGlu receptors can be selectively activated by 3,5-dihydroxyphenylglycine (DHPG) or 3-hydroxyphenylglycine (3-HPG), which cannot readily discriminate between mGlu1 and mGlu5 receptors. 2-Chlorohydroxyphenylglycine (CHPG) selectively activates mGlu5 receptors, but is less potent than DHPG or 1S,3R-ACPD (Schoepp et al., 1999). The phenylglycine derivatives alpha-methyl-4-carboxyphenylglycine (MCPG), 4-carboxyphenylglycine (4CPG), and 4-carboxy-3-hydroxyphenylglycine (4C3HPG) have been widely used to antagonize group-I mGlu receptors with a certain degree of selectivity. All three drugs preferentially antagonize mGlu1 over mGlu5 receptors, but also can recruit mGlu2 and mGlu3 receptors (MCPG as an antagonist, 4C3HPG as an agonist). Novel subtype-selective antagonists are now available that help to efficiently discriminate effects produced by the activation of mGlu1 and mGlu5 receptors. The compounds 2-methyl-4-carboxyphenylglycine (LY367385), (R,S)-1-aminoindan-1,5-dicarboxylic acid (AIDA), and (S)-(+)-2-(3′-carboxybicyclo[1.1.1]pentyl)-glycine (CBPG) behave as competitive mGlu1 antagonists (CBPG is also an mGlu5 receptor agonist), whereas (2S,1′R,2′R)-2-(9-xanthylmethyl)-2-(2′-carboxycyclopropyl)glycine (LY344545) selectively antagonize mGlu5 receptors. 7-(Hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt) and [(3aS,6aS)-6a-Naphtalen-2-ylmethyl-5-methyliden-hexahydro-cyclopental[c]furan-1-on] (BAY36–7620) are nonamino acid molecules acting as noncompetitive antagonists of mGlu1 receptors with a high degree of selectivity (in the low micromolar range) (Annoura et al., 1996; Litschig et al., 1999; Carroll et al., 2001). Interestingly, BAY36–7620 can cross the blood–brain barrier; therefore, it is suitable for in vivo studies (Carroll et al., 2001). A series of phenylpyridine derivatives, including 2-methyl-6-(phenylethynyl)pyridine (MPEP), (E)-2-methyl-6-stryrylpyridine (SIB-1893), and 6-methyl-2-(phenylazo)pyridin-3-ol (SIB-1757), behave as highly potent and selective noncompetitive mGlu5 receptor antagonists, and at least MPEP is proven to be centrally available (Gasparini et al., 1999b; Spooren et al., 2001). mGlu2 and mGlu3 receptors can be activated by 2R,4R-4-aminopyrrolidine-2,4-dicarboxylic acid (2R,4R-APDC), which is so far the most selective agonist at these receptor subtypes. Last generation agonists of mGlu2/3 receptors include the Lilly compounds (1S,2S,5R,6S)-(+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY354740), (−)-2-oxa-4-aminocyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY379268), and (−)-2-thia-4-aminobicyclo[3.1.0] hexane-4,6-dicarboxylic acid (LY389795), which are highly potent (Ki value in the low nanomolar range) and are systemically available. (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV) and its congener (2S,1′S,2′S)-2-(carboxycyclopropyl)glycine (L-CCG-I) have been widely used as mGlu2/3 receptor agonists. However, DCG-IV also can activate NMDA receptors and behaves as a group-III mGlu receptor antagonist, whereas L-CCG-I also can activate mGlu1, −5, and −8 receptors (Brabet et al., 1998; Schoepp et al., 1999). N-acetylaspartylglutamate (NAAG) is an interesting compound because it shows selectivity for mGlu3 over mGlu2 receptors (Wroblewska et al., 1997). mGlu2 and mGlu3 receptors can be antagonized by a series of compounds, of which (2S,4S)-2-amino-4-(2,2-diphenylethyl)pentane-1,5-dioic acid (ADED) and (2S,4S)-2-amino-4-(4,4-diphenylbut-1-yl)pentane1,5-dioic acid (ADBD) (Schoepp et al., 1999) show the highest degree of selectivity. (2S,1′S,2′S)-2-(9-xanthylmethyl)-2-(2′-carboxycyclopropyl)glycine (LY341495) shows a high potency in antagonizing mGlu2/3 receptors, but can recruit all other receptor subtypes at micromolar concentrations (Schoepp et al., 1999). Finally, all group-III mGlu receptors are activated by phosphonoamino acids, such as L-2-amino-4-phosphonobutanoate (L-AP4, the prototypic ligand) and (+)-4-phosphonophenylglycine (PPG), as well as by L-serine-O-phosphate (L-SOP), which is an endogenous compound. All these compounds show a greater affinity for mGlu4, −6, and −8 than for mGlu7. PPG has a greater affinity for mGlu8 than for mGlu4 and mGlu6 receptors (Gasparini et al., 1999a; 2000). Subtype-selective antagonists of group-III mGlu receptors are lacking. (S)-α-methyl-2-amino-4-phosphonobutanoic acid (MAP4) and (RS)-α-methylserine-O-phosphate (MSOP) are generally used to inhibit mGlu4 receptors (Jane et al., 1994; Thomas et al., 1996).
Subtype-selective agonists and antagonists of mGlu receptors

Ag., agonist; Ant., antagonist; C Ant., competitive antagonist; NC Ant., noncompetitive antagonist; DHPG, 3,5-dihydroxyphenylglycine; 3-HPG, 3-hydroxyphenylglycine; AIDA, (R,S)-1-aminoindan-1,5-dicarboxylic acid; LY367385, 2-methyl-4-carboxyphenylglycine; CPCCOEt, 7-(Hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester; BAY36–7620, [(3aS,6aS)-6a-Naphtalen-2-ylmethyl-5-methyliden-hexahydro-cyclopental[c]furan-1-on]; CHPG, 2-chlorohydroxyphenylglycine; MPEP, 2-methyl-6-(phenylethynyl)pyridine; SIB-1757, 6-methyl-2-(phenylazo)pyridin-3-ol; SIB-1893, (E)-2-methyl-6-stryrylpyridine; 2R,4R-APDC, 2R,4R-4-aminopyrrolidine-2,4-dicarboxylic acid; LY354740, (1S,2S,5R,6S)-(+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid; LY379268, (−)−2-oxa-4-aminocyclo[3.1.0]hexane-4,6-dicarboxylic acid; DCG-IV, (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine; NAAG, N-acetylaspartylglutamate; ADED, (2S,4S)-2-amino-4-(2,2-diphenylethyl)pentane-1,5-dioic acid; ADBD, (2S,4S)-2-amino-4-(4,4-diphenylbut-1-yl)pentane-1,5-dioic acid; LY341495, (2S,1′S,2′S)-2-(9-xanthylmethyl)-2-(2′-carboxycyclopropyl)glycine; L-AP4, L-2-amino-4-phosphonobutanoate; SOP, L-serine-O-phosphate; (R,S)-PPG, (+)-4-phosphonophenylglycine; MSOP, (RS)-′-methylserine-O-phosphate; MAP4, (S)-′-methyl-2-amino-4-phosphonobutanoic acid.
ROLE OF GROUP-I mGlu RECEPTORS IN NEURODEGENERATION/ NEUROPROTECTION
Two issues will be addressed in this section: 1) how pharmacologic activation of group-I mGlu receptors affects excitotoxic neurodegeneration, and (2) how endogenous activation of mGlu1 or mGlu5 receptors affects neurodegeneration in a variety of cellular or animal models of CNS disorders. As a corollary of the second point, the authors will comment on the potential use of subtype-selective mGlu1 or mGlu5 receptor antagonists in acute or chronic neurodegenerative disorders.
