
Abstract
In a rat forebrain ischemia model, the authors examined whether loss of cytochrome c from mitochondria correlates with ischemic hippocampal CA1 neuronal death and how cytochrome c release may shape neuronal death. Forebrain ischemia was induced by bilateral common carotid artery occlusion with simultaneous hypotension for 10 minutes. After reperfusion, an early rapid depletion of mitochondrial cytochrome c and a late phase of diffuse redistribution of cytochrome c occurred in the hippocampal CA1 region, but not in the dentate gyrus and CA3 regions. Intracerebroventricular administration of Z-DEVD-FMK, a relatively selective caspase-3 inhibitor, provided limited but significant protection against ischemic neuronal damage on day 7 after reperfusion. Treatment with 3 minutes of ischemia (ischemic preconditioning) 48 hours before the 10-minute ischemia attenuated both the early and late phases of cytochrome c redistribution. In another subset of animals treated with cycloheximide, a general protein synthesis inhibitor, the late phase of cytochrome c redistribution was inhibited, whereas most hippocampal CA1 neurons never regained mitochondrial cytochrome c. Examination of neuronal survival revealed that ischemic preconditioning prevents, whereas cycloheximide only delays, ischemic hippocampal CA1 neuronal death. DNA fragmentation detected by terminal deoxytransferase-mediated dUTP-nick end labeling (TUNEL) in situ was largely attenuated by ischemic preconditioning and moderately reduced by cycloheximide. These results indicate that the loss of cytochrome c from mitochondria correlates with hippocampal CA1 neuronal death after transient cerebral ischemia in relation to both caspase-dependent and -independent pathways. The amount of mitochondrial cytochrome c regained may determine whether ischemic hippocampal CA1 neurons survive or succumb to late-phase death.
Keywords
Although hippocampal CA1 neurons are known to be the most susceptible cell population in response to ischemia and typically succumb over a 48-to 72-hour period (Kirino, 1982; Pulsinelli et al., 1982; Smith et al., 1984), the underlying molecular mechanisms remain unclear. Morphologically, it is controversial whether the delayed hippocampal CA1 neuronal death induced by transient ischemia is apoptotic (Colbourne et al., 1999; Deshpande et al., 1992; Martin et al., 2000; Nitatori et al., 1995). However, given the fact that both proapoptotic and antiapoptotic proteins are expressed in hippocampal CA1 neurons after transient ischemia (Chen et al., 1997; Honkaniemi et al., 1996; Krajewski et al., 1995) and caspase inhibitors can reduce ischemic neuronal damage (Chen et al., 1998; Cheng et al., 1998; Endres et al., 1998), there is no doubt that apoptotic mechanisms are involved in the delayed hippocampal CA1 neuronal death after transient ischemia.
Cytochrome c is known to reside in the space between the outer and inner membranes of mitochondria, where it functions in the respiratory chain by interacting with redox partners of complex III and complex IV (Mathews, 1985). In the cell-free system, extensive studies have shown that cytochrome c enters the cytosol during apoptosis, probably as a result of loss of this protein from mitochondria (Kluck et al., 1997; Liu et al., 1996; Yang et al., 1997). Once released to the cytoplasm, cytochrome c acts as a cofactor in conjunction with Apaf-1, procaspase-9, and adenosine triphosphate (ATP)/dATP to induce activation of caspase-9 and subsequently caspase-3 (Bossy-Wetzel et al., 1998; Slee et al., 1999; Zou et al., 1997). The activated caspases, especially caspase-3, are important in mediating apoptotic cell death (Enari et al., 1998; Nicholson et al., 1995; Sahara et al., 1999). A previous study has shown that caspase-9 was released to the cytoplasm after global cerebral ischemia (Krajewski et al., 1999).
Beyond this scenario, a persistent disappearance of cytochrome c in mitochondria should lead to impairment of complexes III and IV of the respiratory chain (Bossy-Wetzel et al., 1998; Stefanis et al., 1999). Over time, this would lead to a decrease in mitochondrial ATP synthesis. The decreases in mitochondrial ATP synthesis may not only lead to neuronal death but may also play a critical role in determining the shape of cell death (Eguchi et al., 1997; Kass et al., 1996; Leist et al., 1997; Richter et al., 1996). On the other hand, impairment of complexes III and IV of the respiratory chain would maintain complex I and the ubiquinone at complex II in the reduced state. This condition has been shown to favor superoxide formation (Boveris et al., 1976; Cai and Jones, 1998; Luetjens et al., 2000).
Although reports have shown that release of cytochrome c occurs after transient cerebral ischemia (Ouyang et al., 1999; Sugawara et al., 1999), only one study has shown that neuroprotection achieved by ischemic preconditioning was associated with an attenuation of cytochrome c release in the gerbil (Nakatsuka et al., 2000). In the current study, the authors found that loss of cytochrome c from mitochondria correlates with ischemic hippocampal CA1 neuronal death, and inhibition of caspase-3 activation with Z-DEVD-FMK renders only a weaker protection against hippocampal CA1 neuronal damage.
MATERIALS AND METHODS
Forebrain ischemia model
This study was approved by the Committee on the Guidelines for Animal Experiments of Niigata University. For induction of forebrain ischemia, male Wistar rats weighing 250 to 300 g were fasted but allowed access to water for 8 to 12 hours. After the animals were anesthetized with 4% halothane and tracheally intubated, they were mechanically ventilated with 1.5% halothane in 30% O2/balance N2O. Two digital thermistor probes (Multithermistor Meter D321, Technol Seven, Yokohama, Japan) were placed in the rectum and under the left temporalis muscle to monitor core and pericranial temperature. The left femoral artery and the right external jugular vein were cannulated for continuous monitoring of blood pressure and blood sampling, respectively. The bilateral common carotid arteries were isolated from the carotid sheaths using a ventral midline incision. Fifteen minutes before occlusion of the bilateral common carotid arteries, heparin (50 U) was injected intravenously and an arterial blood sample (0.3 mL) was taken for blood gas analysis and plasma glucose assay. Animals were acceptable if blood gas parameters and the levels of plasma glucose met the following criteria: Po2 90 to 140 mm Hg, Pco2 35 to 45 mm Hg, pH 7.35 to 7.45, and glucose 90 to 150 mg/dL.
