
Abstract
The role of c-Fos in neurodegeneration or neuroprotection after cerebral ischemia is controversial. To investigate whether early c-Fos induction after ischemia is associated with neuroprotection, rats were subjected to 10 minutes of transient forebrain ischemia and c-Fos expression was examined. Resistant dentate granule cells and neurons in CA2–4 displayed more robust immunoreactivity than vulnerable neurons in the CA1 region of hippocampus during early hours of reperfusion. By 6 hours after reperfusion, c-Fos immunoreactivity was greatly diminished in all areas of the hippocampus. Administration of N-acetyl-O-methyldopamine (NAMDA), a compound previously shown to protect CA1 neurons against ischemia, increased c-Fos immunoreactivity in the CA1 vulnerable region at 6 hours after ischemia and protected SK-N-BE(2)C neurons from oxygen glucose deprivation. Further in vitro study showed that NAMDA potentiated phorbol-12 myristate-13 acetate (PMA)-induced c-Fos expression, AP1 binding activity, and late gene expression determined by chloramphenicol acetyltransferase (CAT) activity from AP1 containing tyrosine hydroxylase promoter-CAT fusion gene in SK-N-BE(2)C neurons. In vivo and in vitro results showed that a neuroprotectant, NAMDA, in concert with another stimulus (for example, ischemia or PMA) up-regulates c-Fos expression and suggested that the early rise of NAMDA-induced c-Fos expression in vulnerable CA1 neurons may account for neuroprotection by means of up-regulating late gene expression for survival.
In response to cerebral ischemia, neurons in the central nervous system express c-fos, an immediate early gene (IEG) (Kiessling et al., 1993; Kogure and Kato, 1993). The functional significance of c-fos expression after ischemia is controversial. The results of many in vivo and in vitro studies have shown that prolonged c-fos induction precedes neuronal death after ischemia and that c-fos and its gene products are involved in neuronal injury (Gubits et al., 1993; Hafezi et al., 1997; Heurteaux et al., 1993; Kasof et al., 1995; Preston et al., 1996; Smeyne et al., 1993; Wessel et al., 1991). Although c-fos and its gene product are involved in the induction of apoptotic genes that lead to cell death, its expression has been shown to be essential for recovery from ischemia (Akins et al., 1996). For example, it has been reported that suppression of postischemic Fos expression using a c-fos antisense oligonucleotide increased tissue damage (Zhang et al., 1999) and suppressed postischemic nerve growth factor expression in the hippocampus after focal cerebral ischemia (Cui et al., 1999). In addition, exacerbation of methamphetamine-induced neurotoxicity in c-fos null mutant mice and inhibition of apoptosis in a mutant form of c-fos that did not degrade by preteosome in lymphoma cells further support a possible neuroprotective role of c-fos/c-Fos (Deng and Kaufman, 1998; He et al., 1998).
In an animal model of transient global ischemia, CA1 pyramidal neurons in the hippocampus selectively die, whereas CA2–3 and DG neurons are spared (Kirino and Sano, 1984; Pulsinelli, 1985). In similar ischemic models, initial c-fos expression was largely confined to the resistant area, whereas the delayed second wave of IEG expression occurred in the susceptible regions (Dragunow et al., 1993, 1994; Hughes et al., 1998; Wessel et al., 1991). Further, the expression of its gene product, c-Fos, was often limited to the resistant subregions and was lacking in the vulnerable CA1 region (Domanska-Janik et al., 1999; Kiessling et al., 1993; Takemoto et al., 1995). The differential c-Fos expression between surviving and dying neurons at an early postischemic period, in conjunction with its bimodal expression, suggests that the early c-Fos expression may be critical for cell survival and that its lack in CA1 neurons may account for selective vulnerability.
The purpose of the current study was to investigate whether the early c-Fos increase after the postischemic period is associated with neuroprotection. To test the idea, rats were subjected to 10 minutes of global ischemia in the presence and absence of N-acetyl-O-methyldopamine (NAMDA), a neuroprotectant that significantly protected CA1 neurons after cerebral ischemia (Cho et al., 1999), after which the authors investigated whether neuroprotectant NAMDA potentiates early c-Fos expression in vulnerable CA1 neurons in vivo. To understand the functional significance of c-Fos up-regulation by NAMDA, the authors first investigated whether NAMDA protects neurons from oxygen glucose deprivation (OGD) in SK-N-BE(2)C neuron culture and then whether the compound influences downstream effects of c-Fos expression such as AP1 binding and late gene expression in vitro.
MATERIALS AND METHODS
All the materials for cell culture were obtained from Gibco (Grand Island, NY, U.S.A.). Chemicals that are not specified in the methods were obtained from Sigma (St. Louis, MO, U.S.A.). NAMDA was synthesized according to the method described previously by Cho et al. (1999).
Transient forebrain ischemia in rat (four-vessel occlusion model)
Animal surgery was in compliance with Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines set forth in the Public Health Service (PHS) manual Guide in the Care and Use of Laboratory Animals. Male Wistar rats weighing 200 to 250 g (Hill Top, Scottsdale, AZ, U.S.A.) were anesthetized with a mixture of halothane (1%), oxygen, and nitrogen and surgically prepared for four-vessel occlusion ischemia according to the method described by Pulsinelli et al. (1982). The next day, 10 minutes of four-vessel occlusion ischemia was induced by tightening the clasps around the common carotid arteries and the suture. To minimize variability, the following criteria were applied: loss of righting reflex and bilateral pupil dilation during ischemic period, and 20 ± 5 minutes of postischemic coma after 10 minutes of ischemia. The body temperature of all animals was kept at 37.5°C ± 0.5°C by a thermocouple-regulated heating lamp during the ischemia and reperfusion until the animals regained consciousness and reestablished thermohomeostasis.