Pharmacologic activation of group-I mGlu receptors in neurodegeneration/neuroprotection
As activation of mGlu1 or mGlu5 receptors produces excitatory effects in neurons (Pin and Duvoisin, 1995), it is reasonable to assume that these receptors facilitate the induction and/or progression of excitotoxic neuronal death. Early in vivo experiments with 1S,3R-ACPD, which also can activate mGlu2 and −3 receptors, are consistent with this view. 1S,3R-ACPD locally injected into the rat hippocampus or caudate nucleus produces neurodegeneration that requires the endogenous activation of NMDA receptors in the adult, but exclusively depends on intracellular Ca2+ mobilization in neonates (McDonald and Schoepp, 1992; Sacaan and Schoepp, 1992; McDonald et al., 1993). Accordingly, the efficacy of mGlu receptor agonists to stimulate PI hydrolysis is much greater in brain slices from newborn than from adult animals (Nicoletti et al., 1986). In more recent in vivo studies, DHPG has shown neurotoxic and proconvulsant activities when injected intracerebroventricularly (Ong and Balcar, 1997; Camon et al., 1998; Merlin and Wong, 1997; Burke and Hablitz, 1996). The in vitro counterpart of these data has been obtained by using mixed cultures of mouse cortical cells, in which neurons are grown over a monolayer of confluent astrocytes. In this model, a brief pulse with NMDA produces delayed neuronal death that can be detected by assessing trypan blue staining or extracellular lactate dehydrogenase activity 20 to 24 hours after the pulse. 3HPG, DHPG, or quisqualate (a mixed agonist of group-I mGlu and AMPA receptors) coapplied with submaximal concentrations of NMDA amplify excitotoxic neuronal death (Buisson and Choi, 1995; Bruno et al., 1995b). Similar results are obtained with CHPG (Bruno et al., unpublished). Interestingly, at least DHPG and quisqualate retain their neurotoxic effects when applied immediately after the NMDA pulse and maintained in the medium afterwards (Bruno et al., 1995b). Other studies, however, unequivocally show that group-I mGlu receptor activation produces neuroprotection rather than neurotoxicity in a variety of in vitro models, including cultured cerebellar granule cells challenged with glutamate (Pizzi et al., 1993, 1996a, 1999), cultured spinal motor neurons challenged with kainate (Pizzi et al., 2000) or brain slices challenged with excitotoxins or oxygen-glucose deprivation (Opitz and Reymann, 1993; Pizzi et al., 1996b; Schroeder et al., 1999). Possible explanations for these contrasting results have been discussed in a recent review (Nicoletti et al., 1999) and include: (i) the heteromeric composition of NMDA receptors; (ii) the existence of an “activity-dependent switch” from facilitatory to inhibitory group-I mGlu receptors; and (iii) the absence or presence of astrocytes expressing mGlu5 receptors. Two additional aspects will be discussed, i.e. the role of mGlu5 receptors in supporting the survival of developing neurons; and the activity-dependent changes in the subcellular expression of mGlu5 receptors. The presence of the NR2C subunit in NMDA receptors of mature cultured cerebellar granule cells has been considered as a major determinant for the neuroprotective response to group-I mGlu receptor agonists. Accordingly, 3HPG is highly protective against glutamate toxicity in control cultured granule cells, but not in cells treated with NR2C antisense oligonucleotides (Pizzi et al., 1999). This is an interesting observation, particularly because the NR2C subunit is highly expressed in the human brain (Lin et al., 1996; Daggett et al., 1998). The lack of the NR2C in the substantia nigra could predispose nigral neurons to excitotoxic damage. An additional explanation (which is not mutually exclusive with the NR2C hypothesis) is that pharmacologic activation of group-I mGlu receptors may either facilitate or attenuate excitotoxic neuronal death depending on the functional state of the receptors. Dynamic changes in the activity of group-I mGlu receptors have been described by J. Sanchez-Prieto and his collaborators (Herrero et al., 1992, 1994, 1998; Sistiaga et al., 1998, 2000), who examined the modulation of glutamate release in cortico-striatal synaptosomes. In this preparation, a first application of group-I mGlu receptor agonists facilitates glutamate release, whereas a second application (within a restricted timeframe) inhibits release. This “functional switch” in receptor activity involves receptor phosphorylation by PKC, similarly to what happens during classical receptor desensitization, and depends on changes in the G-protein coupling. The same phenomenon has been observed by recording excitatory postsynaptic currents (EPSCs) in CA1 pyramidal cells exposed to DHPG (Rodriguez-Moreno et al., 1998). The authors examined whether the model of the functional switch also can be applied to excitotoxic neuronal death. In mixed cortical cultures, DHPG amplifies NMDA toxicity when applied only once (either before or during the NMDA pulse), but becomes neuroprotective when applied for the second time shortly after a brief preexposure. Neuroprotection is lost when less than 45 minutes are allowed between the first and the second exposure to DHPG, or when DHPG is applied for the first time in combination with PKC inhibitors or mGlu5 receptor antagonists. The release of endogenous glutamate stimulated by the NMDA pulse in culture also is amplified by a unique application, but is reduced by two consecutive applications of DHPG (Bruno et al., 2001). Thus, one expects that the final effect of group-I mGlu receptor agonists on excitotoxic neuronal death is related to the functional state of group-I mGlu receptors, which depends on how frequently these receptors are activated by endogenous glutamate. The authors believe that this dual regulation of excitotoxic neuronal death is exclusively performed by presynaptic mGlu receptors (perhaps coinciding with mGlu5 receptors) regulating glutamate release (Fig. 2). Although classical electron microscopy studies show that mGlu1 and mGlu5 receptors are preferentially localized in the peripheral portions of the postsynaptic densities (Baude et al., 1993; Lujan et al., 1997; Shigemoto et al., 1997), evidence for a presynaptic localization of group-I mGlu receptors is emerging (PJ Conn, unpublished observation), and the possibility exists that mGlu5 receptors may be targeted to nerve endings depending on the availability of particular variants of Homer proteins (see below). The final effect of group-I mGlu receptor agonists on neuronal death may also critically depend on the presence and functional state of glial cells. Interestingly, reactive astrocytes in the injured brain tissue, as well as their equivalent in culture (that is, astrocytes cultivated in chemically defined medium deprived of serum and enriched in endothelial growth factor or transforming growth factor-alpha (TGF-alpha)) express high levels of functional mGlu5 receptors (Miller et al., 1993). The physiologic significance of this phenomenon is unknown, but one can predict that activation of mGlu5 receptors by the endogenously released glutamate controls some aspects related to the responsiveness of reactive astrocytes to neuronal injury. Interestingly, DHPG (which is otherwise neuroprotective) amplifies excitotoxicity when applied to cultured granule cells on which a coverslip with a monolayer of confluent astrocytes has been flipped (G Barbagallo and MV Catania, unpublished observation). Among the putative neurotoxic factors released by astrocytes in response to mGlu5 receptor activation, attention should be focused on nitric oxide. Notably, the neuronal isoform of constitutive nitric oxide synthase (nNOS) is expressed in cultured astrocytes grown in chemically defined medium (Nicoletti et al., 1999) and reactive astrocytes in the spinal cord of individuals who have died from amytrophic lateral sclerosis express high amounts of the beta and gamma variants of nNOS (Catania et al., 2001).