After mean artery blood pressure was lowered to 35 to 40 mm Hg by injecting 0.5 mg phentolamine through the vein catheter, followed by withdrawing and injecting blood from the external jugular vein catheter using a heparinized syringe, both common carotid arteries were occluded with small vascular clips for 10 minutes. The withdrawn blood was held at 37.5°C. Thereafter, the clips were removed and the withdrawn blood was reinfused. Reperfusion in each artery was verified visually. During the period of occlusion, the core temperature was controlled at 37.2°C ± 0.2°C, whereas the pericranial temperature was allowed to decrease gradually from 36.8°C ± 0.2°C at the initiation of occlusion to 36.0°C ± 0.2°C at the end of occlusion. Before anesthesia was discontinued, the vascular catheters were removed and the wounds were sutured. The endotracheal catheter was extubated after spontaneous respiration and righting reflex recovered. The animals were then placed in a warm, humidified chamber (32°C to 33°C) for another 3 hours before being returned to their cages. The animals involved in exposure of the bilateral common carotid arteries without induction of hypotension and bilateral common carotid artery occlusion were used as shams.
Effect of caspase-3 inhibition on ischemic neuronal damage
Because caspase-3 activation has been known to follow cytochrome c redistribution and plays a critical role in apoptotic cell death, the authors examined the extent of protection of caspase-3 inhibition against ischemic neuronal damage. N-benzyloxycarbonyl-asp(OMe)-Glu(OMe)-ValAsp-(OMe)-fluoro-methylketone (Z-DEVD-FMK) (Calbiochem, San Diego, CA, U.S.A.) was dissolved in dimethyl sulfoxide (DMSO) and further diluted in Krebs solution to achieve a concentration of 65 μg/mL. In the Z-DEVD-FMK group (n = 6), Z-DEVD-FMK (0.5 mL/h) was delivered into the left lateral ventricle using a microosmotic pump (Alzet Model 1007D; Alza Corp., Palo Alto, CA, U.S.A.) for 7 days beginning 2 hours before the onset of ischemia. Control animals (vehicle group, n = 6) received infusion of vehicle (DMSO in Krebs solution) at the same rate. Sham animals (sham group, n = 4) received infusion of vehicle without induction of cerebral ischemia.
Ischemic preconditioning and cycloheximide treatment
To see the relation between cytochrome c release and neuronal survival, the authors examined two different neuroprotective approaches (ischemic preconditioning and cycloheximide treatment) on ischemic neuronal damage, cytochrome c release, and DNA fragmentation. For the ischemic preconditioning study, animals were subjected to a 3-minute bilateral common carotid artery occlusion with simultaneous hypotension, as mentioned above, 48 hours before being subjected to the 10-minute ischemia. For cycloheximide treatment, rats were subcutaneously given 1.0 mg/kg cycloheximide 30 minutes after reperfusion, followed by a dose of 0.5 mg/kg each day up to the fourth day, or before the day the animals were killed. Sham animals received saline injection in a manner identical to the cycloheximide-treated animals.
Transcardiac perfusion and brain tissue preparation
At variable time points after reperfusion, animals were reanesthetized by an intraperitoneal injection of sodium pentobarbital (100 mg/kg) and perfused through the ascending aorta with different solutions in accordance with the experimental protocols. At 12, 24, 48, 72, 96, and 168 hours (five or six animals per time point) after reperfusion, animals were perfused with 1 × phosphate-buffered saline (PBS) containing heparin (4 U/mL), followed by 0.01 mol/L periodate/0.075 mol/L lysine/2% paraformaldehyde (PLP) in 0.0375 mol/L phosphate-buffered solution (pH 6.3). The whole brain was removed from the skull and separated through the midline, cut into small pieces, and postfixed in the same fixative for 4 hours. Thereafter, the tissue pieces were washed in gradually increasing concentrations of sucrose in 1 × PBS (10% for 4 hours, 15% and 20% for 12 hours each) and then rapidly frozen in 2-methylbutane chilled at −80°C. Consecutive coronal sections located about 3.5 mm posterior to the bregma were prepared on a microtome and used for microtubule-associated protein-2 (MAP-2) immunohistochemistry, cytochrome c immunofluorescence, and cresyl violet staining. For in situ detection of DNA fragmentation, animals were perfused with 1 × PBS containing heparin (4 U/mL) followed by 20% sucrose in 1 × PBS at 48 or 96 hours after reperfusion. The brains were then removed from the skull and rapidly frozen in 2-methylbutane chilled at −80°C.
Assessment of neuronal damage
Ischemic neuronal damage was visualized by MAP-2 immunostaining. Briefly, coronal sections (16 μm) were postfixed with 4% paraformaldehyde in 1 × PBS for 10 minutes and washed 3 times with 1 × PBS. Endogenous peroxidase was inactivated by incubating the sections with 30% methanol containing 0.1% H2O2 (v/v) for 45 minutes. Sections then were followed by sequential incubations in (1) 2.5% horse serum in 1 × PBS containing 0.2% Tween-20 and 1.5% bovine serum albumin (BSA) for 60 minutes, (2) anti-MAP-2 monoclonal antibody (Boehringer Mannheim, Mannheim, Germany) diluted in 1 × PBS at 4°C overnight, (3) biotinylated horse antimouse IgG for 60 minutes, and (4) avidin-biotin-peroxidase complex in 1 × PBS for 60 minutes. The biotinylated secondary antibody and the biotin-avidin-peroxidase were purchased from Vector Laboratories (Burlingame, CA, U.S.A.). The MAP-2 immunoreaction was visualized by incubating the sections with 0.05% 3, 3′-diaminobenzidine and 0.002% H2O2.
