
Abstract
Apoptosis is one of the two forms of cell death and occurs under a variety of physiological and pathological conditions. Cells undergoing apoptotic cell death reveal a characteristic sequence of cytological alterations including membrane blebbing and nuclear and cytoplasmic condensation. Early activation of an endonuclease has been previously demonstrated after a transient focal ischemia in the rat brain (Charriaut-Marlangue C, Margaill I, Plotkine M, Ben-Ari Y (1995) Early endonuclease activation following reversible focal ischemia. J Cereb Blood Flow Metab 15:385–388). We now show that a significant number of striatal and cortical neurons exhibited chromatin condensation, nucleus segmentation, and apoptotic bodies increasing with recirculation time, as demonstrated by in situ labeling of DNA breaks in cryostat sections. Apoptotic nuclei were also detected in the horizontal limb diagonal band, accumbens nucleus and islands of Calleja. Several necrotic neurons, in which random DNA fragmentation occurs, were also shown at 6 h recirculation, in the ischemic core. Further investigation with hematoxylin/eosin staining revealed that apoptotic nuclei were present in cells with a large and swelled cytoplasm and in cells with an apparently well-preserved cytoplasm. These two types of cell death were reminiscent of those described in developmental cell death. Our data suggested that apoptosis may contribute to the expansion of the ischemic lesion.
Although physiological cell death has been known for decades, interest in the subject was renewed in 1972 when Kerr and co-workers described in detail the ultrastructural changes characteristic of dying cells and coined the term apoptosis to describe this process (Kerr et al., 1972). Cells undergoing apoptosis display cell shrinkage and chromatin condensation associated with cytoplasmic blebbing (Wyllie et al., 1984; Kerr et al., 1987). In contrast, necrosis results when integrity of cytoplasmic membrane becomes compromised.
Several observations suggest that apoptosis plays an important role in the central and peripheral nervous system, where it regulates developmental and physiological cell death (for recent reviews see Raff, 1992; Raff et al., 1993; Vaux, 1993). More recently, apoptosis has also been demonstrated to play a role in pathological processes in the adult CNS. Thus, morphological criteria suggest that dentate granule cells undergo apoptosis after adrenalectomy (Sloviter et al., 1993) and CA3 pyramidal neurons after intraamygdaloid injection of kainate (Pollard et al., 1994).
The death of neurons that follows focal or global ischemia of the brain has been commonly described as necrosis rather than apoptosis. For example, morphological evidence of apoptosis was not present in ischemically injured rat hippocampus (Deshpande et al., 1992). However, transient ischemia in rodents is known to result in a delayed and relatively selective loss of specific neuronal populations (Pulsinelli et al., 1982). Recently, activation of apoptosis has been suggested to be involved in this delayed cell death based on protection offered by protein synthesis inhibitors (Goto et al., 1990; Shigeno et al., 1990; Papas et al., 1992). This was described as a particularly interesting feature of apoptosis in many situations (Wyllie et al., 1984) and has often been adduced as evidence for the active nature of apoptosis as compared with necrosis, referred to as accidental cell death. In addition to the collapse of chromatin into highly condensed electron-dense masses, the appearance of the ladder of DNA fragments due to the activation of a Ca2+-dependent endogenous endonuclease (Arends et al., 1990) was described as the primary biochemical event occurring in apoptosis. Direct evidence of oligonucleosome degradation of DNA from ischemic cortex and striatum has been recently demonstrated 24 and 48 h after global (Héron et al., 1993; MacManus et al., 1993) and focal (Linnik et al., 1993; Tominaga et al., 1993; MacManus et al., 1994; Charriaut-Marlangue et al., 1995) ischemia.
In the present study, we demonstrated that at earlier intervals after a reversible MCA occlusion, striatal and cortical neurons showed primarily morphological features of apoptotic cell death. Most cells exhibiting chromatin condensation and DNA fragmentation are localized to the inner boundary of the infarcted tissue. In contrast, we also found that some vulnerable neurons display necrotic cell death mainly located in the ischemic core. These data indicate that a significant apoptotic component may precede necrosis, and should offer new approaches of potential therapeutic intervention in stroke.
EXPERIMENTAL PROCEDURES
Focal ischemia model
Experiments were performed in strict accordance with National Institutes of Health guidelines. Male Sprague–Dawley rats (300–350 g) were anesthetized with chloral hydrate (400 mg kg−1, i.p.). Both common carotid arteries (CCAs) were isolated through a ventral midline neck incision, and loose ligatures were placed around them. The left middle cerebral artery (MCA) was exposed via a temporal craniectomy and was occluded at its proximal site with a Zen-type temporary clip (13 × 0.4 mm, Ohwa Tsuho Co., Ltd., Tokyo, Japan). Simultaneously, the CCAs were occluded. One hour after the onset of ischemia, the MCA clip was removed and recirculation within the CCAs was allowed for 3 (n = 3), 6 (n = 10), and 18 h (n = 10). Restoration of blood flow in all three arteries was observed under a microscope. During the surgical procedure, and until recovery from anesthesia, body temperature was maintained at 37–38°C.
Tissue preparation
For Hoechst 33258 and hematoxylin and eosin (H&E) staining, rats (6 h postischemia) were perfused via the ascending aorta while they were under deep anesthesia with FAM (formaldehyde, acetic acid, methanol; 1/1/8) as previously described(Lekieffre et al., 1992). Brains were left in situ for 24 h at 4°C, and then removed and stored in FAM for 1 week. They were then embedded in paraffin. Coronal sections were cut (at 10 μm) at the level of the septohippocampal nucleus (Paxinos and Watson, 1982).
For cresyl violet staining and in situ DNA nick-end labeling, rats were decapitated at 6 and 18 h recirculation. Whole brains were rapidly frozen in isopentane (−40°C), and subsequently stored at −70°C. Coronal cryostat sections (15-μm thick), at different levels, bregma 2.7 to −0.3 mm (Paxinos and Watson, 1982), were prepared on gelatin-coated slides, fixed with 4% paraformaldehyde, washed, dehydrated in graded alcohols, and frozen (−70°C) until use.