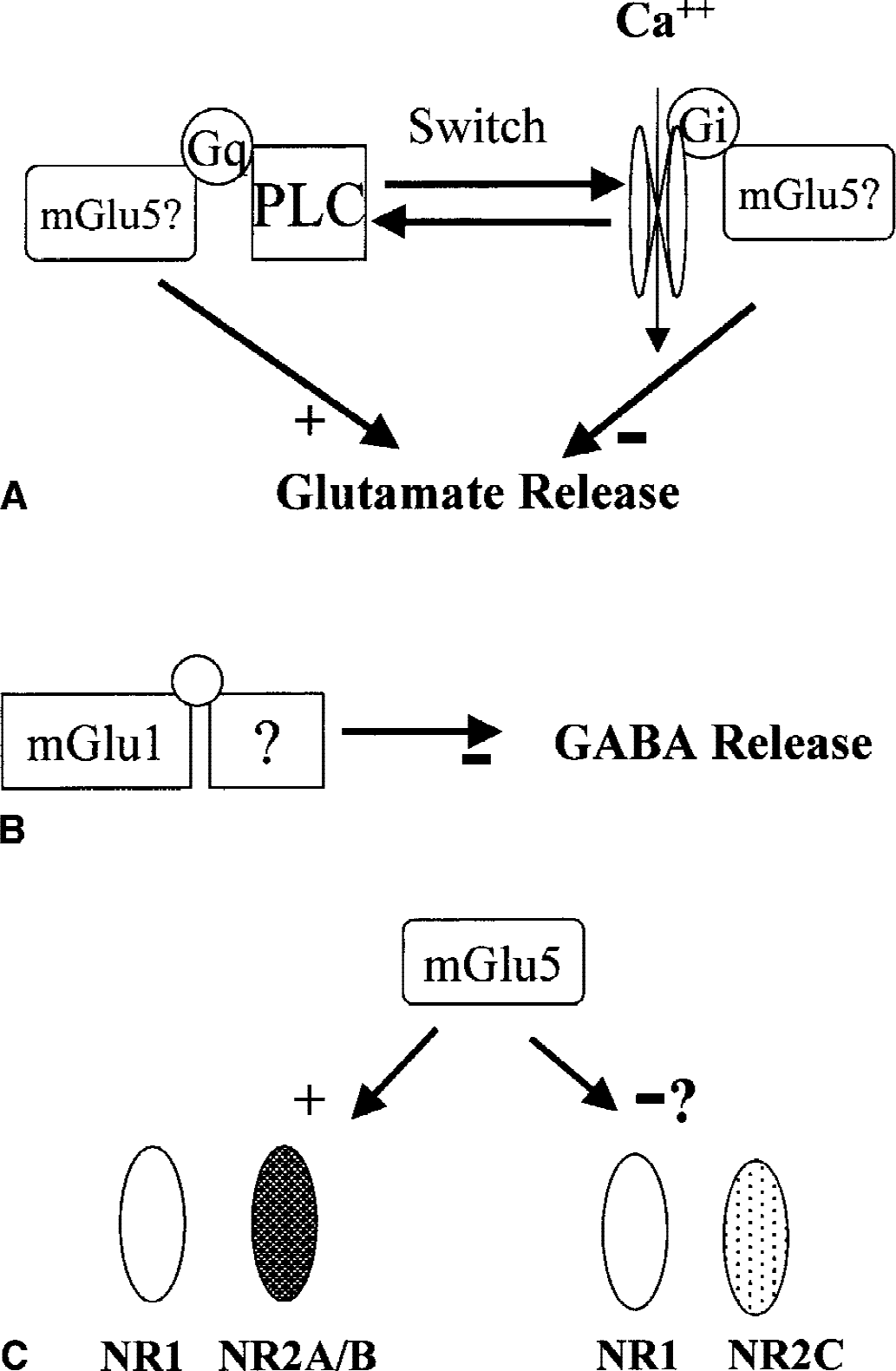
Interestingly, pharmacologic activation of mGlu5 receptors protects neurons against apoptosis by trophic deprivation. Copani et al. (1998) have shown that in granule cells grown under suboptimal conditions—that is, in medium containing 10 rather than 25 mmol/L K+)—the onset of apoptotic death coincides with the developmental decline in mGlu5 receptors, and cells are rescued by the addition of group-I mGlu receptor agonists. In addition, pharmacologic activation of group-I mGlu receptors is protective in cortical or hippocampal neurons deprived of trophic support (Sagara and Schubert, 1998); hence, one expects that the final effect of mGlu5 receptor agonists on neuronal death depends on the proportion of neurons that primarily die in response to the toxic insult and those that die because of the lack of trophic inputs. Obviously, this is impossible to predict in human pathology.
Finally, the diversity in the response to group-I mGlu receptor agonists may depend on the subcellular localization of mGlu1 or mGlu5 receptors in a particular neuronal population. Group-I mGlu receptors interact with members of a family of synaptic proteins named Homer, which bind to a proline-rich motif of the C-terminus of mGlu receptors through their PDZ domain. The longer crosslinking-capable isoforms of Homer—that is, Homer-1b, −1c, −2 and −3—can allow mGlu receptor clustering and a functional coupling of mGlu1 or mGlu5 receptors with other cell receptors, ion channels, or cytoskeleton proteins (Brakeman et al., 1997; Tu et al., 1999; Kammermeier et al., 2000; Fagni et al., 2000; Minakami et al., 2000). Homer-1a, the short homer isoform that is encoded by an early gene switched on by synaptic hyperactivity, is unable to form crosslinking with other Homer proteins and can act as a negative dominant by disrupting mGlu receptor clusters (Xiao et al., 2000). A number of studies show that Homer proteins critically affect the cell surface targeting of mGlu receptors (Ciruela et al., 2000; Ango et al., 2000). In cultured granule cells, for example, cotransfection of mGlu5 receptors with Homer-1a disrupts receptor clustering but allows the axonal targeting of the receptor (Ango et al., 2000). Thus, synaptic hyperactivity (with ensuing induction of Homer-1a) may substantially affect the subcellular distribution of group-I mGlu receptors, and therefore the functional response to mGlu1 or mGlu5 receptor agonists.
All these factors make the final response to a group-I mGlu receptor agonist highly unpredictable and rule out a possible clinical application for these drugs in neurodegenerative disorders. This pessimistic view is strengthened by the low central bioavailability of group-I mGlu receptor agonists and by their propensity to induce seizures.
Effect of endogenous activation of group-I mGlu receptors on neurodegeneration: mGlu1 and mGlu5 receptor antagonists as neuroprotective agents
mGlu1 receptor antagonists and neuroprotection.
In spite of the contrasting effects of group-I mGlu receptor agonists, mGlu1 receptor antagonists are always neuroprotective, indicating that endogenous activation of mGlu1 receptors contributes to the induction and/or progression of neuronal death. Both competitive and noncompetitive mGlu1 receptor antagonists (such as AIDA, LY367385, 4-carboxypheylglycine, and CPCCOEt) (Pellicciari et al., 1995; Clark et al., 1997; Watkins and Collingridge, 1994; Litshig et al., 1999) reduce excitotoxic neuronal death in mouse cortical cultures challenged with NMDA (Strasser et al., 1998; Bruno et al., 1999; Battaglia et al., 2001) and also are effective in protecting striatal neurons against NMDA or quinolinic acid toxicity (Bruno et al., 1999; Orlando et al., 2001; Battaglia et al., 2001). Neuroprotection by LY367385, AIDA, and CBPG also is observed in models that are closer to human pathology, such as oxygen glucose deprivation in mouse cortical cultures or rat organotypic hippocampal cultures, and the 4-vessel occlusion model in gerbils (Pellegrini-Giampietro et al., 1999a, 1999b; Bruno et al., 1999). Notably, no mGlu receptor ligands have been proven to be effective in animal models of permanent or transient focal ischemia. Interestingly, mGlu1 receptor expression is reduced after ischemic preconditioning in gerbil hippocampus (Sommer et al., 2000).
Recent studies have gained new insights into the mechanism whereby mGlu1 receptor antagonists attenuate neuronal death. mGlu1 receptors are expressed in GABAergic neurons, and their activation depresses inhibitory synaptic transmission (Baude et al., 1993; Gereau and Conn, 1995; Morishita et al., 1998; Bushell et al., 1999; Paquet and Smith, 2000); hence, Battaglia et al. (2001) have hypothesized that mGlu1 receptor antagonists are neuroprotective by enhancing GABA release (Fig. 2). This hypothesis is supported by the following observations: 1) neuroprotection by LY367385 or CPCCOEt against NMDA toxicity in culture is obliterated by GABA or SKF89976A (an inhibitor of GABA transporter) and is prevented by bicuculline and 2-hydroxysaclophen, which antagonize GABAA and GABAB receptors, respectively; 2) the same cocktail of GABA receptor blockers prevents the protective activity of CPCCOEt against excitotoxic death of striatal neurons in in vivo experiments; 3) toxic concentrations of NMDA can stimulate GABA release in the caudate nucleus of freely moving rats only if LY367385 or CPCCOEt are present in the perfusate; and 4) in cortico-striatal slices, DHPG depresses GABAergic IPSCs through the activation of mGlu1 receptors (Battaglia et al., 2001). This contrasts with the protective action of mGlu5 receptor antagonists, which does not involve a GABAergic component (Battaglia et al., 2001). Based on these findings, one expects that mGlu1 receptor antagonists are particularly helpful in the treatment of CNS disorders that are primarily caused by an impairment of inhibitory synaptic transmission, such as temporal lobe epilepsy associated with Ammon horn sclerosis and mossy fiber sprouting. Under these particular conditions, seizure activity results from the development of recurrent excitatory synapses within neighbouring dentate gyrus granule cells that overcome the control of inhibitory GABAergic synapses (McNamara, 1999). Seizure activity causes further damage of granule cells, thus generating a vicious cycle that might be interrupted by enhancing GABA release with mGlu1 receptor antagonists. Interestingly, the expression of mGlu1a receptors is up-regulated in the molecular layer of the dentate gyrus of patients with temporal lobe epilepsy, as well as in the dentate gyrus of kainate-treated and kindled rats (Blumcke et al., 2000). The relevance of mGlu1 receptors in the pathophysiology of temporal lobe epilepsy is strengthened by the evidence that DHPG induces long-lasting epileptiform discharges in the hippocampus (Merlin, 1999; Galoyan and Merlin, 2000), whereas mGlu1 antisense oligonucleotides are protective against hippocampal kindling (Greenwood et al., 2000). In addition, the mGlu1 receptor antagonists LY367385 and AIDA reduce sound-induced seizures in DBA/2 mice and in genetically epilepsy prone rats, and exert antiepileptic activity in lethargic mice (Chapman et al., 1999).