Tissue preparation
Animals were anesthetized with sodium pentobarbital (120 mg/kg) and perfused transcardially with saline containing 0.5% sodium nitrite and 10 U/mL heparin sulfate followed by 4% cold formaldehyde in 0.1 mol/L sodium phosphate buffer (pH 7.2). The brains were further postfixed for 1 hour and stored in a 30% sucrose solution overnight. Using a sliding microtome, the dorsal hippocampus was sectioned at a 40-μm thickness for immunocytochemistry.
Immunocytochemistry
Immunocytochemistry was performed using an avidin-biotin peroxidase (ABC, Vectastain) method as described previously (Cho et al., 1999). Briefly, sections were washed in 0.1 mol/L phosphate-buffered saline (PBS), incubated in 0.1 mol/L PBS containing 1% bovine serum albumin and 0.2% Triton-X-100 for 30 minutes, and subsequently incubated overnight with specific c-Fos antibody (Oncogene, 1:10,000 dilution). The next day, the sections were incubated with appropriate secondary IgG (1:200 dilution) for 1 hour and avidin-biotin peroxidase for 1 hour in a humidified chamber. Phosphate-buffered saline (0.1 mol/L, pH 7.4) containing 0.5% bovine serum albumin was used to wash sections on slides between all steps. The antigen–antibody complexes were visualized by incubation for 5 minutes in 0.05% 3,3′-diaminobenzidine and 0.003% H2O2. These temporal conditions were strictly observed in all studies.
Cell culture
The immortalized human neuroblastoma SK-N-BE(2)C cells were grown and maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and penicillin–streptomycin at 37°C in a humidified incubator with 5% CO2. When the cells became 80% to 90% confluent, they were subcultured 1 to 5.
In vitro oxygen glucose deprivation
Oxygen glucose deprivation was produced by exposing SK-N-BE(2)C neurons to D-glucose-deficient Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and penicillin–streptomycin. The culture was immediately placed in a modular chamber (Billups-Rothenberg) that was flushed with 95% N2/5% CO2 gas mixture for 10 minutes either in the presence or absence of NAMDA. The hypoxic chamber was kept at 37°C in a tissue culture incubator for 24 hours, and surviving neurons were counted by the trypan blue exclusion method.
Western blot hybridization
Cells (1 × 106) were washed with PBS, collected, and centrifuged at 1,000 g for 5 minutes. To obtain total cell lysate, 0.6 mL RIPA buffer (1 × PBS, 1% NP40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, with freshly added 100 ng/mL phenylmethylsulfonylfluoride, 30 μL/mL aprotinin, 10 μL/mL 100 mmol/L sodium orthovanadate) was added to cells in a 10-cm cell culture plate. Collected cells were incubated for 30 to 60 minutes on ice and centrifuged at 15,000 g for 20 minutes at 4°C. Protein concentrations were determined (Bio-Rad, Hercules, CA, U.S.A.), and 30 to 50 μg protein was loaded for sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Electrophoresis was performed and proteins were transferred to nitrocellulose membrane using an electroblotting apparatus. Membranes were blocked overnight in tris-buffered saline (TBS) containing 0.1% Tween-20 and 5% dry milk and incubated first with specific c-Fos (Oncogene, 1:1,000) antibody and then horseradish peroxidase-conjugated secondary antibodies for 1 hour. Membranes were washed three times (30 minutes each) with TBS containing 0.1% Tween-20 and 0.5% dry milk between each step, and bands were visualized using the ECL+Plus Western blotting detection system (Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.).
Gel mobility shift assay for AP1
Micropurification of nuclear proteins from cells was performed according to Roy et al. (1991). Briefly, 4 mL NE1 (250 mmol/L sucrose, 15 mmol/L spermine, 0.5 mmol/L spermidine, 1 mmol/L dithiothreitol, 0.4 mmol/L phenylmethylsulfonylfluoride, 26 mmol/L KCl, and 2 mmol/L MgCl2) were added to cells (5–10 × 108) and then homogenized gently in a Dounce tissue grinder. To free nuclei, the cells were further homogenized with added Nonidet p40 (0.5%), washed twice, and pelleted at 3,000 g. One packed-cell-volume of NE2 (NE1 buffer containing 350 mmol/L KCl) was added to resuspend nuclei. The resuspended nuclei were incubated for 5 minutes on ice and gently homogenized by performing 20 strokes with Dounce tissue grinder. The homogenate was centrifuged for 90 minutes at 4°C (180,000 g) and the supernatant was dialyzed for 45 minutes against 50 mol/L KCl, 4 mmol/L MgCl2, 20 mmol/L K3PO4 (pH 7.4), 1 mmol/L β-mercaptoethanol, and 20% glycerol. Extracted protein concentrations were determined with a colorimetric assay (Bio-Rad).
The oligonucleotide containing the tyrosine hydroxylase (TH) AP1 motif (synthesized by Genelink) was identical to the regions of the TH 5′ upstream sequence ranging from −189 to −212 with the following sequence: 5′-TGAGGGTGATTCAGAGGCAGGTGC-3′ and 3′-ACTCCCACTAAGTCTCCGTCCACG-5′. The sense and antisense oligonucleotides were mixed in a 1:1 molar ratio and end-labeled with 32P using bacteriophage T4 polynucleotide kinase (Roche Molecular Biochemicals, Indianapolis, IN, U.S.A.) according to Sambrook et al. (1989). In a 20-μL volume, nuclear protein (3 μg) was incubated at room temperature for 20 minutes in binding buffer (50 mmol/L Tris [pH 7.5], 500 mmol/L NaCl, 5 mmol/L dithiothreitol, 5 mmol/L EDTA, 20% glycerol) in the presence of poly (dI-dC) (1 μg) with 32P-labeled probe. The samples were loaded on an acrylamide gel and run at 100 V in a low ionic strength buffer (0.25 × TBS) for 2 hours. The gels were dried and exposed to X-Omat films (Kodak, Rochester, NY, U.S.A.). For supershift assays, nuclear extracts were mixed with appropriate antibodies (c-Fos, Jun-D; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.). The mixture were incubated on ice for 20 minutes and then subjected to a gel retardation assay as described above.