For counting intact neurons, adjacent sections (8 (m) were stained with 0.1% Luxol fast blue for 12 hours followed by 0.1% cresyl violet for 20 minutes. The number of viable cell bodies in the whole CA1 pyramidal cell layer was counted by a masked experimenter and expressed as the number of cells per millimeter length of the CA1 pyramidal cell layer (neuronal density).
Immunofluorescence for detecting mitochondrial cytochrome c
A mouse monoclonal antibody (6H2.B4; Pharmingen, San Diego, CA, U.S.A.) was used against the native form of cytochrome c (mitochondrial cytochrome c) in situ. Sections (12 μm) were washed 4 times with 1 × PBS for 5 minutes each; incubated for 20 minutes in 1 × PBS containing 0.2% gelatin, 1.5% BSA, and 0.1% Tween-20; blocked with 2.5% goat serum diluted in 1 × PBS containing 0.1% Tween-20 and 1.5% BSA for 60 minutes; incubated with a mouse anticytochrome c (20 (g/mL) diluted in 1 × PBS containing 1.5% BSA and 0.1% Tween-20 at 4°C for 44 hours; and washed 6 times (5 minutes each) with 10% sucrose in 1 × PBS. The sections then were incubated in FITC-conjugated goat antimouse IgG (American Qualex, San Clemente, CA, U.S.A.) diluted in 1 × PBS containing 1.5% BSA and 0.1% Tween-20 for 8 hours at 4°C, washed 6 times in 1 × PBS, mounted in 50% glycerol in 1 × PBS, and finally visualized under a Nikon fluorescence microscope (Optiphot-2; Nikon, Tokyo, Japan). Sections obtained from animals at 24 hours after reperfusion also were examined by a laser scanning confocal microscopy (MRC 1024; Bio-Rad Laboratories, Hercules, CA, U.S.A.). Negative controls included the omission of the primary or the secondary antibody.
In situ labeling of DNA fragmentation
By using an apoptosis in situ detection kit (Wako Pure Chemical Industries, Osaka, Japan), terminal deoxytransferase-mediated dUTP-nick end labeling (TUNEL) was performed on nonfixed sections (12 μm). Sections were postfixed with 4% paraformaldehyde in 1 × PBS for 15 minutes at room temperature, followed by ethanol:acetic acid (2:1) for 5 minutes at −20°C, and then penetrated with 3% Triton X-100 in 1 × PBS for 60 minutes at room temperature. Sections then were incubated in TUNEL reaction solution for 60 minutes at 37°C. After washing, sections were incubated in 3% H2O2 in 1 × PBS for 10 minutes to quench endogenous peroxidase and incubated with anti-FITC-conjugated peroxidase for 60 minutes at 37°C. TUNEL-positive cells were visualized with diaminobenzidine. Similar to assessment of intact neurons, TUNEL-positive cells counted from the whole CA1 pyramidal cell layer were expressed as the number of positive cells per millimeter length of the pyramidal cell layer.
Preparation of cell lysates
At 4, 12, 24, and 72 hours after reperfusion, animals (n = 4 per time point) were anesthetized with an intraperitoneal injection of sodium pentobarbital (100 mg/kg) and decapitated. Brains were quickly removed, and hippocampi were dissected in lysis buffer. Sham control animals underwent exposure of bilateral common carotid arteries but were not subjected to occlusion of bilateral common carotid arteries and hypotension. Cytosolic protein extracts were prepared on ice by Dounce homogenization of tissues in a lysis buffer containing 15 mmol/L Tris-HCl (pH 7.50), 250 mmol/L sucrose, 2 mmol/L ethylenediamine tetraacetic acid (EDTA), 1 mmol/L ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 1 mmol/L MgCl2, 1 mmol/L DL-dithiothreitol (DTT), 50 mmol/L NaF, 100 μmol/L Na3VO4, 2 mmol/L sodium pyrophosphate, 100 μg/mL phenylmethylsulfonyl fluoride (PMSF), and 10 μg/mL each pepstatin, leupeptin, and aprotinin (all from Sigma, St. Louis, MO, U.S.A.). Cell lysates were centrifuged at 32,900 g for 60 minutes, and the supernatants were stored at −80°C until use. Protein concentrations were determined using the method of Lowry, with BSA as a standard.
Western blot analysis of cytochrome c
Cytosolic protein samples (50 μg) were run on 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred onto a nitrocellulose membrane (Bio-Rad) at 80 V for 75 minutes. Blots were probed with a mouse monoclonal antibody (7H8.2C12, Pharmingen) against the denatured form of cytochrome c at a dilution of 1:5,000 at 4°C for 45 minutes. After the primary antibody incubation, the membrane was washed and incubated with a horseradish peroxidase-conjugated goat antimouse IgG (American Qualex) for 40 minutes at room temperature. The immunoreaction was visualized using Amersham enhanced chemiluminescence (Amersham Pharmacia Biotech, Buckinghamshire, U.K.). After this detection, the bound primary and secondary antibodies were stripped by incubating the membrane in stripping buffer (100 mmol/L 2-mercaptoethanol, 2% sodium dodecyl sulfate, 62.5 mmol/L Tris-HCl, pH 6.7) at 50°C for 30 minutes. The membrane was reprobed with a mouse cytochrome c oxidase subunit IV antibody (Molecular Probes, Eugene, OR, U.S.A.) against cytochrome oxidase at a dilution of 1:5,000 at 4°C for 45 minutes and then visualized with a procedure identical to that for cytochrome c.