A potential drawback associated with the use of mGlu1 receptor antagonists is the induction of ataxia. mGlu1a receptors are located in the dendritic spines of Purkinje cells, which receive excitatory inputs from both parallel and climbing fibers (Nusser et al., 1994). MGlu1 knockout mice are ataxic and do not develop long-term depression (LTD) at the parallel fiber-Purkinje cell synapses (Aiba et al., 1994). In two patients in remission from Hodgkin disease, paraneoplastic ataxia has been associated to neutralizing autoantibodies directed against mGlu1a receptors; injection of these antibodies in mice induces reversible ataxia (Sillevis Smitt et al., 2000). In vivo studies with centrally available mGlu1 receptor antagonists are necessary to establish whether pharmacologic inhibition of mGlu1 receptors impairs motor coordination in experimental animals.
mGlu5 receptor antagonists and neuroprotection.
The noncompetitive mGlu5 receptor antagonists—MPEP, SIB-1893, and SIB-1757—are currently used as powerful tools to examine how endogenous activation of mGlu5 receptors affects neurodegeneration and neuronal function in general. However, the recent report that relatively high concentrations of MPEP or SIB-1893 (20 or 100 μmol/L) reduce NMDA currents in cultured cortical neurons (O'Leary et al., 2000; Movsesyan et al., 2001) casts doubts on the usefulness of these pharmacologic agents, and therefore deserves some comments. In oocytes expressing NR1 and NR2A or NR2B subunits, in which no partnership exists between NMDA and mGlu5 receptors, MPEP has no effect on NMDA currents. A slight inhibition (22% of controls) is only observed in cells expressing NR1 + NR2B subunits, but this effect is not statistically significant (Gasparini et al., 1999b). Neither SIB-1893 nor SIB-1757 has any significant antagonistic activity at recombinant NMDA receptors (Varney et al., 1999). The inhibitory effects of MPEP or SIB-1893 on NMDA currents in cortical neurons might reflect different regulatory properties of native versus recombinant NMDA receptors, or more likely, the existence of the functional link between NMDA and mGlu5 receptors (see below). One cannot exclude that inhibition of NMDA currents by MPEP and SIB-1893 derives from their ability to antagonize mGlu5 receptors that are endogenously activated by the glutamate released extracellularly or trapped in the plasma membrane. The authors can safely rely on experiments in which MPEP or SIB-1893 is used at concentrations less than 20 μmol/L, although it is their belief that even at 20 μmol/L the two drugs predominantly, if not exclusively, antagonize mGlu5 receptors.
MPEP, SIB-1757, and SIB-1893 are highly potent (substantial activity in the nanomolar range) in reducing NMDA toxicity in mixed cultures of cortical cells, and relatively low doses of MPEP (5 to 20 nmol) are protective against NMDA or quinolinic acid toxicity when locally injected into the rat caudate nucleus (Bruno et al., 2000b). To strengthen the specificity of this effect, it is noteworthy that iso-MPEP (an isomer of MPEP that does not interact with mGlu5 receptors) fails to protect against NMDA toxicity (Bruno et al., 2000b). Neuroprotection by MPEP in vitro and in vivo is not affected by GABAergic drugs (Battaglia et al., 2001) and can be ascribed to the ability of the drug to disrupt the functional coupling between mGlu5 and NMDA receptors. A growing body of evidence indicates that mGlu5 receptors positively modulate NMDA receptors (Awad et al., 2000; Ugolini et al., 1999; Salt and Binns, 2000; Doherty et al., 2000; Jia et al., 1998; Pisani et al., 1997), and that the two receptors are coexpressed in most of the neurons. The lack of both mGlu5 and NMDA receptors in Purkinje cells supports this view. A chain of anchoring proteins may physically link NMDA and mGlu5 receptors. The NR2 subunits of NMDA receptors interact with a series of “membrane-associated guanlyl kinase” (MAGUK) proteins (such as PSD-95, PSD-93, and SAP-102), which, through their PDZ domains, may assemble with the SHANK proteins. These proteins can interact with the long isoforms of Homer proteins, thus linking NMDA receptors with mGlu5 receptors (Tu et al., 1999). One mechanism by which mGlu5 receptors positively modulate NMDA receptors is mediated by the formation of diacylglycerol with ensuing activation of PKC. Protein kinase C can phosphorylate NMDA receptors, thus relieving the Mg2+ blockade of the NMDA-gated ion channel (Chuang et al., 2000). This modulation mediated by PKC has some structural requirements. In recombinant cells, only NMDA receptors containing the NR2A or NR2B subunits are positively modulated by PKC, whereas receptors containing the NR2C or NR2D subunits are not (Kutsuwada et al., 1992; Mori et al., 1993) (Fig. 2). The Ca2+ released from intracellular stores through the InsP3-sensitive channels (that also bind to Homer proteins) can induce changes in NMDA receptor activity after binding to calmodulin. Ca2+-calmodulin can compete with alfa-actinin-2 for a common binding site on the NR1 subunit, thus affecting the anchorage of NMDA receptors to the cytoskeleton (Niethammer et al., 1996; Wyszynski et al., 1997; Dunah et al., 2000). The interaction between NMDA and mGlu5 receptors is reciprocal. Activation of NMDA enhances mGlu5 receptor responses by preventing the homologous desensitization of mGlu5 receptors (De Blasi et al., 2001). This effect is mediated by calcineurin, which is activated by Ca2+ and dephosphorylates mGlu5 receptors at a PKC phosphorylation site (Alagarsamy et al., 1999). Thus, a vicious cycle exists in which NMDA and mGlu5 receptors potentiate each other. This particular form of receptor crosstalk has important implications in synaptic plasticity and in neurotoxicity. The expression of long-term potentiation is mediated by an increased activity of AMPA receptors, which results, at least in part, from the phosphorylation of Ser 831 of the GluR1 subunit by PKC and CaM kinase II (Scannevin and Huganir, 2000). A coactivation of mGlu5 and NMDA receptors might be required for a sustained activity of PKC and CaM kinase II, and, therefore, for the induction of a long-lasting, nondecremental form of long-term potentiation (Jia et al., 1998). A similar scenario can be proposed for the induction and progression of excitotoxic damage (Fig. 3). In cultured cortical cells challenged with NMDA, for example, it is known that the amount of glutamate released during and after the NMDA pulse contributes to the development of neuronal damage (Monyer et al., 1992). One can speculate that the combined activation of NMDA and mGlu5 receptors leads to the phosphorylation of AMPA receptors, and that activation of “hyperactive” AMPA receptors by the endogenous glutamate facilitates the progression of excitotoxic damage. Pharmacologic blockade of mGlu5 receptors during the induction phase of excitotoxic death would “isolate” NMDA receptors from its natural partner, thus reducing the strength of the excitotoxic insult below the death threshold. If this model is correct, then one expects that mGlu5 receptor antagonists are highly effective as neuroprotective agents in all CNS disorders in which activation of NMDA receptors is critically involved in the pathophysiology of neurodegeneration. Focal ischemia is one of these disorders, but no data on the effect of mGlu5 receptor antagonists are currently available. This may simply suggest that results are not encouraging. In the gerbil model of transient global ischemia, which is more refractory to NMDA receptor antagonists, MPEP is shown to be more effective than AIDA in increasing neuronal survival (Rao et al., 2000). However, this is not confirmed in another study in which only mGlu1 receptor antagonists and not MPEP are neuroprotective in ischemic gerbils and in cultured neurons subjected to oxygen–glucose deprivation (D. Pellegrini-Giampietro and F. Moroni, unpubished observation). The idea that mGlu5 receptor antagonists are promising drugs in the treatment of stroke is met with skepticism because of the heterogeneity of neuronal death, which includes necrosis (and/or apoptosis) directly caused by the shortfall in oxygen and glucose supply, and apoptosis secondary to the loss of synaptic inputs or neurotrophic factors (Lee et al., 1999). Knowing that activation of mGlu5 receptors supports neuronal survival during early development (Copani et al., 1998), it is easy to predict that mGlu5 receptor antagonists are detrimental for neurons that develop apoptosis by trophic deprivation in response to brain ischemia (Allen et al., 2000). One also should take into account that mGlu5 receptors are expressed in endothelial cells (Lin and Maiese, 2001; Morley et al., 1998), where their physiologic role is still unknown. Clearly, more studies are needed to clarify this issue.