Histology
Cryostat coronal sections were stained with cresyl violet. On each section, striatal and cortical areas of infarction were measured using an image analyzer (Imstar, Paris, France). Volumes of infarction were calculated by integrating the necrotic areas.
Coronal sections (deparaffinized in xylene) were stained with H&E to identify morphological features of the lesion as described by Garcia et al. (1993).
Apoptosis assays
In situ labeling of fragmented DNA. Frontal cryostat sections were used for this study and processed according to the transferase dUTP nick-end labeling (TUNEL) technique described in Gavrieli et al. (1992). Briefly, sections were incubated with terminal deoxynucleotidyl transferase (0.3 U/μl; Gibco) and biotinylated dUTP (40 μM; Boehringer). The reaction product was visualized with streptavidin-biotin-peroxidase complex (Vectastain Elite ABC Kit, Vector Laboratories) and diaminobenzidine. In contrast, normal nuclei, which had relatively insignificant numbers of DNA 3′-OH ends, did not stain with this technique. Cell exhibiting necrotic morphologies, containing detectable concentrations of DNA ends, showed a more diffuse labeling as compared with apoptotic cells. For negative controls, incubation of sections was done in absence of either the enzyme or the nucleotide. Labeled (apoptotic and necrotic) cells were counted by the use of a 100 × oil immersion objective. We quantified apoptotic (AC, condensed chromatin) and necrotic cells (NC, uncondensed chromatin) in ischemic core and penumbra from ipsilateral ischemic hemisphere (n = 5) on morphological criteria already described herein. Data are presented as mean ± SD and as a ratio [R = AC/(AC + NC)], at 6 h after MCA and CCAs occlusion.
Nuclear staining. Nuclei of cells from deparaffinized coronal sections were stained using the specific DNA stain Hoechst 33258 (bisbenzimide, Sigma) as previously described (Dessi et al., 1995; Forloni et al., 1993). Briefly, sections were incubated with Ho 33258 (0.4 μg/ml) 10 min at room temperature, washed three times in distilled water, and then mounted with glycerol. Microscopy analysis was performed under conditions of normal illumination and fluorescent light.
RESULTS
Transient MCA occlusion induces a necrotic volume
One-hour MCA and CCAs occlusion followed by 6-h reperfusion resulted in the presence of dying neurons as shown after staining with cresyl violet in the frontoparietal cortex and in the caudate putamen as shown in Fig. 1A. With an 18-h recirculation delay, the necrotic volume increased both in the striatum and cortex (46 ± 2 and 212 ± 13 mm 3, respectively, n = to), although the border between this volume of dying neurons and the adjacent brain is less conspicuous with Nissl stain (Fig. 1B). A 24-h reperfusion resulted in the same but more delineated necrotic volume (not shown). The unilateral nature of the infarct allowed us to use the contralateral hemisphere as an internal control for the following experiments.
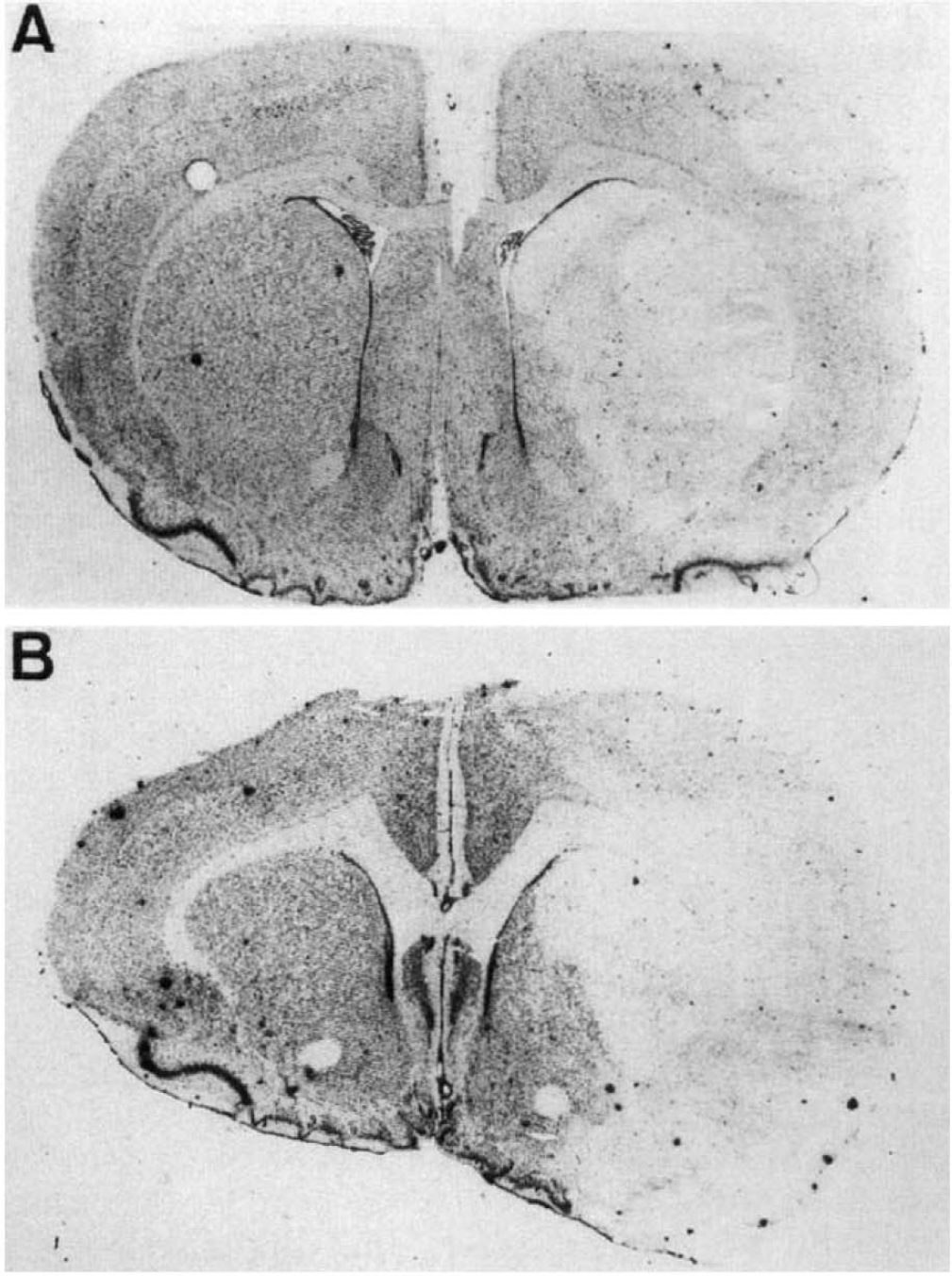
Ischemic lesion at two recirculation times after 1 h MCA occlusion (Cresyl violet, original magnification ×8).