Statistical analysis
Quantitative data are expressed as mean ± standard deviation and analyzed using one-way analysis of variance after normality of distribution was proved. Analysis of variance was followed by Dunnett's post hoc test for multiple comparisons. P < 0.05 was considered statistically significant.
RESULTS
Delayed hippocampal CA1 neuronal death after 10-minute forebrain ischemia
Loss of MAP-2 immunoreactivity has been shown as an early indication of ischemic neuronal injury in the gerbil ((Kitakawa et al., 1989; Matesic and Lin, 1994). In the sections obtained from sham rats, both somata and dendrites were extensively stained (Fig. 1A). The easily noticeable change in MAP-2 immunoreactivity 24 hours after reperfusion is that the dendritic staining was intermittent (Fig. 1B). By 72 hours after reperfusion, extensive dendritic and somatic loss of MAP-2 immunoreactivity appeared in the hippocampal CA1 region (Fig. 1C). On day 7 after reperfusion, MAP-2 immunoreactivity was almost completely lost in both somata and dendrites, except for few neurons that survived the ischemic insult (Fig. 1D). A time-dependent loss of hippocampal CA1 neurons after the 10-minute ischemia as counted from cresyl violet-stained sections is shown in Fig. 2.
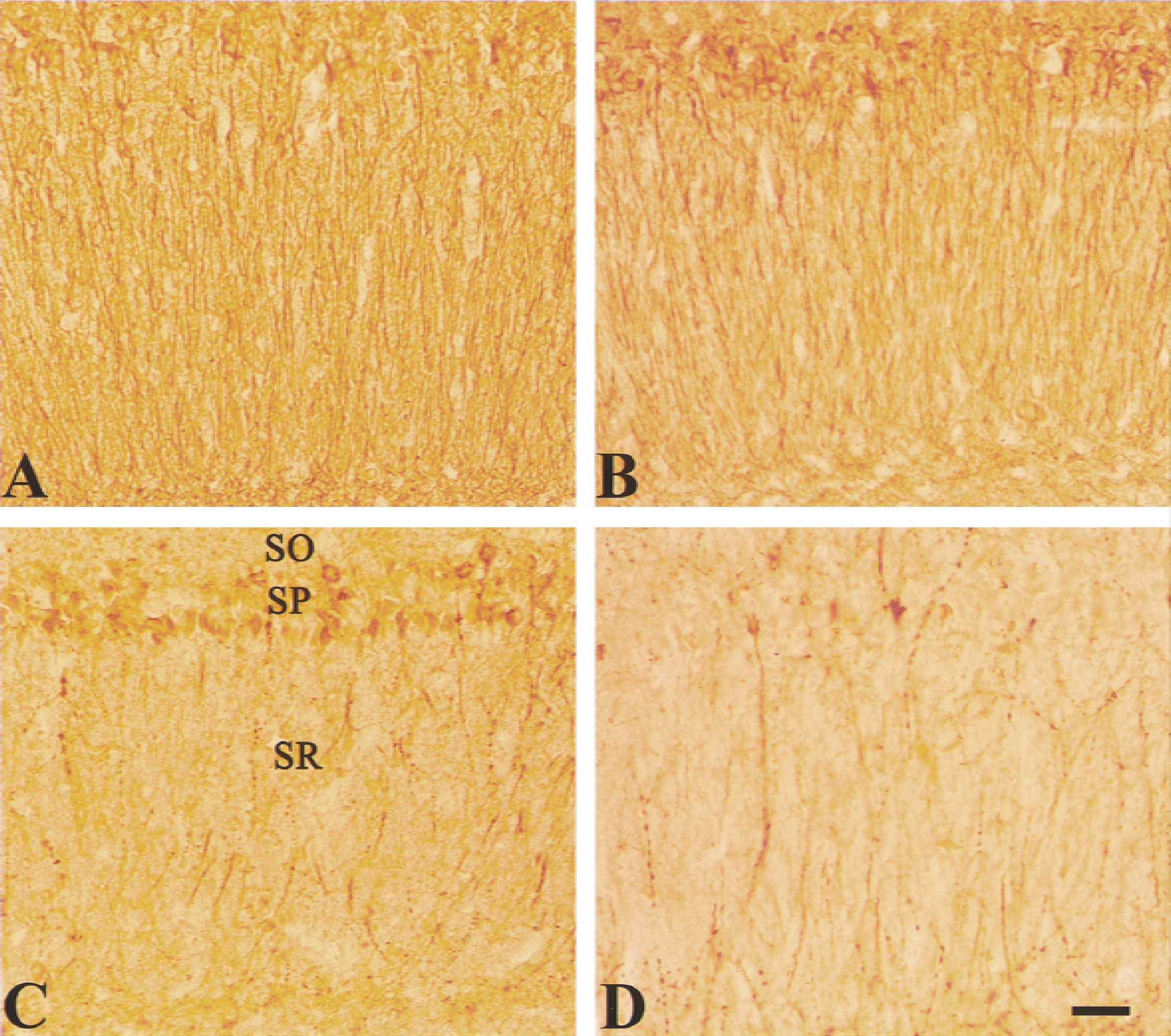
Microtubule-associated protein-2 (MAP-2) immunostaining shows delayed hippocampal CA1 neuronal damage after 10 minutes of forebrain ischemia.