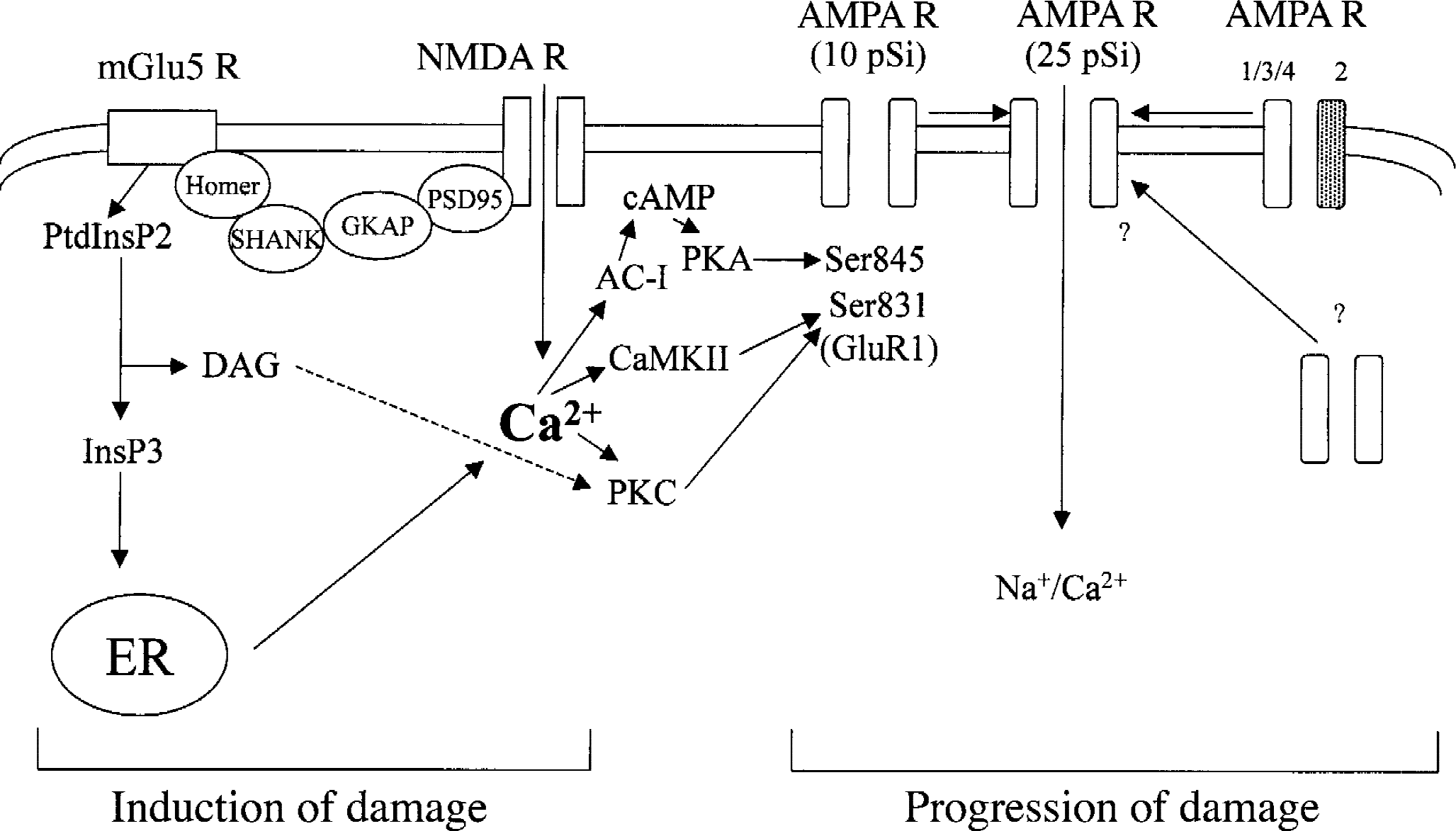
Speculative model on the interaction between N-methyl-d-aspartate (NMDA) and metabotropic glutamate 5 (mGlu5) receptors in the induction of excitotoxic damage (from Soderling and Derkach, 2000). The two receptors are physically linked by a chain of anchoring proteins, and act synergistically to activate a number of Ca2+-regulated proteins, such as Ca2+/calmodulin-dependent protein kinase II (CaMKII), protein kinase C (PKC), and type-I adenylyl cyclase (AC-I). This leads to the phosphorylation of specific Ser residues in the GluR1 subunit of alpha-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, thus increasing the activity of AMPA receptors. The possibility that AMPA receptors lose the GluR2 subunit (Pellegrini-Giampietro et al., 1992) or move from the cytosol to the membrane also is considered. Activation of AMPA receptors by endogenous glutamate may contribute to the progression of excitotoxic damage. ER, endoplasmic reticulum; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; DAG, diacylglycerol.
The authors believe that mGlu5 receptor antagonists may be beneficial during those conditions in which a toxic insult homogeneously impairs the survival of a specific neuronal population (that is, when the component of apoptosis by trophic deprivation is lacking). This is typified by toxicologic parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or by some amphetamine derivatives (such as metamphetamine), which selectively destroy the nigro-striatal dopaminergic pathway. The authors recently have found that systemically administered MPEP or SIB-1893 (but not CPCCOEt) is protective against degeneration of striatal dopaminergic terminals produced by three consecutive injections of metamphetamine in mice. MPEP also can reduce the formation of reactive oxygen species induced by metamphetamine in the striatum of freely moving mice without interfering with the uptake of metamphetamine into dopaminergic terminals (G. Battaglia et al., manuscript in preparation). This raises the intriguing possibility that endogenous activation of mGlu5 receptors contributes to the oxidative damage of the nigro-striatal dopaminergic pathway in response to amphetamine derivatives. mGlu5 receptor antagonists also can act as symptomatic drugs in experimental models of nigro-striatal degeneration. Activation of mGlu5 receptors induces direct excitation, and selectively potentiates NMDA currents in neurons of the subthalamic nucleus (Awad et al., 2000). As an increased activity of the subthalamic nucleus is implicated in the pathophysiology of bradykinesia, one can predict that mGlu5 receptor antagonists can improve motor symptoms resulting from the loss of nigro-striatal dopaminergic fibers. It would be interesting to study the efficacy of mGlu5 antagonists in the MPTP model of parkinsonism in mice or primates.
The evidence that activation of mGlu5 receptors enhances NMDA responses in striatal medium spiny neurons (Pisani et al., 2000) and restores NMDA toxicity in the striatum of decorticated animals (Orlando et al., 2001) suggests a potential application for mGlu5 receptor antagonists in the experimental treatment of Huntington chorea. However, the role of excitotoxic mechanisms in the pathophysiology of Huntington disease (DiFiglia, 1990) has been questioned by the unexpected finding that transgenic mice carrying an expanded CAG repeat in the Huntington gene are less sensitive to kainic acid toxicity (Hansson et al., 1999). It is certainly interesting to examine whether an early treatment with a centrally available mGlu5 receptor antagonist (like MPEP or SIB-1893) delays the onset of neurologic symptoms and reduces the neuropathologic hallmarks of the disease in these animals. Whether or not mGlu5 receptor antagonists may be beneficial in the experimental treatment of experimental amyotrophic lateral sclerosis (ALS) is currently unknown. Immunocytochemical analysis shows that spinal cord motoneurons express mGlu1 but not mGlu5 receptors (Alvarez et al., 2000), and this may explain the resistance of this particular neuronal type to NMDA toxicity. However, mGlu5 receptors may still be linked to the pathophysiology of ALS because they are heavily expressed on reactive astrocytes, which, in a mouse model of ALS, play an active role in motoneuron degeneration (Levine et al., 1999). Catania et al. (2001) have shown that reactive astrocytes in the ventral horns and white matter of autoptic spinal cord samples from sporadic or familiar ALS patients express high levels of nNOS. Activation of mGlu5 receptors in reactive astrocytes might provide a potential source for the Ca2+-stimulated formation of NO, thus contributing to neuronal damage through a process of glial-neuronal interaction.
Finally, studies performed on cortical cultures show that MPEP, SIB-1893, and SIB-1757 are highly potent against neurotoxicity induced by the active fragment of ß-amyloid peptide (fragment 25–35) (Bruno et al., 2000b). These experiments are performed in the presence of a cocktail of NMDA and AMPA receptor antagonists (MK-801 and DNQX), thus providing a model in which NMDA receptors do not contribute to neuronal death. Interestingly, inhibition of mGlu1 receptors by AIDA does not reduce but rather exacerbates ß-amyloid toxicity in cortical cells (Allen et al., 1999). Although the mechanism(s) underlying the neuroprotective effect of mGlu5 receptor antagonists is unknown, this evidence encourages the use of MPEP or its congeners in experimental animal models of Alzheimer disease, such as mice carrying a double mutation in the amyloid precursor protein and presenilin-1 genes.