In situ labeling of DNA fragmentation
Our previous study indicated that cell death induced by I-h transient focal ischemia leads to DNA laddering and early endonuclease activation as shown by high-molecular-weight DNA fragments (Charriaut-Marlangue et al., 1995). We have now analyzed a series of parameters generally adopted to identify the apoptotic cell death: morphological appearance of pyknotic nuclei (condensed chromatin) and apoptotic bodies formation. With the TUNEL method (Gavrieli et al., 1992), which allows detection of DNA breaks in cell nuclei, we observed a selective nuclear staining of scattered apoptotic cells mainly located in the head of the caudate putamen (CPu) and the horizontal limb diagonal band, 3 h after MCA occlusion (Fig. 2A and B). High magnification in light microscopy analysis showed chromatin condensation and numerous dense masses of membrane-bounded apoptotic bodies, then surface protuberances were separated with sealing of cytoplasmic membrane (arrowheads in Fig. 2A and B). At 6-h recirculation, apoptotic cells increased and were detected in clusters in the head of CPu showing chromatin condensation around the margin of the nucleus (Fig. 2C) and apoptotic bodies (Fig. 2D). Scattered apoptotic neurons with different types of chromatin condensation (forming either ring or patches) were also observed in the cortex (Fig. 3A-C). However, the presence of several necrotic cells was also detected in the striatum and the cortex at 6-h recirculation. Necrotic cells show a large, diffuse, light brown color in both cell nucleus and cytoplasm, without apoptotic bodies (arrows in Figs. 2D and 3C), suggesting random DNA fragmentation and fragility of the nuclear membrane. Coronal sections from rats killed at 18- and 24-h recirculation, labeled with TUNEL staining, showed apoptotic and necrotic nuclei in spongy tissue with nonspecific staining of cellular debris (not shown). Sometimes, nuclear labeling (in two or three cells) was detected in the contralateral hemisphere (in the lateral septum) of 3-h postischemic rats (in two animals) but not later.
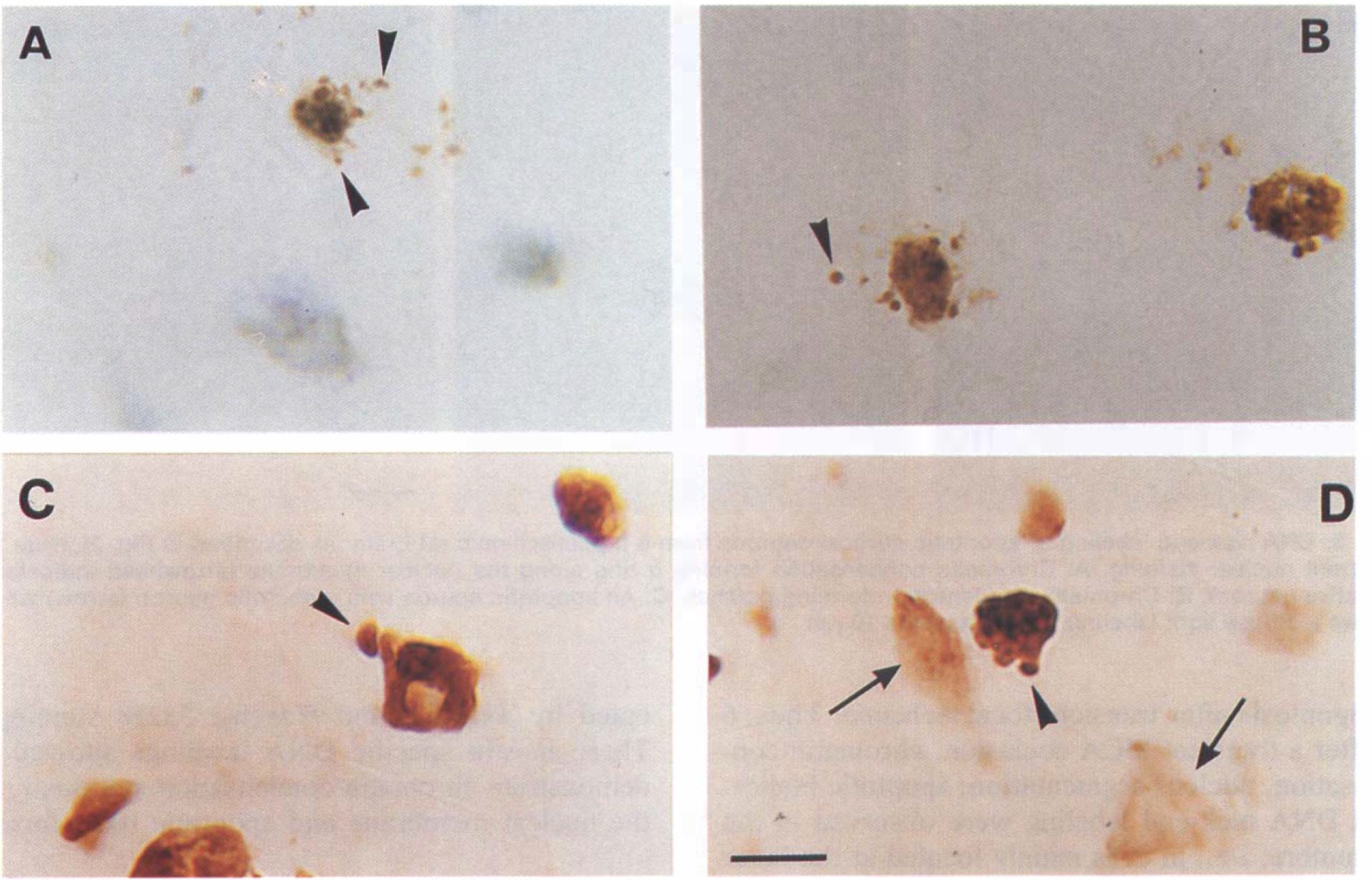
DNA nick-end labeling of apoptotic neurons from 3
DNA nick-end labeling of apoptotic cortical neurons from 6 h postischemic rat brain (as described in Fig. 2). Note the different nuclear staining.