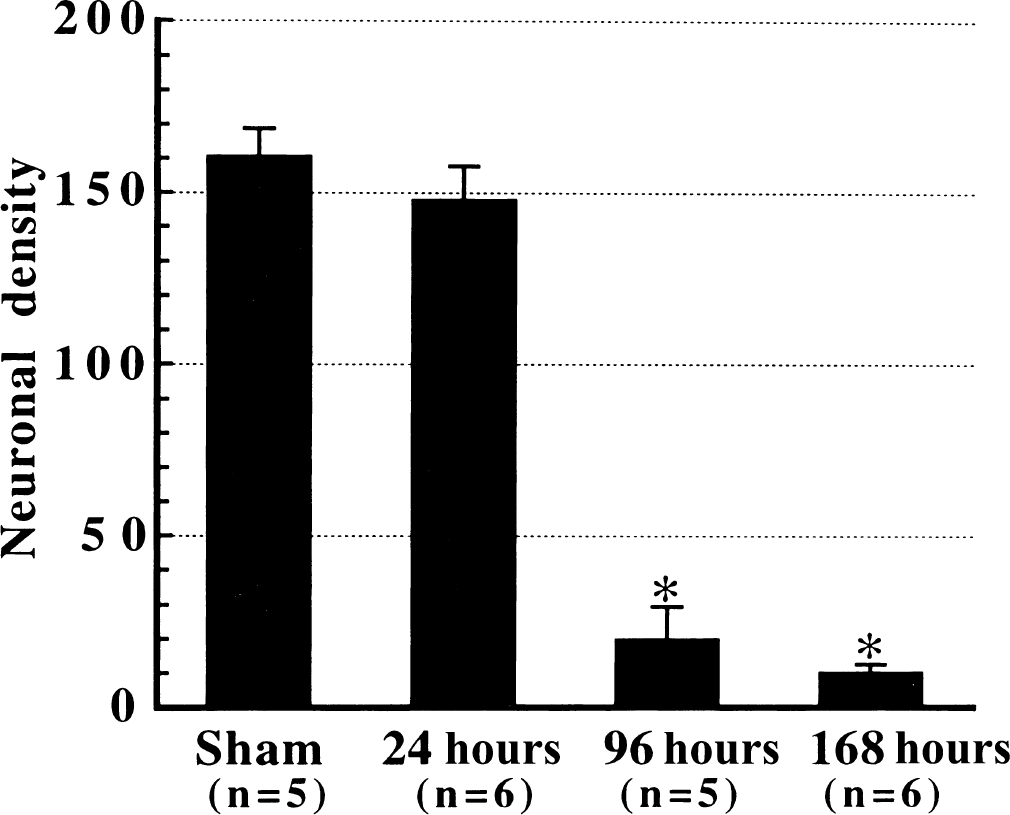
A time-dependent loss of hippocampal CA1 neurons after 10 minutes of forebrain ischemia. Intact neurons were counted from the whole CA1 pyramidal layer in cresyl violet-stained sections (8 μm) and expressed as the number of neurons per millimeter hippocampal CA1 pyramidal layer (neuronal density). Most hippocampal CA1 neurons began dying 24 hours after reperfusion. * P < 0.001 versus sham.
Redistribution of cytochrome c selectively occurs in the hippocampal CA1 region
The distribution of cytochrome c was examined by immunofluorescence using an antibody against mitochondrial cytochrome c and by Western blotting analysis. Immunofluorescent staining (Fig. 3) revealed that neurons in the sections prepared from sham animals had a bright, punctate cytoplasmic distribution of cytochrome c fluorescence, clearly excluded from the nuclear space in the hippocampal CA1 region and in other hippocampal regions. After reperfusion, the mitochondrial cytochrome c rapidly decreased in the CA1 region, whereas the pattern of distribution remained unchanged until 24 hours after reperfusion, indicating that cytochrome c was rapidly degraded after it was released into the cytoplasm, as reported previously (Neame et al., 1998). By 72 hours after reperfusion, the punctuated mitochondrial pattern of cytochrome c changed to a diffuse cytoplasmic pattern; cytochrome c entered into the nuclear space or distributed around the condensed nucleus in some hippocampal CA1 neurons, whereas mitochondrial cytochrome c disappeared in most of the hippocampal CA1 neurons. Neither the intensity nor the distribution pattern of mitochondrial cytochrome changed in the CA3 region. Occasionally, a slight loss of cytochrome c occurred in the dentate gyrus. Confocal microscopic examination confirmed the punctuated distribution of cytochrome c in sham-controlled animals (Fig. 4A) and revealed that near-complete loss of cytochrome c occurred not only in the soma but also in the dendrites of CA1 pyramidal cells at 24 hours after reperfusion (Fig. 4B).