Transient transfection analysis
A fusion construct containing 8.9 kb of a rat TH 5′ upstream DNA sequence (Min et al., 1994) and a reporter CAT gene was transiently transfected in SK-N-BE(2)C neurons by the calcium phosphate coprecipitation method as previously described (Kim et al., 1993). When cells reached approximately 50% confluence, they were transfected with 2 μg DNA overnight. Transfection was terminated by changing media and cells were treated according to experimental conditions overnight, harvested, suspended in 60 μL 0.25 mol/L Tris-HCl (pH 8.0), exposed to 3 freeze–thaw cycles, and then heated at 60°C for 10 minutes to inactivate endogenous acetylase. CAT activities were measured by counting radioactivity from the reaction mixture of 0.5 μCi [14C] chloramphenicol, n- butyryl coenzyme A (200 μmol/L), and 10 to 20 μL cell extract incubated at 37°C for 30 minutes.
Data analysis
For Western blot analysis and gel mobility shift assay, a representative autoradiogram was presented from at least two independent experiments, which showed similar results. Results for c-Fos positive neuron counts, c-Fos Western blot analysis, and CAT activity measurements were presented as mean ± SD and were analyzed using analysis of variance and the post hoc Newman–Keuls multiple comparison test.
RESULTS
Ischemia induces c-Fos in hippocampus during an early reperfusion period
The temporal profile of c-Fos expression during first 6 hours of cerebral reperfusion was investigated in the hippocampus of rats subjected to 10 minutes of four-vessel occlusion ischemia. Compared with the control hippocampus, c-Fos immunoreactivity was most prominent in dentate granule cells 1 hour after ischemia and reperfusion(Fig. 1). Three hours after ischemia and reperfusion, ischemia induced c-Fos expression in all subregions of hippocampus, and the expression was substantially reduced by 6 hours. Closer examination of c-Fos immunoreactivity 3 hours after ischemia revealed that there was less c-Fos expression in the vulnerable CA1 region compared with the resistant CA3 region and dentate granule cells. The data showed that ischemia induces differential temporal and spatial c-Fos expression, with more rapid and robust immunoreactivity in surviving hippocampal neurons.
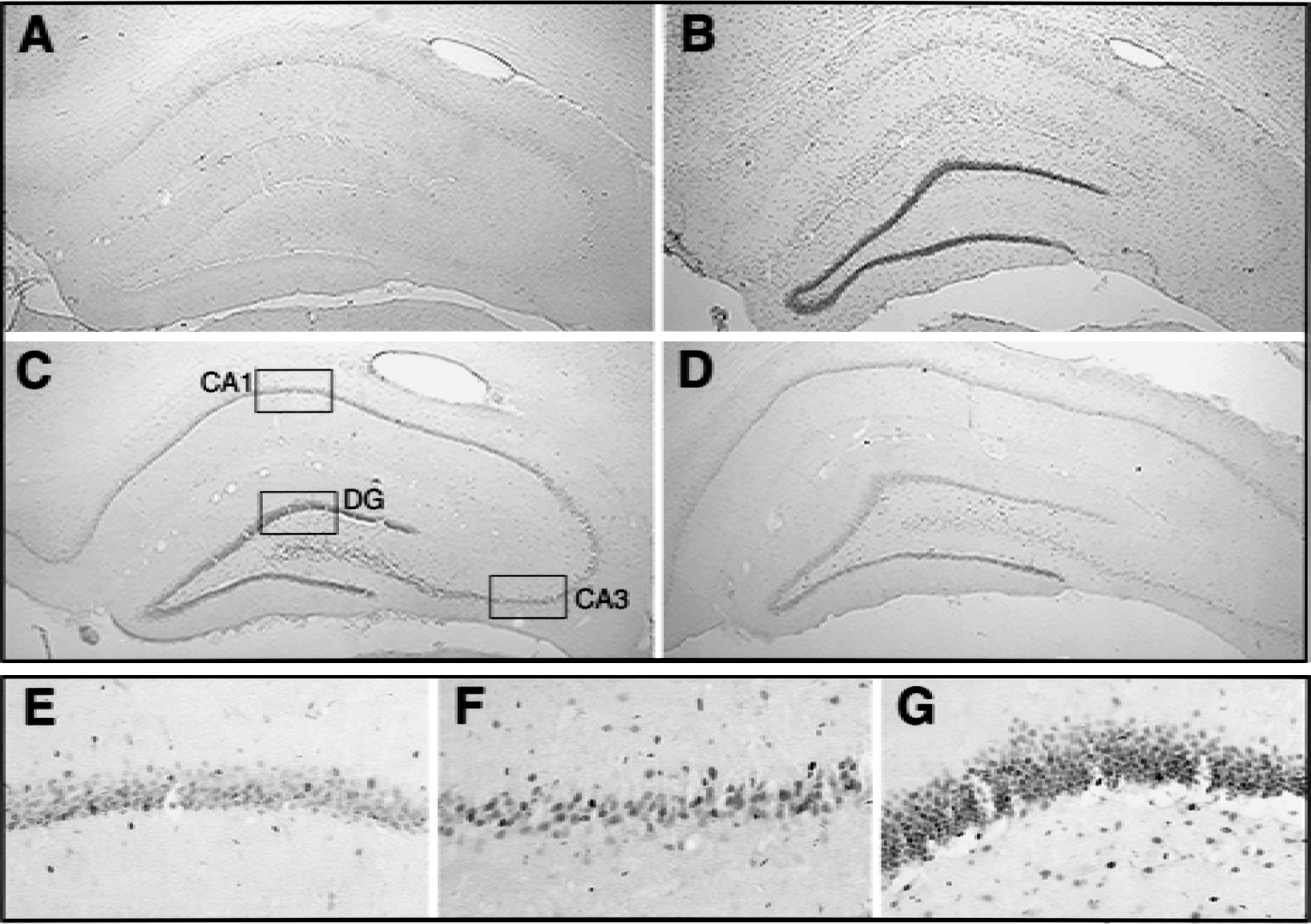
c-Fos immunoreactivity in rat hippocampus after ischemia.