Mapping of in situ DNA fragmentation
Figure 4 shows the localization of apoptotic neurons (with chromatin condensation and/or apoptotic bodies as determined with TUNEL staining) within the ipsilateral ischemic hemisphere at 3- and 6-h reperfusion after I-h MCA occlusion, in coronal sections from bregma 2.7 to −0.3 mm (Paxinos and Watson, 1982). Only a few apoptotic neurons shown at 3 h (Fig. 4A) were present in the border of CPu, in the frontoparietal cortex, in the lateral septum (Fig. SA) and the horizontal limb diagonal band. Furthermore, two to four apoptotic cells were also observed in the ipsilateral corpus callosum (Fig. 5B), but not seen at later recirculation intervals, At 6-h recirculation (Fig. 4B), numerous clustered apoptotic neurons were concentrated in the head of the CPu, the accumbens nucleus, the olfactory tubercle, and in the islands of Calleja (Fig. 5C). Quantification of apoptotic (AC) and necrotic (NC) cells indicates that the ratio ([R = AC/(AC + NC)] see Experimental Procedures) is ∼0.9 ± 0.05 in the head of CPu and the inner borders of necrotic volume as compared with a ratio of 0.5 ± 0.06 in the ischemic core (Table 1). In the cortex, only scattered apoptotic neurons were observed with several necrotic neurons. Our results indicated that apoptosis was mainly observed in ventrocaudal areas, whereas necrosis was shown in rostrodorsal areas.
Quantification of ipsilateral apoptotic (AC) and necrotic (NC) nuclei in different areas in coronal sections, taken at the level of the septohippocampal nucleus (d level in Fig. 4), from rats (n = 5) subjected to 1-h MCA and CCA occlusion followed by 6-h reperfusion
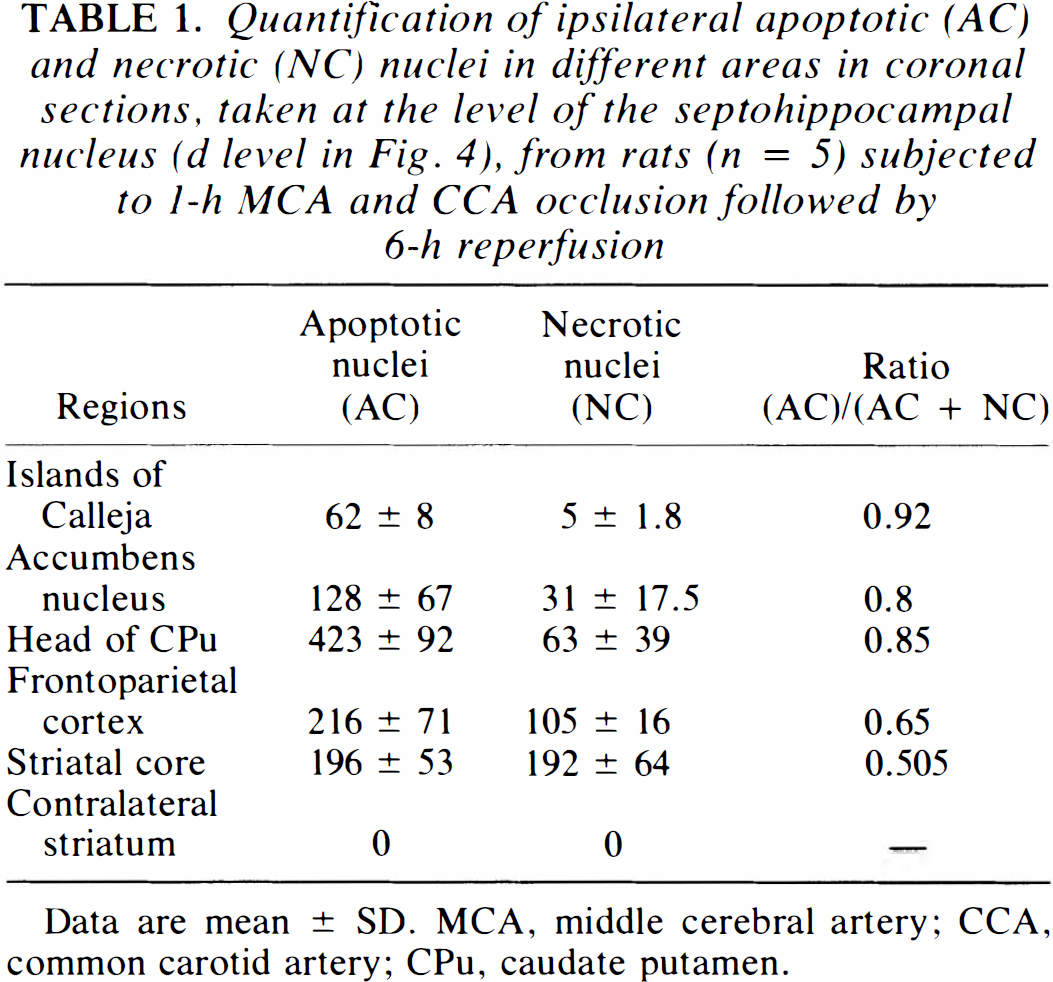
Data are mean ± SD. MCA, middle cerebral artery; CCA, common carotid artery; CPu, caudate putamen.