Role group-II mGlu receptors in neuroprotection
mGlu2/3 receptor agonists are protective in experimental paradigms of neurodegeneration.
Activation of mGlu2/3 receptors produces inhibitory effects mediated by a reduction in intracellular cyclic adenosine monophosphate levels or by a decreased activity of voltage-gated Ca2+ channels (Pin and Duvoisin, 1995). This has encouraged the search for potent and selective agonists of potential use as neuroprotective agents. The search led to the discovery of a number of compounds, such as LY354740 and LY379268, which are potent and systemically active. LY354740 is currently in clinical trials but, curiously, it has been indicated for the treatment of anxiety rather than for the treatment of stroke or other neurodegenerative disorders (Helton et al., 1998; Klodzinska et al., 1999). The recent finding that mGlu3 receptors expressed by astrocytes control the production of putative neurotrophic agents (such as nerve growth factor or TGF-ß) leads to a serious reconsideration of these particular receptor subtypes in the therapy of neurodegenerative disorders. Initial studies on the role of mGlu2/3 receptors in neurodegeneration were performed in in vitro models—mixed cortical cultures, cultures of mesencephalic neurons, and cultures of cerebellar granule cells—using “old generation” agonists, such as DCG-IV, 4C3HPG, L-CCG-I, and 1S,3R-ACPD (Pizzi et al., 1993; Ambrosini et al., 1995; Bruno et al., 1994, 1995a, Buisson et al., 1995; Thomsen et al., 1996). In cortical cultures, these drugs were neuroprotective against neurotoxicity induced by a brief NMDA pulse and, interestingly, they retained their activity when applied up to 1 hour after the NMDA pulse (Bruno et al., 1995a). The authors have shown that group-II mGlu receptor agonists also are protective against neuronal toxicity induced by a prolonged exposure to kainic acid (Bruno et al., 1995a), although in another study this effect was restricted to the small subpopulation of cultured neurons that respond to kainate with an enhanced influx of Co2+ (Gottron et al., 1995). More recently, neuroprotection studies have been extended to “second generation” mGlu2/3 receptor agonists, such as 2R,4R-APDC, LY354740, LY379268, and LY389795. All these drugs were proven to protect neurons against NMDA toxicity in culture (Battaglia et al., 1998; Kingston et al., 1999a, 1999b; Behrens et al., 1999; D'Onofrio et al., 2001). However, the relatively low potency of LY354740 and LY379268 in the study of Behrens et al. (1999) led to the suggestion that neuroprotection was mediated by the activation of mGlu4 or mGlu8 receptors, which can be recruited by micromolar concentrations of both compounds (Schoepp et al., 1999). An alternative explanation is that native mGlu2/3 receptors have a lower affinity to LY354740 or LY379268 or that some unknown factor limits the access of these compounds to mGlu2/3 receptors in cortical cells. Initially, it was believed that mGlu2/3 receptor agonists were neuroprotective by reducing the release of endogenous glutamate. It was consistent with this hypothesis that an enhanced glutamate release occurring during or after the NMDA pulse contributes to the progression of neuronal damage (Monyer et al., 1992). However, although DCG-IV could reduce the release of glutamate in cultured cortical cells challenged with NMDA, this effect was lost when DCG-IV was applied 1 hour after the NMDA pulse, although the drug was still neuroprotective (Bruno et al., 1995a). The idea that some additional mechanisms could take place was supported by the evidence that the protein synthesis inhibitor, cycloheximide, prevented neuroprotection by DCG-IV in cultured cortical cells (Bruno et al., 1997). Unexpectedly, cycloheximide was interfering with a novel form of glial–neuronal interaction that involved mGlu3 receptors present in astrocytes (Petralia et al., 1996). The medium collected from cultured astrocytes challenged with any group-II mGlu receptor agonist (including the mGlu3-selective agonist, NAAG) was highly protective when transferred to cultured neurons challenged with NMDA (Bruno et al., 1997; Wroblewska et al., 1997). This protection was abolished when protein synthesis in astrocytes was inhibited by cycloheximide or when the glial medium was heated (Bruno et al., 1997). Thus, it appeared that activation of glial mGlu3 receptors promoted a novel form of glial–neuronal interaction mediated by the synthesis and release of a putative neuroprotective factor (Fig. 4). This is fully consistent with more recent evidence that LY354740 and LY379260 are much more effective against excitotoxic death in mixed cultures than in pure neuronal cultures (Kingston et al., 1999b). Different factors including nerve growth factor and protein S-100ß (Ciccarelli et al., 1999) are released from astrocytes in response to mGlu3 receptor agonists, but neuroprotection appears to be mediated by the synthesis and release of TGF-ß. Application of neutralizing anti–TGF-ß1 or anti–TGF-ß2 antibodies prevents neuroprotection induced by mGlu2/3 receptor agonists in mixed cultures and abolishes the protective activity of the glial medium. In addition, mGlu2/3 receptor agonists (including LY379268) enhance the de novo synthesis and release of TGF-ß1 and TGF-ß2 in cultured astrocytes (Bruno et al., 1998; D'Onofrio et al., 2001). The transduction pathway activated in response to glial mGlu3 receptors does not involve a reduction in cyclic adenosine monophosphate formation (as one would expect), but rather activation of the mitogen-activated protein (MAP) kinase and the PI-3-kinase pathways. Inhibitors of these pathways (that is, the compounds PD98059 and LY294002) reduce the ability of LY379268 to enhance the de novo synthesis of TGF-ß in astrocytes and to protect neurons grown in mixed cultures against excitotoxic death (D'Onofrio et al., 2001). This mechanism has been confirmed in in vivo studies, where LY379268 enhances the expression of TGF-ß1 in the corpus striatum and cerebral cortex and protects striatal neurons against NMDA toxicity through a mechanism that involves the activation of the mitogen-activated protein kinase pathway (D'Onofrio et al., 2001). Mitogen-activated protein kinases, P90rsk and P70S6k, which regulate gene expression and protein synthesis, could represent the downstream effectors of the de novo synthesis of TGF-ß (Fig. 4). The evidence that glial mGlu3 receptors regulate the production of TGF-ß extends the potential application of group-II mGlu receptor agonists to multiple forms of neuronal degeneration. Tumor growth factor-ß is protective against neuronal degeneration induced by excitotoxins, oxygen-glucose deprivation, ß-amyloid peptide, and the HIV capside protein gp 120 (Chao et al., 1994; Prehn et al., 1994, 1996; Copani et al., 1995a, b , 1999; Henrich-Noak et al., 1996; Meucci and Miller, 1996; Prehn and Miller, 1996; Tomoda et al., 1996; Ren et al., 1997; Bruno et al., 1998; Flanders et al., 1998; Ruocco et al., 1999; Scorziello et al., 1997). Its mechanism of action includes induction of serpins (Buisson et al., 1998; Docagne et al., 1999), induction of cell-cycle inhibitors (Datto et al., 1995; Copani et al., 1999), and inhibition of cyclooxygenase-2 (Pruzanski et al., 1998) (Fig. 4). Finally, the existence of this particular form of glial–neuronal interaction may explain the unexpected protective activity of mGlu2/3 receptor agonists against neuronal apoptosis induced by staurosporine (Buisson et al., 1995; Kingston et al., 1999a), or by ß-amyloid peptide in the presence of ionotropic glutamate receptor antagonists (Copani et al., 1995a). Interestingly, the latter effect is seen in mixed cultures of cortical cells, but not in cultured cerebellar granule cells, where only few astrocytes are present (Copani et al., 1995a).