NAMDA potentiates stimulus-induced c-Fos expression in vivo and in vitro
The authors investigated whether treatment with neuroprotectant NAMDA increases c-Fos expression in the vulnerable CA1 region during an early postischemic period. Although there was no visible c-Fos expression in control rats that were treated with either saline or NAMDA, ischemia induced modest c-Fos expression in the CA1 hippocampus at 6 hours after ischemia (Fig. 2). Treatment with NAMDA (triple injection of 10 mg/kg each at 0, 0.5, and 2 hours of reperfusion) potentiated ischemia-induced c-Fos expression in the vulnerable region. The data showed that NAMDA up-regulated ischemia-induced c-Fos expression.
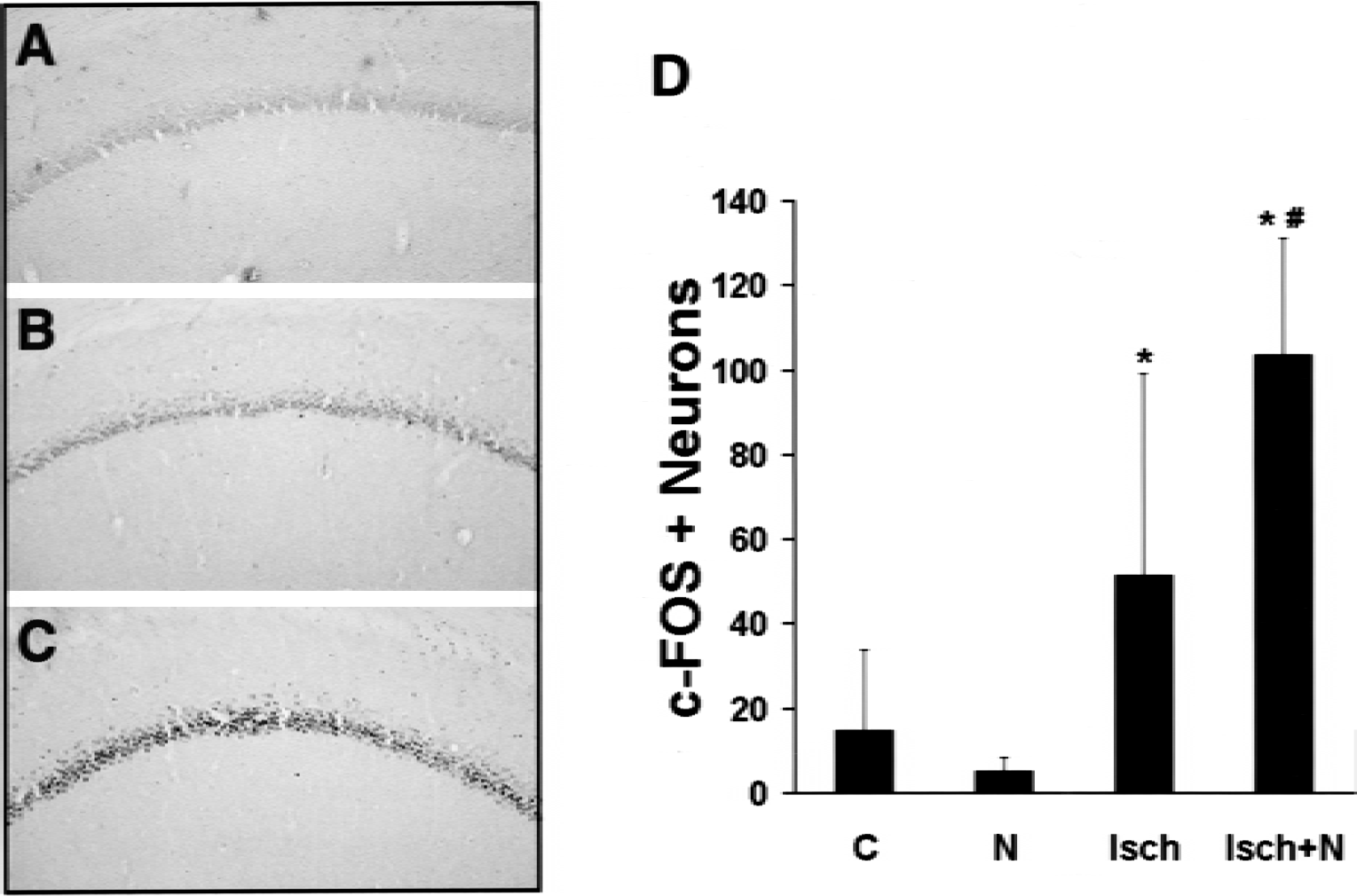
Potentiation of ischemia-induced c-Fos immunoreactivity by N-acetyl-O-methyldopamine (NAMDA) in CA1 neurons.