Schematic diagrams to illustrate the regional distribution of apoptotic cells, as demonstrated by the in situ DNA nick-end labeling, in ischemic rats at 3 (,  ,
,  ,
,  areas respectively (see also Table 1). Acb, accumbens nucleus; cc, corpuscallosum; iCj, islands of Calleja; LH, lateral septum; HDB, horizontal limb diagonal band.
areas respectively (see also Table 1). Acb, accumbens nucleus; cc, corpuscallosum; iCj, islands of Calleja; LH, lateral septum; HDB, horizontal limb diagonal band.
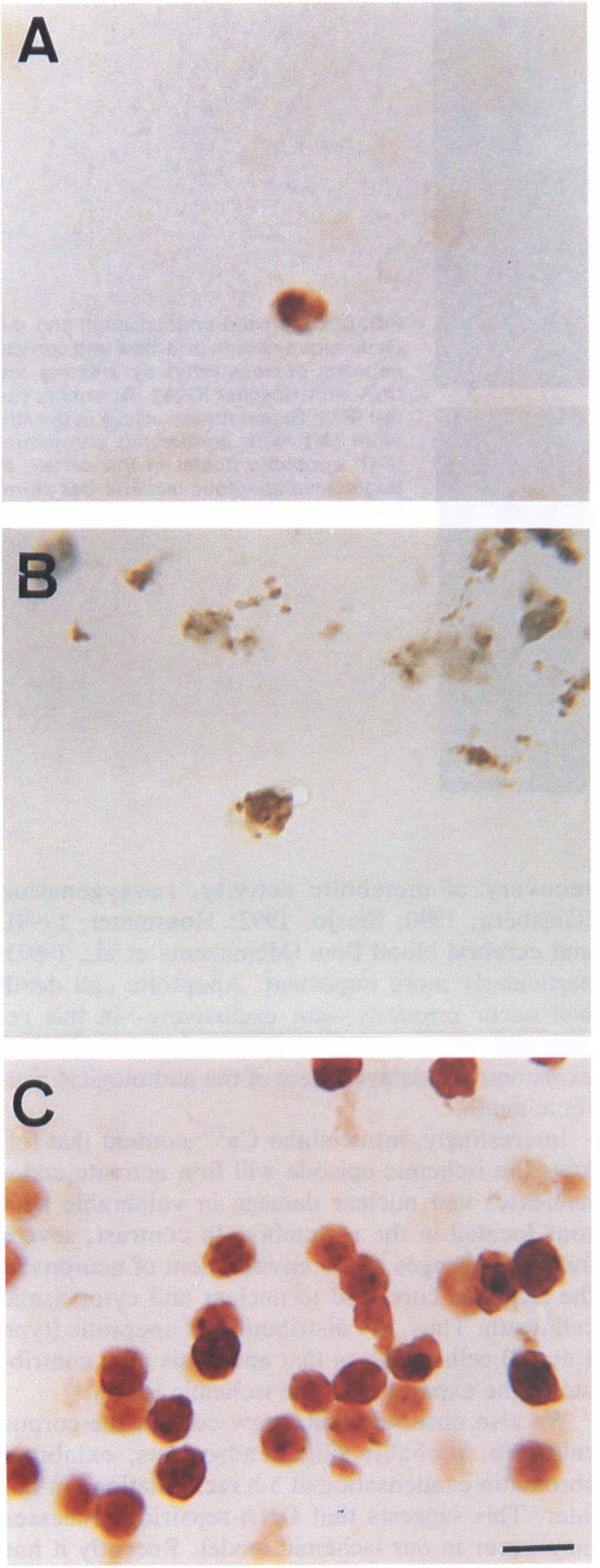
DNA nick-end labeling of apoptotic neurons located in corpus callosum
Chromatin condensation and segmentation of the nucleus
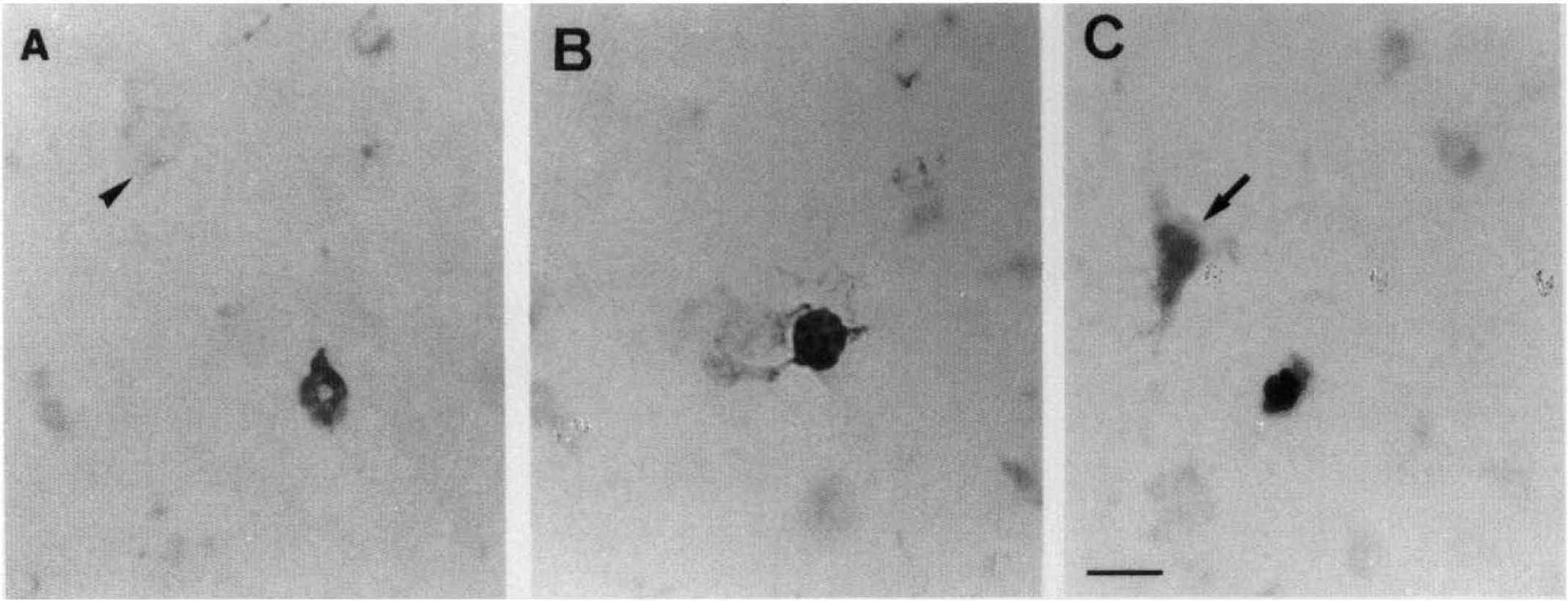
Figure 6 shows typical morphology of apoptotic nuclei stained with the DNA-binding fluorophore Hoechst 33258, located in the striatum (Fig. 6B) and the cortex (Fig. 6C and D) in rats n = 4 at 6-h recirculation. The nuclei appear slightly smaller than normal nuclei (Fig. 6A), probably because apoptotic cells have a rounded shape (Oberhammer et al., 1994), whereas normal cells are flat. Chromatin has condensed and aggregated at the nuclear membrane as indicated by a bright fluorescence at the periphery (Fig. 6B) followed by nuclear fragmentation and apoptotic body formation, as shown in Fig. 