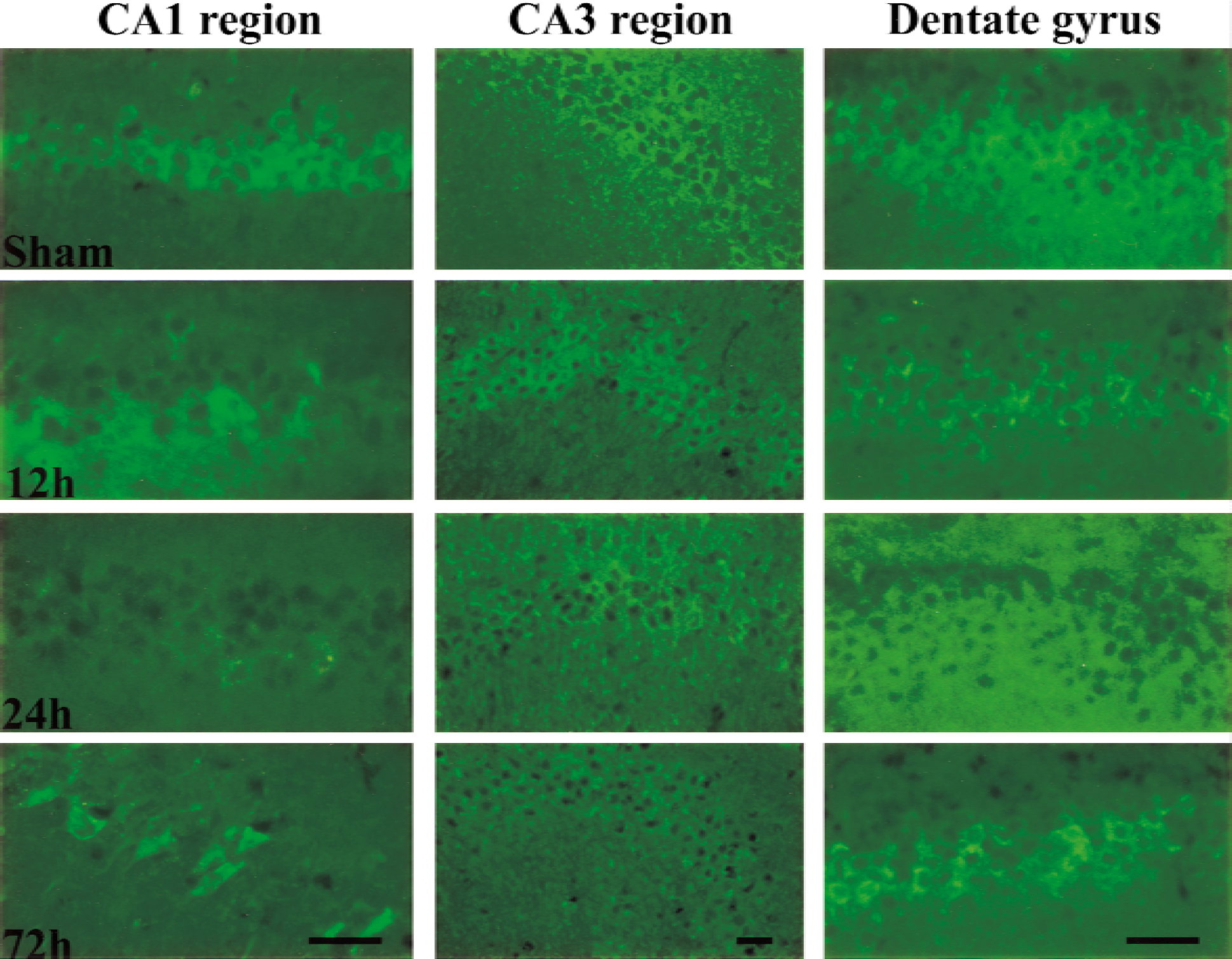
Dramatic changes in mitochondrial cytochrome c immunofluorescence occurred in the CA1 but not in the dentate gyrus and CA3 regions after transient cerebral ischemia. Typical organellar cytochrome c immunofluorescence occurred in the CA1, dentate gyrus, and CA3 region (top row) in a sham control animal. At 12 hours after reperfusion, most CA1 neurons showed partial or nearly complete loss of cytochrome c in the CA1 region, and almost all CA1 pyramidal cells lost mitochondrial cytochrome c 24 hours after reperfusion. By 72 hours after reperfusion, a diffuse distribution of cytochrome c appeared in the CA1 region but not the other two regions. Scale bars = 40 μm.
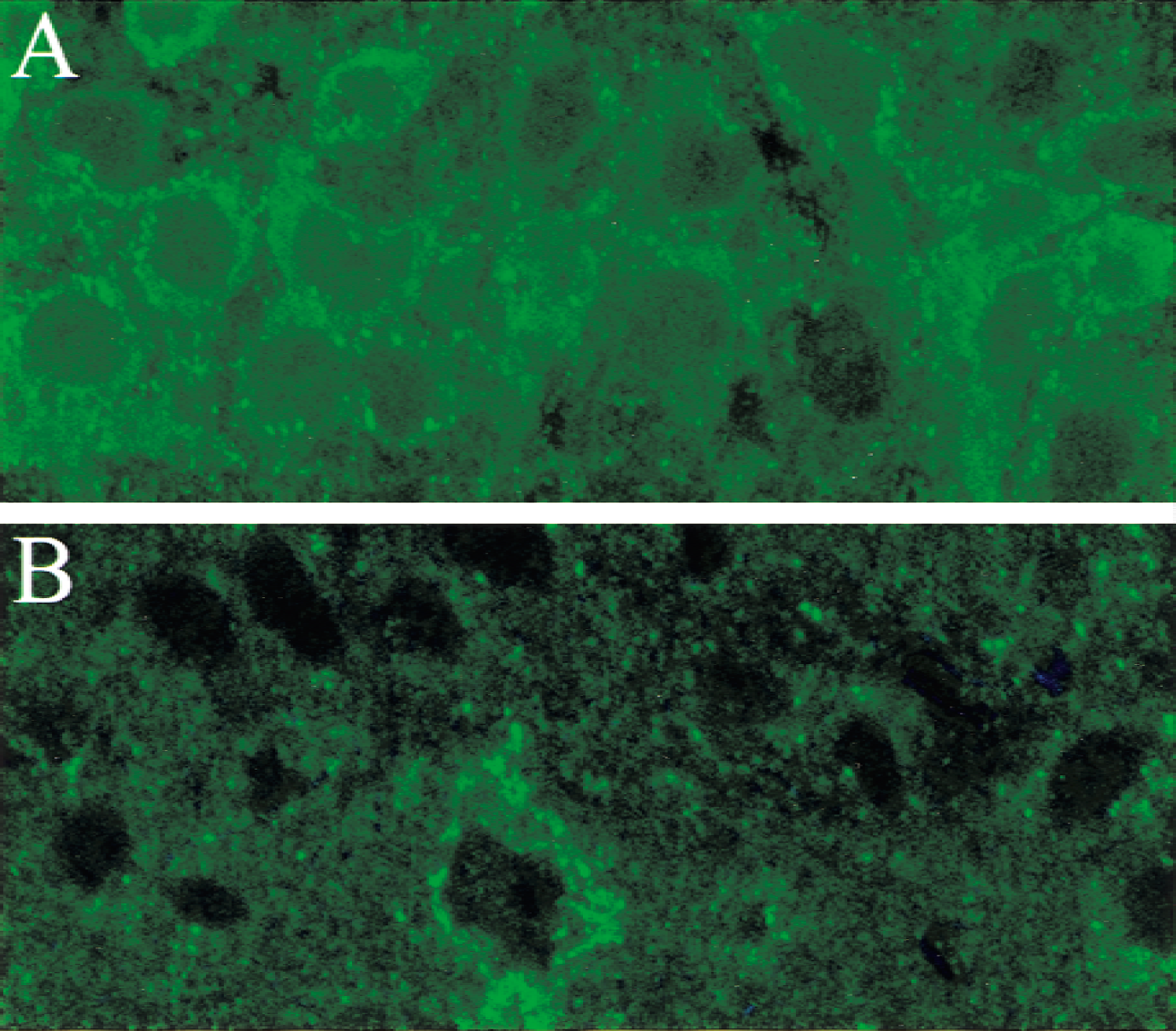
Confocal microscopic images show cytochrome c immunofluorescence in the hippocampal CA1 region in sham and ischemia animals.