NAMDA protects neurons from oxygen glucose deprivation in vitro
To confirm the neuroprotective action of NAMDA in vitro, SK-N-BE(2)C neurons were subjected to 24 hours of OGD either in the presence or absence of 5 mmol/L NAMDA. Compared with control cultures, 24 hours of OGD resulted in neuronal death (Fig. 3). However, treating the neurons with 5 mmol/L NAMDA at the time of OGD rescued neurons significantly. Data showed that NAMDA protects SK-N-BE(2)C neurons from OGD, supporting previously shown in vivo neuroprotection by NAMDA against ischemia.
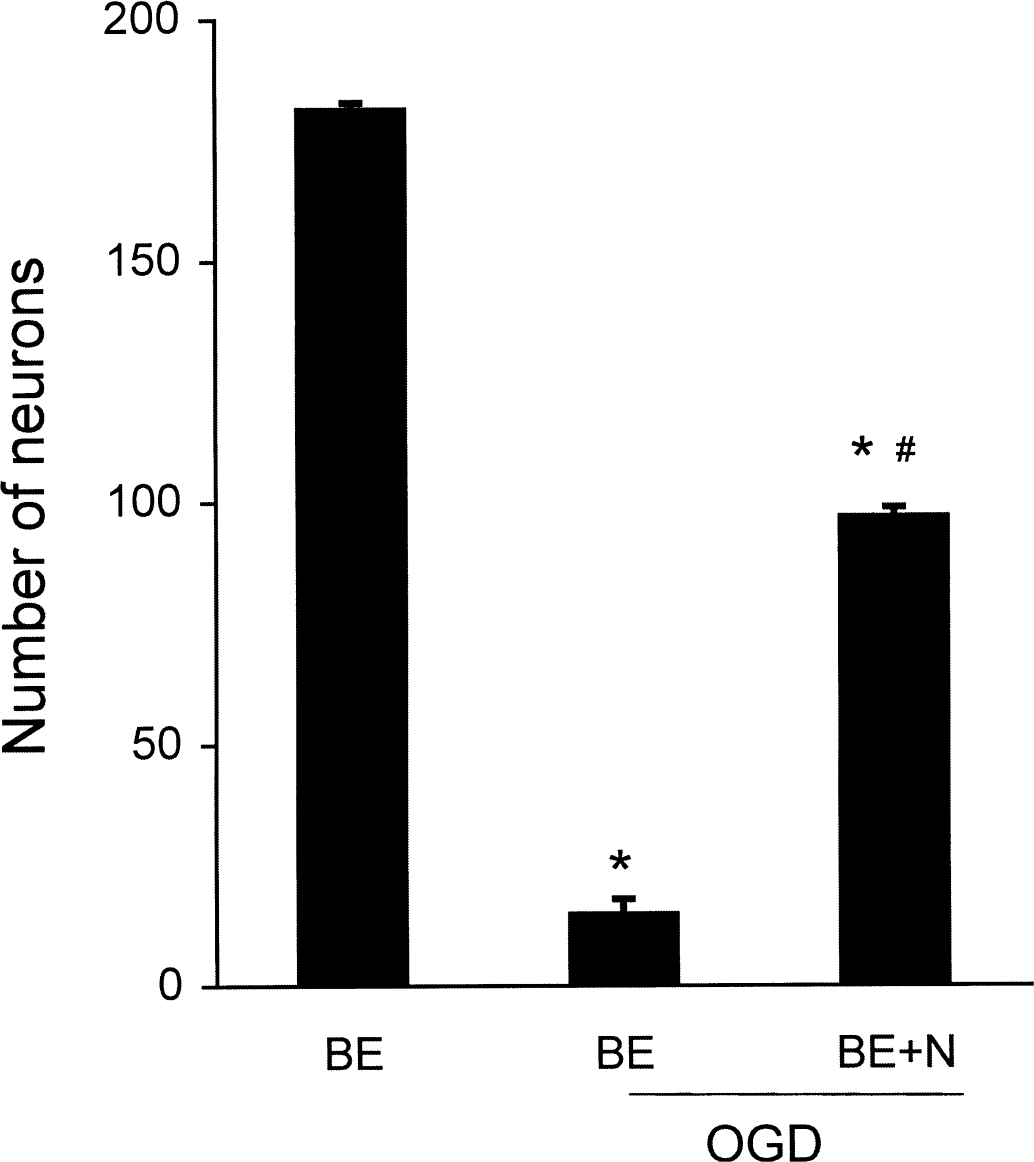
In vitro neuroprotection by N-acetyl-O-methyldopamine (NAMDA). Number of surviving SK-N-BE(2)C neurons after 24 hours of oxygen glucose deprivation (OGD) either in the presence or absence of 5 mmol/L NAMDA (* P < 0.01 vs. control; #P < 0.01 vs. OGD). OGD, 24 hours of OGD; OGD+N, 24 hours of OGD with 5 mmol/L NAMDA. BE, SK-N-B(2) C neurons.
NAMDA potentiates stimulus-induced c-Fos expression
To confirm NAMDA's ability to potentiate c-Fos expression in vitro, human neuroblastoma SK-N-BE(2)C cells were stimulated with phorbol-12 myristate-13 acetate (PMA) in the presence or absence of NAMDA and c-Fos expression was analyzed. The culture treated PMA but neither control nor the culture treated with NAMDA alone induced c-Fos expression (Fig. 4). The PMA-induced c-Fos expression was significantly potentiated by cotreatment with NAMDA. Taken together, the data showed that NAMDA potentiated c-Fos expression induced by ischemia (in vivo) or PMA (in vitro).