6C. Sometimes, the condensation of the chromatin occurs in the center of the nucleus (Fig. 6D), indicating perinucleolar aggregation of the chromatin, thus suggesting dissolution of the nucleolus (Oberhammer et al., 1993). H&E staining revealed both necrotic and apoptotic cell death in the cortex and the striatum after MCA occlusion (Fig. 7). The light microscopic identification of apoptosis depended on the recognition of nuclei containing pyknotic chromatin (arrows in Fig. 7A), sometimes showing segmentation of the nucleus (Fig. 7C). Presence of small rounded masses, suggesting the formation of apoptotic bodies, around the nuclear membrane was also identified (Fig. 7B). However, cytoplasmic staining indicated that two types of cell death occur in dying neurons with condensed chromatin: cells with a large and swollen cytoplasm, suggesting intracellular vacuolization (small arrows in Fig. 7B) and cells with an apparently well-preserved cytoplasm (arrowheads in Fig. 7C). In contrast, necrotic swollen cells showed diffuse chromatin staining with apparent nuclear membrane disruption and vacuolization in the cytoplasm (open arrow).
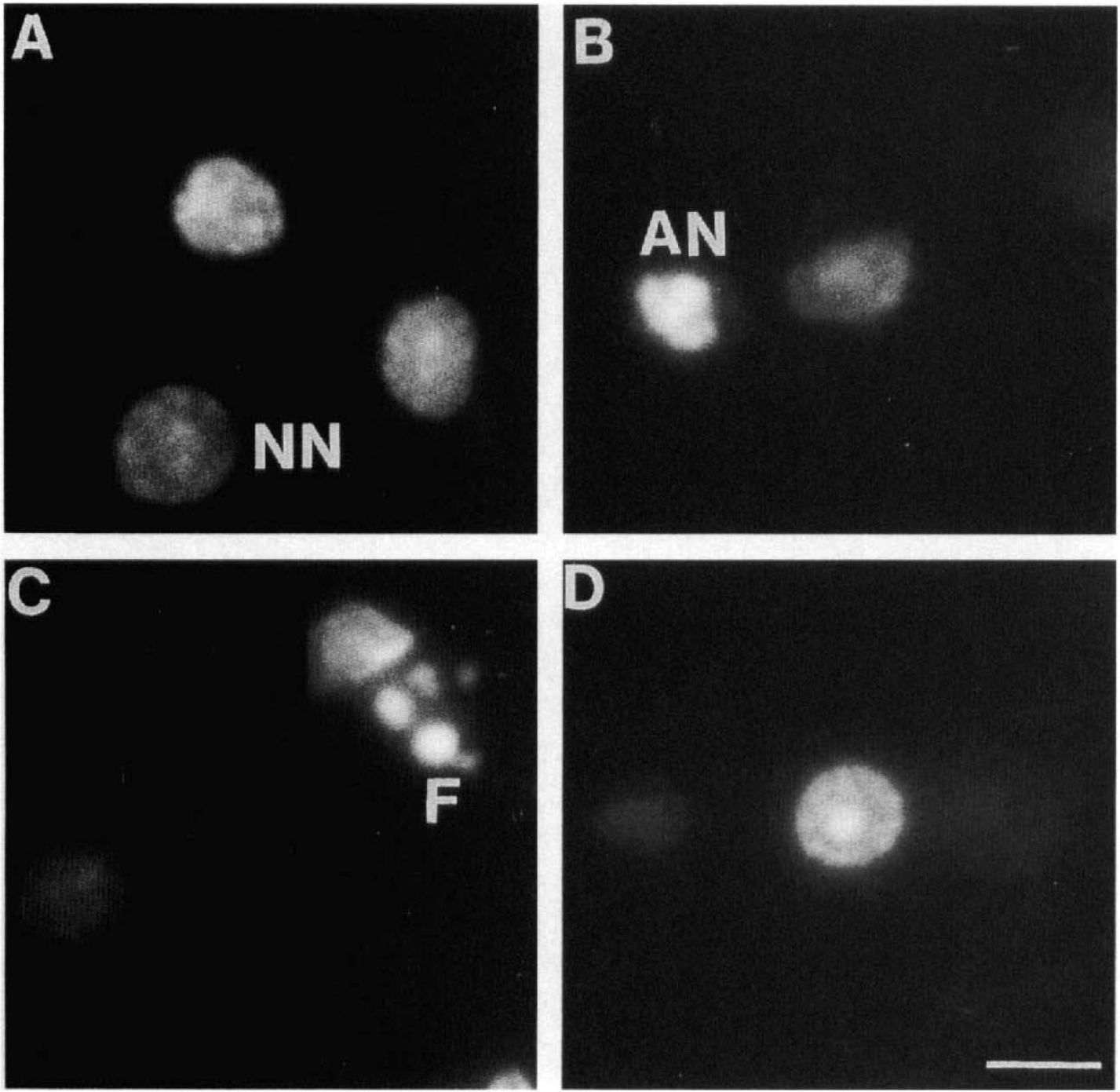
Chromatin condensation and nucleus segmentation in striatal and cortical neurons demonstrated by staining the DNA with Hoechst 33258.