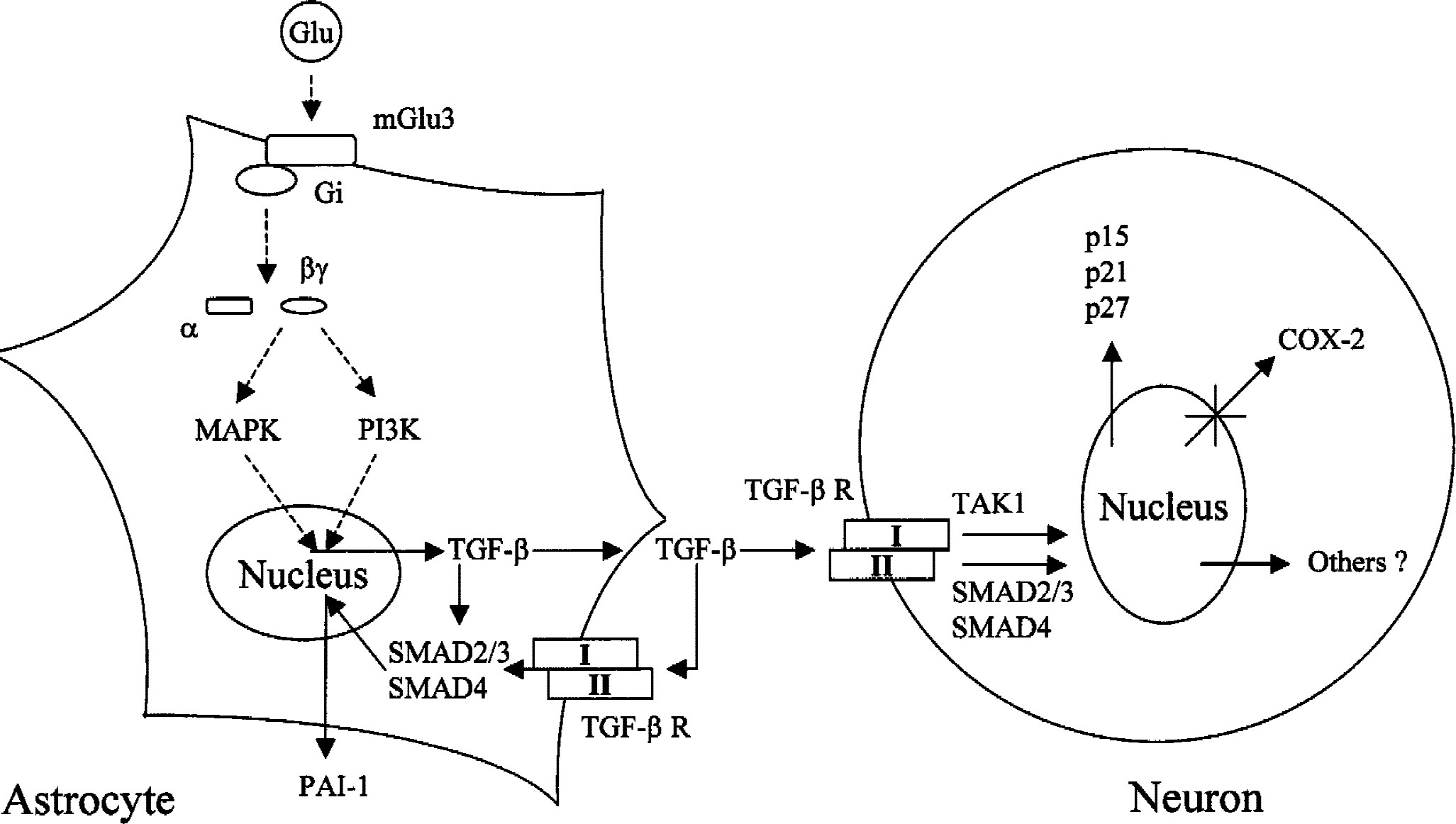
The neuroprotective effect of group-II mGlu receptors is mediated by glial metabotropic glutamate 3 (mGlu3) receptors and requires glial production of transforming growth factor-β (TGF-β), through the activation of mitogen-activated protein kinase (MAPK) and PI-3-K pathways. TGF-β released from astrocytes may exert its neuroprotective effect by an autocrine/paracrine mechanism on the glial cell inducing an overexpression of the serpin PAI-1 (plasminogen activator inhibitor type 1), which is then released from the astrocyte (left panel), and by acting in neurons through a set of high affinity serine-threonine kinases (TGF-ßRI and TGF-ßRII), which signal to the nucleus through the activation of SMAD2/3 and SMAD4 transcriptional factors or through the activation of transforming growth factor kinase 1 (TAK1). Downstream events include the induction of cell cycle proteins (p15, p21, and p27) or the inhibition of cyclooxygenase-2 (COX-2) expression. Other factors, still unknown, might contribute to TGF-β–induced neuroprotection (right panel).
mGlu2/3 receptor agonists as potential drugs in acute and chronic neurodegeneration.
Because of their particular mechanism of action, mGlu2/3 receptor agonists become serious candidates as neuroprotective agents. First, these drugs produce a “delayed rescue” effect in culture (Bruno et al., 1995a), which suggests a favourable therapeutic window in the treatment of brain ischemia. Second, the mechanism mediated by the release of neurotrophic factors extends the potential spectrum of these drugs to most of the acute and chronic neurodegenerative disorders, and may circumvent the major limitation associated with the systemic injection of neurotrophic factors, that is, the low penetration across the blood–brain barrier. Third, the impact of these drugs on fast excitatory synaptic transmission should be minimal because presynaptic mGlu2/3 receptors are less important than group-III mGlu receptors in the regulation of glutamate release, and glial mGlu3 receptors are not involved in the regulation of synaptic transmission. Finally, glial mGlu3 receptors appear to be localized in the vascular side of glial cell membranes (Shigemoto R, unpublished observation); therefore, they should not be saturated by the synaptically released glutamate. How are these predictions met by current evidence in in vivo models of neuronal degeneration?
The first evidence that activation of mGlu2/3 receptors is neuroprotective in vivo was provided by a series of studies performed in the laboratory of Dr. H. Shinozaki in Japan. Intracerebroventricular injection of DCG-IV was shown to protect vulnerable neurons against kainate toxicity (Miyamoto et al., 1994). This finding was remarkable because DCG-IV also can activate NMDA receptors, albeit at greater concentrations (Ishida et al., 1993). Data with “second generation” agonists on in vivo excitotoxicity are controversial. D'Onofrio et al. (2001) have shown that LY379268 protects striatal neurons against NMDA toxicity when injected locally or systemically, whereas no effect of LY354740 on striatal NMDA toxicity was reported by Behrens et al. (1999). A greater potency of LY379268 at native mGlu2/3 receptors or, more likely, the different doses of drugs used in the two experiments (10 nmol of LY354740 vs. 25 nmol of LY379268) may account for this difference. The effect of mGlu2/3 receptor agonists on ischemic neuronal damage has been studied in animal models of global or focal ischemia. In the gerbil model of global ischemia, intracerebroventricular injection of 4C3HPG protects hippocampal CA1 neurons when administered before or immediately after the induction of ischemia (Henrich-Noack et al., 1998). This drug, however, also behaves as a mGlu1 receptor antagonist (see above). Systemically injected LY354740 (Bond et al., 1998) or LY379268 (Bond et al., 1999) also are neuroprotective in the ischemic gerbil and in a neonatal rat model of hypoxia–ischemia (Cai et al., 1999). The protective activity of LY379268 against ischemic degeneration of CA1 pyramidal cells in gerbils is not mediated by an increase in the expression of TGF-ß (Bond et al., 1999, 2000). Interestingly, the hippocampus is the only brain region examined where local infusion of mGlu2/3 receptor agonists does not enhance the expression of TGF-ß, raising the possibility that the response of astrocytes to these drugs is region-specific (D'Onofrio et al., 2001). LY354740 and LY379268 do not reduce the infarct size in rat models of focal ischemia, that is, electrocoagulation, (Lam et al., 1998) and endothelin-1–induced (Bond et al., 1999) middle cerebral artery occlusion. This casts doubt on the possible efficacy of mGlu2/3 receptor agonists in the experimental therapy of stroke.
Recently, group-II mGlu receptors have been proposed as targets for the therapy of Parkinson disease. Overactive glutamatergic afferents from the subthalamic nucleus could cause both an excitotoxic loss of dopaminergic neurons in the substantia nigra and hyperactivation of GABAergic neurons in the globus pallidus, which leads to a reduction of motor activity (Albin et al., 1989). Drugs that can reduce glutamate release by activating presynaptic mGlu2/3 receptors might at one time delay the degeneration of substantia nigra neurons and improve motor activity (Konieczny et al., 1999; Wolfarth et al., 2000). No studies have been performed on the efficacy of systemically active mGlu2/3 receptor agonists in animal models of Huntington disease. However, the observation of an early reduction of mGlu2 receptors in the striatum of transgenic mice carrying expanded CAG repeats in the Huntington gene (Cha et al., 1998) raises the intriguing possibility that the loss of these receptors contributes to the pathophysiology of neuronal damage in Huntington chorea. The evidence that mGlu2/3 receptor agonists protect cultured spinal motoneurons against kainate toxicity (Pizzi et al., 2000) encourages the use of these drugs in animal models of amyotrophic lateral sclerosis, such as transgenic mice carrying mutations of superoxide dismutase. Group-II mGlu receptor agonists are protective against seizures in experimental animal models of epilepsy. Microinjections of 4C3HPG and 1S,3S-ACPD into the inferior colliculus reduce sound-induced seizures in genetically epilepsy-prone rats (Tang et al., 1997). Systemic injection of LY354740 exerts protective activity against seizures induced by ACPD, pentylenetetrazole, or picrotoxin (Monn et al., 1997; Klodzinska et al., 1999, 2000; Suzuki et al., 1999). It would be interesting to examine whether activation of mGlu2/3 receptors protects against neuronal damage that is secondary to neuronal hyperactivity, as observed in models of temporal lobe epilepsy.