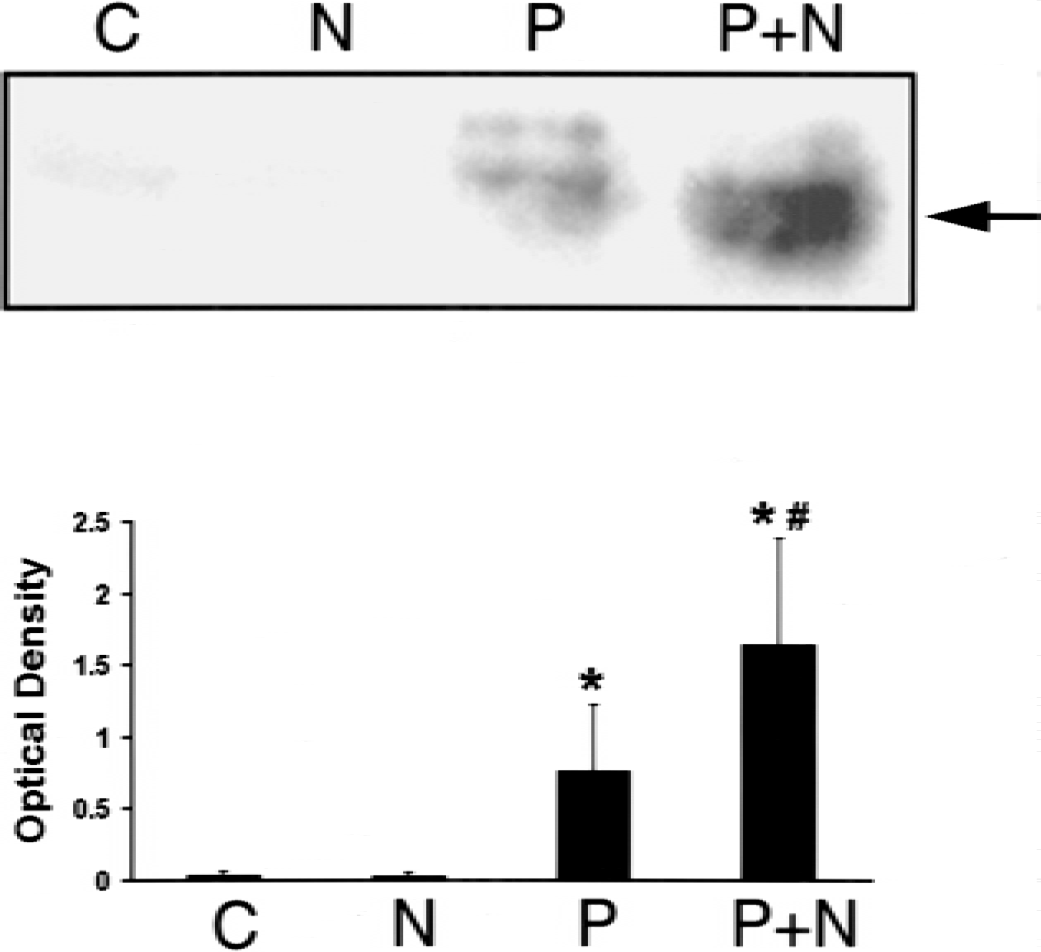
Potentiation of phorbol-12 myristate-13 acetate (PMA)-induced c-Fos expression by N-acetyl-O-methyldopamine (NAMDA) in SK-N-BE(2)C neurons.
NAMDA potentiates PMA-induced AP1 binding activity and AP1 containing late gene expression in neurons
The authors investigated whether up-regulation of c-Fos expression by NAMDA affects AP1 binding activities in PMA-stimulated SK-N-BE(2)C neurons. Control cells did not exhibit any detectable AP1 binding activity, and NAMDA treatment alone had no effect on AP1 binding. However, PMA substantially increased AP1 binding activity, and the PMA-induced AP1 binding activity was further augmented by NAMDA (Fig. 5). The DNA–protein complex was shifted by c-Fos, and the shift was more pronounced in cultures cotreated with NAMDA and PMA, indicating that c-Fos is the major component of the AP1 complex.