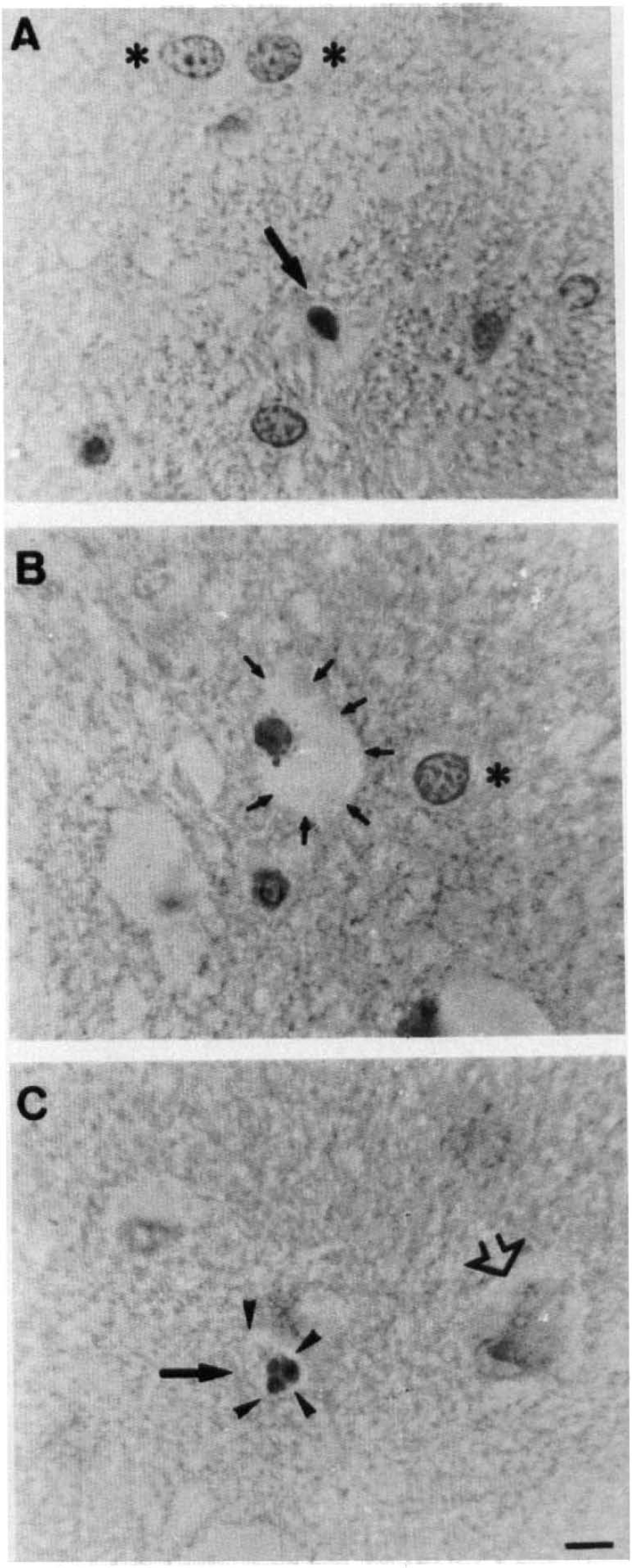
Chromatin condensation and nucleus segmentation as demonstrated with H&E staining. Three types of cell degeneration were observed: apoptotic cells (chromatin condensation) with a normal cytoplasm (arrows)
DISCUSSION
In this article we have demonstrated that a significant number of cells in the striatum, cortex, accumbens nucleus, and islands of Calleja die by a process that has the morphological characteristics of apoptosis after transient focal ischemia. Thus, 6 h after a transient MCA occlusion, chromatin condensation, nucleus segmentation, apoptotic bodies, and DNA nick-end labeling were observed in the penumbra, i.e., in cells mainly located at the inner borders of the necrotic volume.