GROUP-III mGlu RECEPTOR AGONISTS AS NEUROPROTECTIVE DRUGS
The pharmacology of group-III mGlu receptors (mGlu4, −6, −7, and −8) is less well developed than the pharmacology of the other subtypes because of the requirement for an omega-phosphonic group in agonist molecules, such as L-AP4 or PPG (see above). In addition, the strong acidic moiety limits the penetration of the drugs across the blood–brain barrier, and no centrally available agonists or antagonists have been developed so far. Thus, most of the available information derives from in vitro studies or from the effect of intracerebral injections of group-III mGlu receptor agonists. Group-III mGlu receptor agonists protect cultured cortical cells and cultured cerebellar granule cells against excitotoxic death (Bruno et al., 1995a, 1996, 2000a; Gasparini et al., 1999a; Lafon-Cazal et al., 1999) and apoptosis induced by the active fragment of ß-amyloid peptide (Copani et al., 1995a). In addition, group-III mGlu receptor agonists are protective against “mechanical injury” in neuronal cultures (Faden et al., 1997). In mixed cortical cultures challenged with NMDA, neuroprotection by L-AP4 or PPG is mediated by mGlu4 receptors, because it is no longer observed in cultures prepared from mGlu4 knockout mice (Bruno et al., 2000a). Interestingly, cultures from mGlu4 knockout mice are more sensitive to NMDA toxicity and release greater amounts of glutamate under basal conditions and in response to NMDA. High concentrations of PPG, which can recruit mGlu7 receptors, reduce NMDA-stimulated glutamate release in cultures lacking mGlu4 receptors, but never less than the levels observed in wild-type cultures challenged with NMDA (Bruno et al., 2000a). These data suggest that mGlu4 receptors are critically involved in the homeostatic regulation of glutamate release, whereas mGlu7 receptors are recruited only in response to high levels of extracellular glutamate. In vivo studies support a role for mGlu4 receptors in the control of glutamate release and excitotoxic neuronal death. mGlu4 receptor knockout mice are refractory to the protective activity of low doses of PPG coinjected with NMDA into the caudate nucleus. In addition, these animals show a much greater stimulation of striatal glutamate release by NMDA in microdialysis studies (Bruno et al., 2000a). Thus, mGlu4 receptor agonists should effectively prevent the component of excitotoxic neuronal death mediated by an excessive release of glutamate. Other possible mechanisms underlying neuroprotection include the inhibition of NMDA responses by postsynaptic group-III mGlu receptors (Martin et al., 1997) and the modulation of nitric oxide formation (Maiese et al., 1995, 2000). (R,S)-PPG improves the functional recovery of CA1 pyramidal cells of hippocampal slices subjected to a severe hypoxic/hypoglycemic insult (Henrich-Noak et al., 2000; Sabelhaus et al., 2000), providing evidence that is indicative of a protective activity against ischemic brain damage. Studies on experimental animal models of transient or focal ischemia await the synthesis of potent and centrally available receptor agonists.
Interestingly, activation of group-III mGlu receptors supports the survival of cultured cerebellar granule cells undergoing apoptosis by trophic deprivation (Graham and Burgoyne, 1994). This effect cannot be explained by any of the mechanisms outlined above, and suggests that activation of group-III mGlu receptors supports neuronal survival through some additional intracellular mechanisms. The authors recently have found that activation of group-III mGlu receptors stimulates the PI-3-kinase pathway in cultured cerebellar granule cells (Iacovelli et al., manuscript in preparation). The PI-3-kinase pathway has an established role in supporting cell survival and is activated in response to neurotrophic agents (such as insulin-like growth factor-I) that prevent apoptosis of cultured cerebellar granule cells (D'Mello et al., 1997). Whether this intracellular response is exclusive of group-III mGlu receptors expressed on cultured granule cells or can be extended to other neuronal types remains to be established.
Similar to group-I antagonists or group-II agonists, group-III mGlu receptor agonists exert anticonvulsant activity (Abdul-Ghani et al., 1997; Tang et al., 1997; Chapman et al., 1999; Gasparini et al., 1999a; Claudio et al., 2000). Although these effects are common to many drugs, a growing body of evidence suggests that mGlu4 receptors are directly involved in the pathophysiology of epilepsy (Lie et al., 2000).
Pentylenetetrazol-induced seizures lead to an increase in (3H)-L-AP4 binding in the cerebral cortex (Thomsen, 1999), whereas electrical kindling is associated with a lower ability of group-III mGlu receptor agonists to inhibit transmission at the lateral perforant path–dentate gyrus synapses (Klapstein et al., 1999). Human studies show that mGlu4 receptors are up-regulated in dentate granule cells and CA4 neurons in surgical hippocampal specimen obtained from patients with temporal lobe epilepsy (Lie et al., 2000), whereas a reduced function of L-AP4–sensitive mGlu receptors is observed in the epileptic sclerotic hippocampus (Dietrich et al., 1999). An attractive hypothesis is that seizure vulnerability of distinct neuronal populations is regulated by the levels of expression and function of mGlu4 receptors. A reduced activity of mGlu4 receptors may predispose vulnerable neurons to excitotoxic degeneration, thus contributing to the development of Ammon horn sclerosis. Interestingly, mGlu4 knockout mice show an increase in seizure susceptibility, and the human mGlu4 receptor gene falls within a susceptible focus for juvenile myoclonic epilepsy in the chromosome 6, band p21.3 (Wong et al., 2001). mGlu7 and mGlu8 also have been implicated in the pathophysiology of epilepsy. Although no association between polymorphisms in genes encoding mGlu7 and mGlu8 receptors and idiopathic generalized epilepsy has been found so far (Goodwin et al., 2000), mGlu7 knockout mice show increased seizure susceptibility (H. van der Putten, personal communication).
CONCLUSIONS
Based on recent studies with subtype-selective ligands, the authors can comment on which mGlu receptor subtype should be targeted by neuroprotective drugs in different acute or chronic neurodegenerative disorders (Table 2). For all of the reasons outlined above, mGlu1/5 receptor agonists should not be used as neuroprotective agents. mGlu1 receptor antagonists have a potential application in the treatment of brain ischemia or might be used for the prophylaxis of neuronal damage induced by synaptic hyperactivity, as occurs in temporal lobe epilepsy. Ataxia may represent a serious side effect of these drugs. mGlu5 receptor antagonists should be particularly indicated in all conditions in which excitotoxicity resulting from a sustained activation of NMDA receptors contributes to neuronal death. The impairment of apoptosis by trophic deprivation may limit the clinical efficacy of these drugs in focal ischemia. In the authors' opinion, mGlu5 receptor antagonists may be beneficial in the experimental treatment of chronic neurodegenerative disorders, such as Huntington chorea, ALS, or Alzheimer disease (see above). Inhibition of glial mGlu5 receptors could contribute to the neuroprotective activity of these drugs in chronic disorders. mGlu2/3 receptor agonists induce neuroprotection through two distinct mechanisms—the inhibition of glutamate release and the production of neurotrophic factors (such as TGF-ß) by astrocytes. The latter mechanism suggests a broad application for these drugs in neurodegenerative disorders and may allow circumventing the major limitation associated with the peripheral administration of neurotrophic factors, that is, their poor penetration across the blood–brain barrier. mGlu4/7/8 receptor agonists induce neuroprotection by reducing glutamate release. These drugs have a potential application in the experimental treatment of seizure disorders. mGlu4 and mGlu7 receptors seem to be seriously implicated in the pathophysiology of epilepsy.
Possible applications of subtype-selective mGlu receptor agonists or antagonists in human pathology

mGlu, metabotropic glutamate; GABA, gamma aminobutyric acid; NMDA, N-methyl-D-aspartate; ALS, amyothrophic lateral sclerosis.