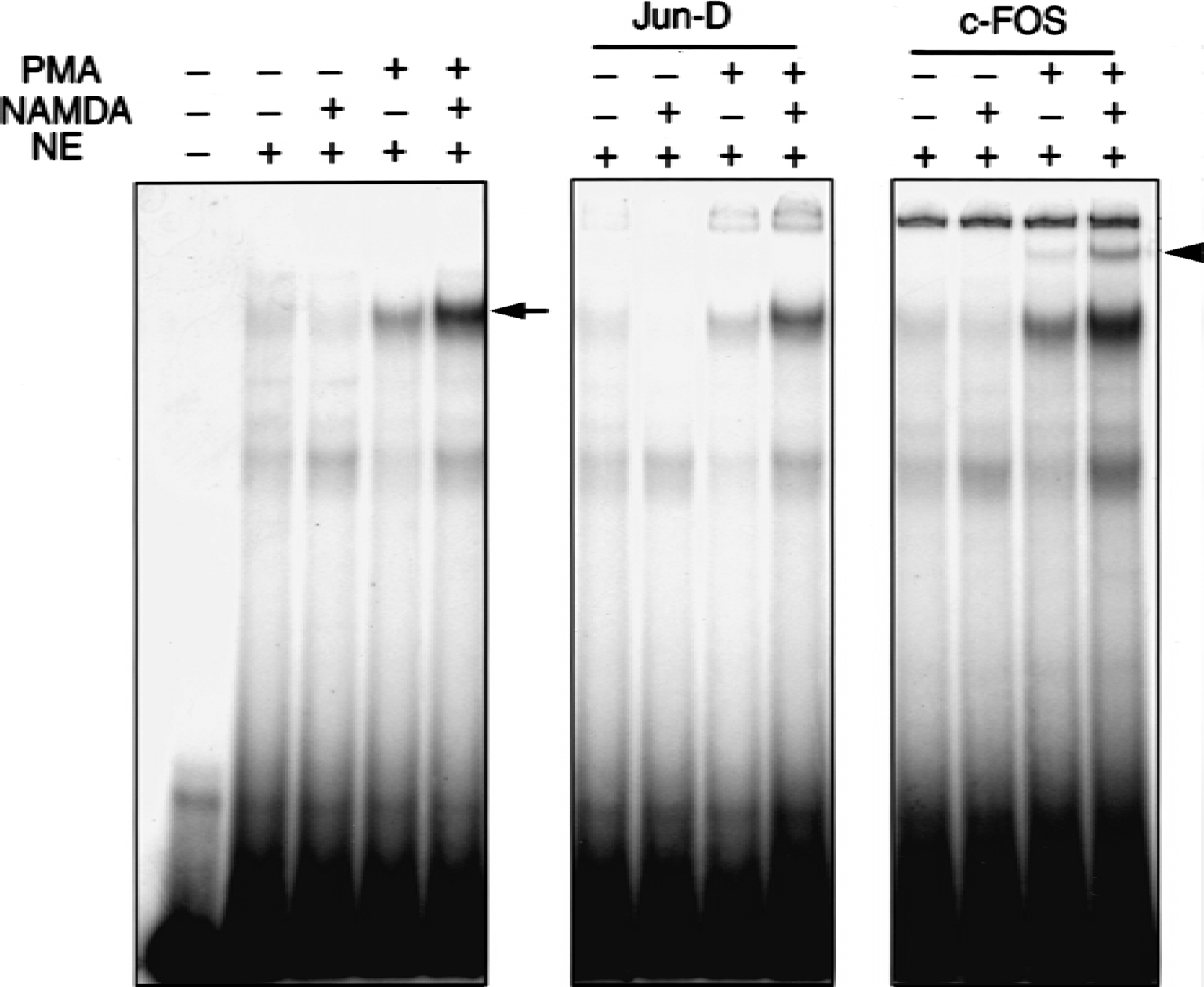
Effect of N-acetyl-O-methyldopamine (NAMDA) on AP1 binding in SK-N-BE(2)C neurons. Cultures were treated for 6 hours with phorbol-12 myristate-13 acetate (PMA; 80 nmol/L) or NAMDA (5 mmol/L) singly or in combination, and 5 μg nuclear extract (NE) was loaded in each lane for gel mobility shift assay. NAMDA potentiated PMA-induced AP1 binding activity (arrow), and the shifted DNA–protein complex by c-Fos antibody (arrowhead) was more pronounced by PMA + NAMDA cotreatment (arrowhead).
The promoter of rat TH contains cis-acting elements, including an AP1 site, that regulate gene transcription (Yoon and Chikaraishi, 1992). To investigate whether the potentiation of AP1 binding activity by NAMDA is functional, the authors transiently transfected a plasmid containing 8.9 kb upstream promoter of TH and reporter CAT gene (p8.9TH-CAT) and measured CAT activities in PMA-stimulated SK-N-BE(2)C cells. Compared with control, cultures treated with NAMDA alone did not affect CAT activity. However, treatment with PMA increased CAT activity by threefold. Furthermore, cultures cotreated with PMA and NAMDA significantly potentiated PMA-induced CAT activity (Fig. 6). Taken together, the data show that NAMDA potentiates PMA-induced AP1 binding activity and AP1 containing late gene expression in neurons.