Most investigations concerning cerebral ischemia have attributed the death of neurons to passive, necrotic processes. In contrast, in recent years many reports described how DNA fragmentation indicative of apoptosis may be contributing to the death of neurons after global (Héron et al., 1993; MacManus et al., 1993) and focal (Linnik et al., 1993; MacManus et al., 1994) ischemia. However, in these studies, the presence of oligonucleosomal-size fragments on agarose gels was only detected 24 and 48 h after the onset of ischemia. The identification of programmed cell death (apoptosis) in situ via the specific labeling of nuclear DNA fragmentation (Gavrieli et al., 1992) allowed confirmation of DNA cleavage in specific vulnerable neurons located in the CA1 pyramidal layer and in the striatum, respectively, 24 and 48 h after global ischemia (Héron et al., 1993; MacManus et al., 1993). Our first objective in this work was to identify if degenerating cells presented the morphological features of apoptosis, i.e., chromatin condensation and apoptotic bodies after transient focal ischemia, and if pathological changes might be observed at earlier times of recirculation in rat brain subjected to transient MCA occlusion. We first demonstrated that at 3- and 6-h recirculation, although no DNA degradation was detected on agarose gels (Charriaut-Marlangue et al., 1995), DNA fragmentation and chromatin condensation was identified in dying neurons as indicated by TUNEL and Hoechst 33258 stainings. These in situ specific DNA labelings allowed to demonstrate chromatin condensation marginally to the nuclear membrane and apoptotic body formation, which consisted of membrane-bounded dense masses. These changes suggest that ischemic insult induces clumping of chromatin and DNA fragmentation. The presence of some nuclear staining (exhibiting apoptosis but not necrosis feature) in the contralateral septum at 3 h recirculation and not later, may be an indication of programmed cell death in brain. In association with chromatin condensation, absence of a failure of organelle integrity was also described in apoptosis (Wyllie et al., 1980). Although electron microscopy could not reveal the ultrastructure, further identification was given by H&E staining, demonstrating that degenerating cells showing chromatin condensation may exhibit cytoplasmic vacuolization in some cases (Fig. 7). These results suggest that three types of cell death develop after transient focal ischemia with similar morphological features, as previously described by Garcia et al. (1993) after MCA occlusion in the rat brain. They are also reminiscent of the three types of death described by Clarke (1990) and reviewed by Server and Mobley (1991) during developmental cell death. Type 1, which was first described as “shrinkage necrosis” (Kerr, 1965; Kerr, 1971) and was then renamed apoptosis by Kerr et al. (1972) and nuclear cell death by Clarke (1990), is characterized by condensation of both the nucleus and the cytoplasm. Type 2 is characterized by the formation of numerous autophagic vacuoles. The nuclei of autophagic dying cells are in some cases pyknotic, which can occur either at early (Clarke and Hornung, 1989) or late (Beaulaton and Locksin, 1982) stages of degeneration, but the pyknosis is not so prevalent or striking as it is in type 1 cell death. Type 3 is typified by a general disintegration of the dilated cellular organelles with no clumping of the cell chromatin and no blebbing of the plasma membrane, reminiscent of necrotic cell death. However, the latter type of cell death has also been reported in the developing nervous system (Clarke, 1990). Type 1 (apoptosis) and type 3 (necrosis) have also been observed together in several neonatal systems including spinal motoneurons of the chick (Chu-Wang and Oppenheimer, 1978). Similar morphological changes have also been described in adult tissues after seizure-induced damage. In the epileptic brain, after an intraamygdaloid injection of kainate, CA3 pyramidal neurons displayed different morphological cell death reminiscent of types 1 and 2 (Pollard et al., 1994).
At an early recirculation interval, apoptotic cells (types 1 and 2) are primarily localized to the inner boundary zones of the infarct, whereas necrotic cells are mainly distributed within the ischemic core. It is suggested that after ischemia, ionic homeostasis falls in the region of the ischemic core, leading principally to necrotic cell death, whereas in the penumbra, a “gray zone”—which varies from species to species and depends on the severity of the insult—there is a block of synaptic transmission (reviewed in Hammond et al., 1994) but ionic homeostasis is maintained (Astrup et al., 1981), and recovery of metabolic activity, reoxygenation (Ginsberg, 1990; Siesjo, 1992; Hossmann, 1994), and cerebral blood flow (Memezema et al., 1992), particularly more important. Apoptotic cell death will occur primarily—not exclusively—in this region and may represent only a transient step, thus explaining the delayed state of the pathological neuronal death.
Interestingly, intracellular Ca2+ content that follows the ischemic episode will first activate endonucleases and nuclear damage in vulnerable neurons located in the penumbra. In contrast, severe injurious changes in the environment of neurons of the ischemic core lead to nuclear and cytoplasmic cell death. Thus, the distribution of apoptotic (type 1 and 2) cells suggests that apoptosis may contribute to the expansion of the ischemic lesion.
We also observed that a few cells in the corpus callosum, probably oligodendrocytes, exhibited chromatin condensation at 3-h recirculation but not later. This suggests that DNA-repairing processes may occur in our ischemic model. Recently it has been reported that endo–exonucleases, which are multifunctional enzymes, repair DNA but degrade genomic DNA when it is damaged beyond repair (Fraser, 1994). Normal oligodendrocyte death in developing postnatal rat optic nerves has been described by Barres et al. (1992) and reviewed by Raff et al. (1993). Furthermore, they and others described that growth factors and cytokines, produced from neighboring cells (astrocytes), promote both the survival (Barres et al., 1992) and the proliferation (Noble et al., 1988) of oligodendrocyte precursor cells.
Cells undergoing apoptosis may involve one of several parallel transduction pathways that converge in the expression of a set of death genes, ced-3 and ced-4, and TRPM-2 (Ellis et al., 1991). These genes encode for proteins that are now elucidated in vertebrate tissues, such as bcl-2 (Hockenbery et al., 1990), p53 (Yonish-Rouach et al., 1991; Lowe et al., 1993), and c-myc (Lotem and Sachs, 1992). Very recently, transgenic mice in which neurons overexpress bcl-2 were demonstrated to be more resistant to permanent focal ischemia (Martinou et al., 1994; Chen et al., 1995). If neurons are actively contributing to their own cell death, that may open unique therapeutic approaches and drug intervention for the preservation of cells in stroke and neurodegenerative disorders, in keeping with the new pharmacological strategies suggested to treat cancer (Carson and Ribeiro, 1993; Karp and Broder, 1994) or the aging process (Tomei et al., 1994).
Footnotes
Acknowledgment:
We thank I. Jorquera, F. Borrega, and M. Allix for helpful technical assistance and S. Guidasci for the photographs. This work was supported by INSERM and DRET.