The levels of cytochrome c in cytosolic extracts were increased 4 hours after reperfusion and decreased beginning 24 hours after reperfusion, whereas cytochrome c oxidase (subunit IV) was nearly absent (Fig. 5). The absence of cytochrome c oxidase in cytosolic extracts confirmed that the cytosolic extracts were free of mitochondrial contamination.
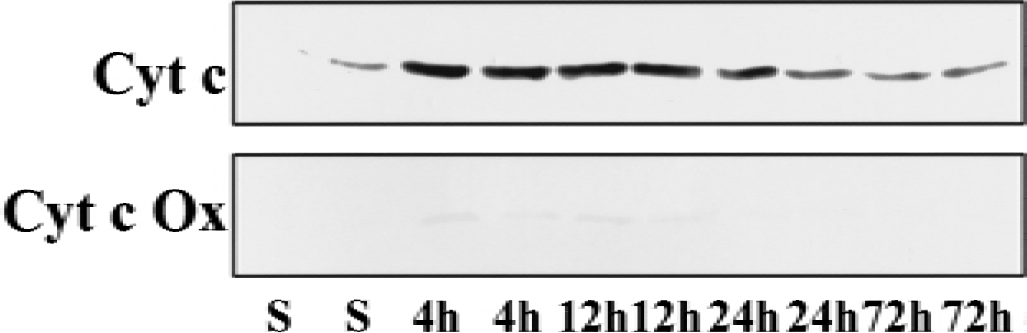
Western blotting analysis of cytochrome c (Cyt c) and cytochrome c oxidase subunit IV (Cyt c Ox) in the cytosolic fraction (50 μg protein per lane) of hippocampus from sham control rats (S) or ischemic rats at 4, 12, 24, and 72 hours after reperfusion. Each lane contains a sample from a single rat. Levels of Cyt c peaked 4 hours after reperfuion. Cyt c Ox was nearly absent in all samples.
Inhibition of caspase-3 activation with Z-DEVD-FMK provides limited protection against ischemia
Previous studies have shown that intracerebroventricular administration of Z-DEVD-FMK at doses less than or the same as the dose used in the current study almost completely inhibited caspase-3 activation (Chen et al., 1998; Gillardon et al., 1999). As shown in Fig. 6, the protective effect of Z-DEVD-FMK examined on day 7 after reperfusion was unexpectedly weak, indicating that caspase-3 activation is involved in but is not dominant in ischemic hippocampal CA1 neuronal death.