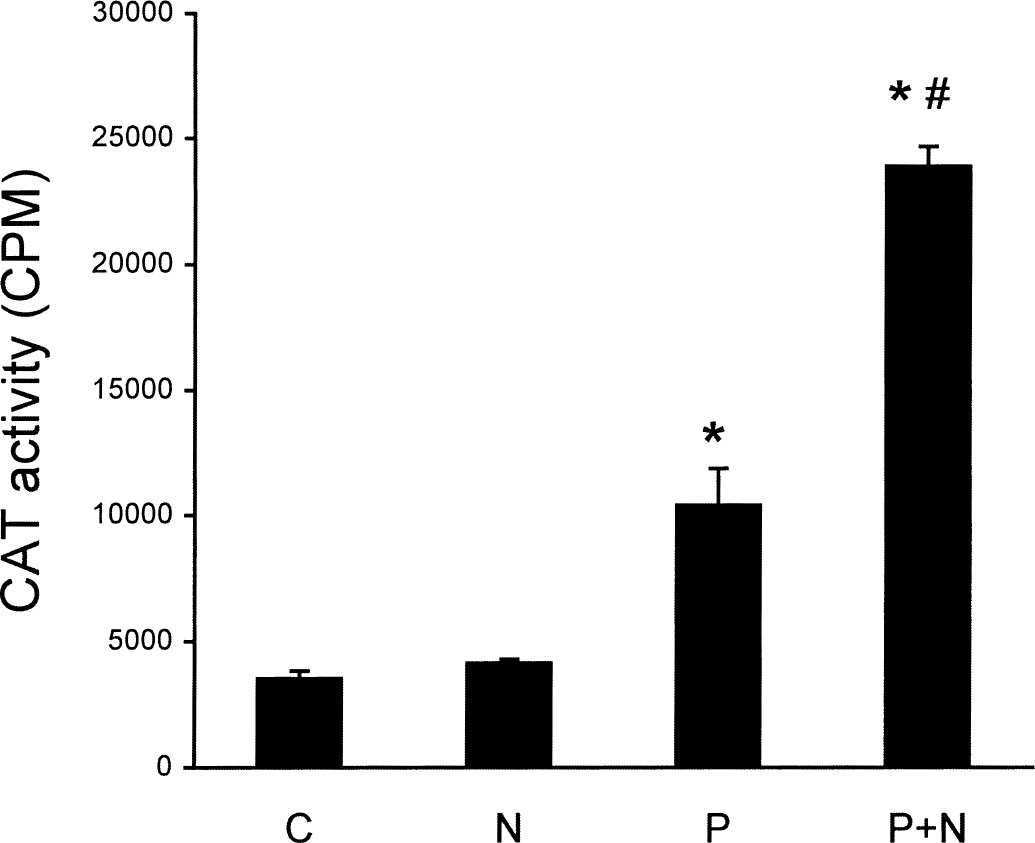
Effect of N-acetyl-O-methyldopamine (NAMDA) on late gene expression in SK-N-BE(2)C cells by transient transfection of TH-CAT construct. Cells were treated overnight with phorbol-12 myristate-13 acetate (PMA; 80 nmol/L) or NAMDA (5 mmol/L) singly or in combination. PMA significantly increased CAT activity, and the increase was potentiated by cotreatment with NAMDA (n = 4, mean ± SD; * P < 0.01 vs. control [C]; #P < 0.01 vs. PMA [P]). N, NAMDA; P+N, PMA + NAMDA. CPM, counts per minute.
DISCUSSION
Using a neuroprotectant NAMDA, the authors investigated a role of early c-Fos induction in CA1 neurons after cerebral ischemia. Compared with saline-treated ischemic animals, treatment with NAMDA increased the number of c-Fos immunoreactive neurons in the CA1 vulnerable region. NAMDA also potentiated PMA-induced c-Fos expression in SK-N-BE(2)C neurons, and the potentiation was accompanied by increased PMA-induced AP1 binding activity and AP1 containing late gene expression in the neurons.
Although severe ischemia caused c-Fos expression during a late reperfusion period, its expression has been shown to occur biphasically with mild ischemia (Takemoto et al., 1995). In the current 10-min four-vessel occlusion ischemic rat model, c-Fos expression was detected during an early reperfusion period. Consistent with findings by others (Domanska-Janik et al., 1999; Kiessling et al., 1993; Takemoto et al., 1995), the current authors observed stronger c-Fos expression in the resistant subregions, especially dentate granule cells, but moderate c-Fos expression in CA1 neurons. Interestingly, NAMDA potentiated ischemia-induced c-Fos expression in CA1 neurons and PMA-induced c-Fos expression in neuron cultures. Because administration of NAMDA after ischemia was previously shown to protect CA1 neurons (Cho et al., 1999), c-Fos expression is positively correlated with surviving neurons, and NAMDA potentiates c-Fos expression in vulnerable CA1 neurons, these observations indicate that the potentiation of c-Fos expression may be an underlying neuroprotective mechanism after ischemia.
The significance of c-Fos protein expression underlies the fact that it forms transcription factor–AP1 complex by dimerization with Jun family protein and that it regulates late gene expression. In cerebral ischemia, increased AP1 DNA binding in the hippocampus, thalamus, and striatum during early hours of reperfusion has been reported (Yoneda et al., 1994). However, the increased AP1 binding is transient in the vulnerable CA1 neurons but persistent in the resistant CA3 and dentate gyrus (Yoneda et al., 1997). Interestingly, AP1 binding in vulnerable CA1 neurons is potentiated by applying neuroprotective measures such as ischemic tolerance, hypothermia, shorter ischemic duration, and neuroprotectant bifemelane (Kapinya et al., 2000, Yoneda et al., 1997, 1998a, 1998b). The authors report here that neuroprotectant NAMDA also potentiated PMA-induced AP1 binding in vitro. These findings imply that potentiation of AP1 binding activity by means of increased c-Fos expression may account for NAMDA's ability to protect neurons against ischemia.
Alteration of gene expression responding to cerebral ischemia is not confined to IEGs. For example, heat shock proteins also are rapidly induced in the same time frame as IEGs and may be neuroprotective (Sharp and Sagar, 1994). Other genes that are altered after ischemia include basic fibroblast growth factor, nerve growth factor, brain-derived neurotrophic factors, and neurotrophins (Finklestein et al., 1988, 1990; Hsu et al., 1993, 1994; Lindvall et al., 1992; Takeda et al., 1993). The mechanism responsible for the possible induction of these late genes by IEGs is not known. However, temporal changes in binding activities of transcription factors that are related to IEGs such as c-Fos/AP1 may be involved in late gene expression. The results of the current study showed that increased c-Fos/AP1 binding by NAMDA influences expression of one of the late genes, such as AP1 containing TH gene. Thus, a candidate neuroprotective measure such as NAMDA administration might block neuronal injury by up-regulating survival genes by means of IEG activation.
In summary, the authors showed that potentiation of early c-Fos expression after cerebral ischemia may be associated with neuroprotection by means of up-regulating AP1 binding and late gene expression that is essential for neuronal survival. To elucidate the role of potential target genes for neuroprotection, further studies on the identification of late genes and products that are regulated by increased AP1 binding after ischemia are required.