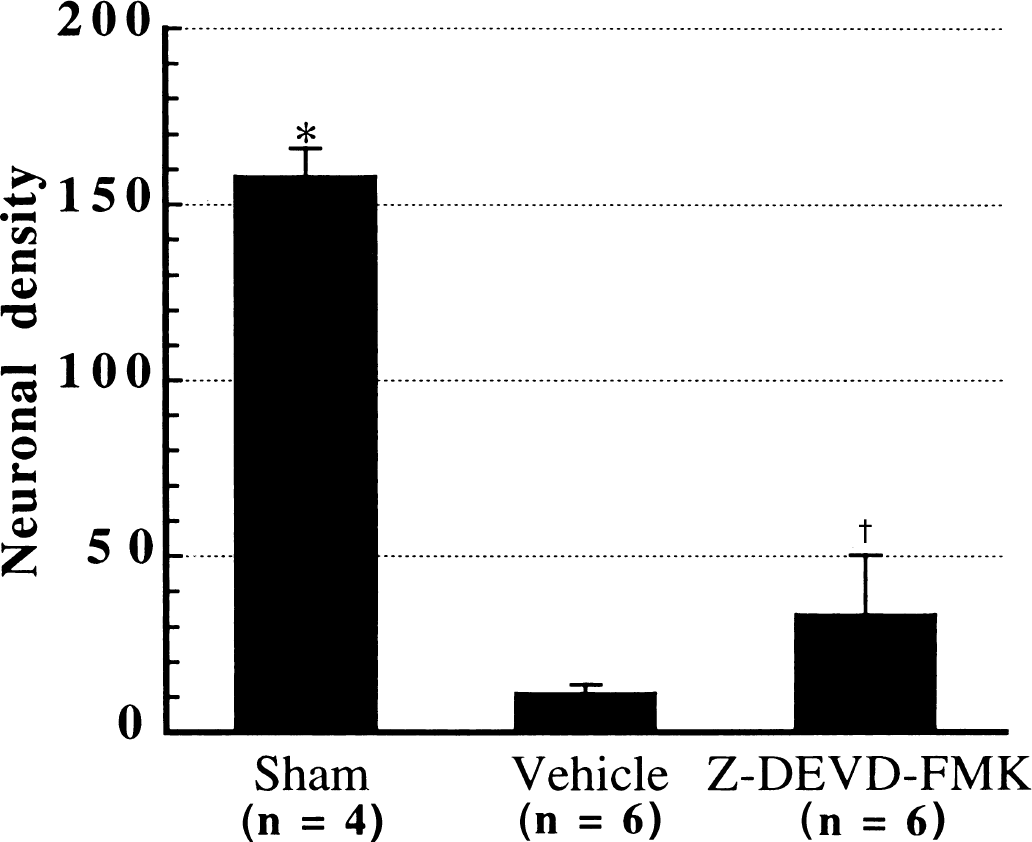
The effect of caspase-3 inhibition on hippocampal CA1 neuronal survival on day 7 after reperfusion. Z-DEVD-FMK was administered into the left lateral ventricle by a microosmotic pump for 7 days beginning 2 hours before the onset of ischemia. Control animals received vehicle infusion at the same rate. Cell counts were made in cresyl violet-stained sections (8 μm) in the left hippocampus. Bars indicate standard deviation. * P < 0.001 versus either vehicle or Z-DEVD-FMK group; †P < 0.01 versus vehicle group (Dunnett's post hoc test).
Ischemic preconditioning attenuates both early and late phases of cytochrome c redistribution
Ischemic preconditioning is a phenomenon whereby a brief episode of sublethal ischemia (Kirino et al., 1991; Kitakawa et al., 1990) or other stresses (Takahata and Shimoji, 1986) produces strong protection against a subsequent ischemic insult. The demonstration of ischemic preconditioning in cultured neurons clearly indicates that ischemic preconditioning in vivo may be caused not by alterations in blood flow, body temperature, or systemic response to ischemia but rather by alterations in brain parenchyma (Grabb and Choi, 1999). In the rat 2-VO model, a 3-minute ischemic episode performed 48 hours before a 10-minute ischemia provided drastic protection against ischemic insult caused by the 10-minute ischemia as examined on day 7 after reperfusion (Fig. 7). Examination of cytochrome c distribution in the preconditioned animals 24 and 72 hours after reperfusion revealed that both the early and the late phases of cytochrome c redistribution were attenuated (Fig. 8).
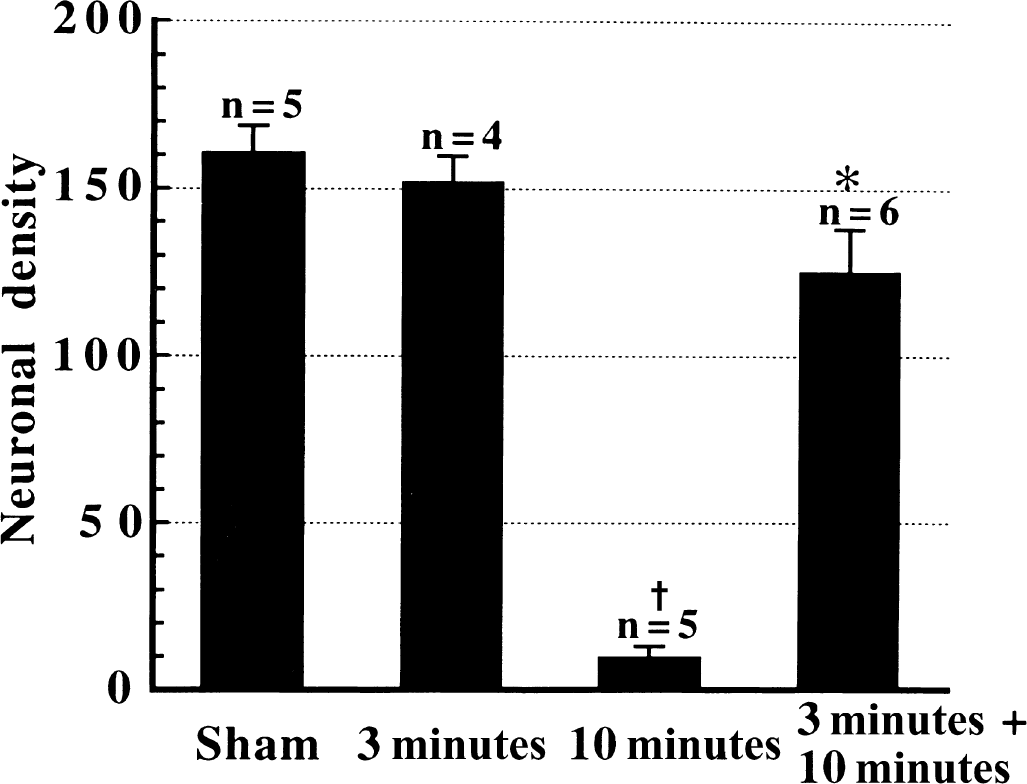
Ischemic preconditioning provides a drastic protection against a subsequent 10-minute ischemic insult. Ischemic neuronal damage was examined on day 7 after the 10-minute ischemic insult. The ischemic preconditioning itself, which was induced by a 3-minute ischemia 48 hours before the 10-minute ischemia, did not cause detectable neuronal death. * P < 0.001 versus 10-minute ischemia; †P < 0.001 versus sham or 3-minute ischemia.
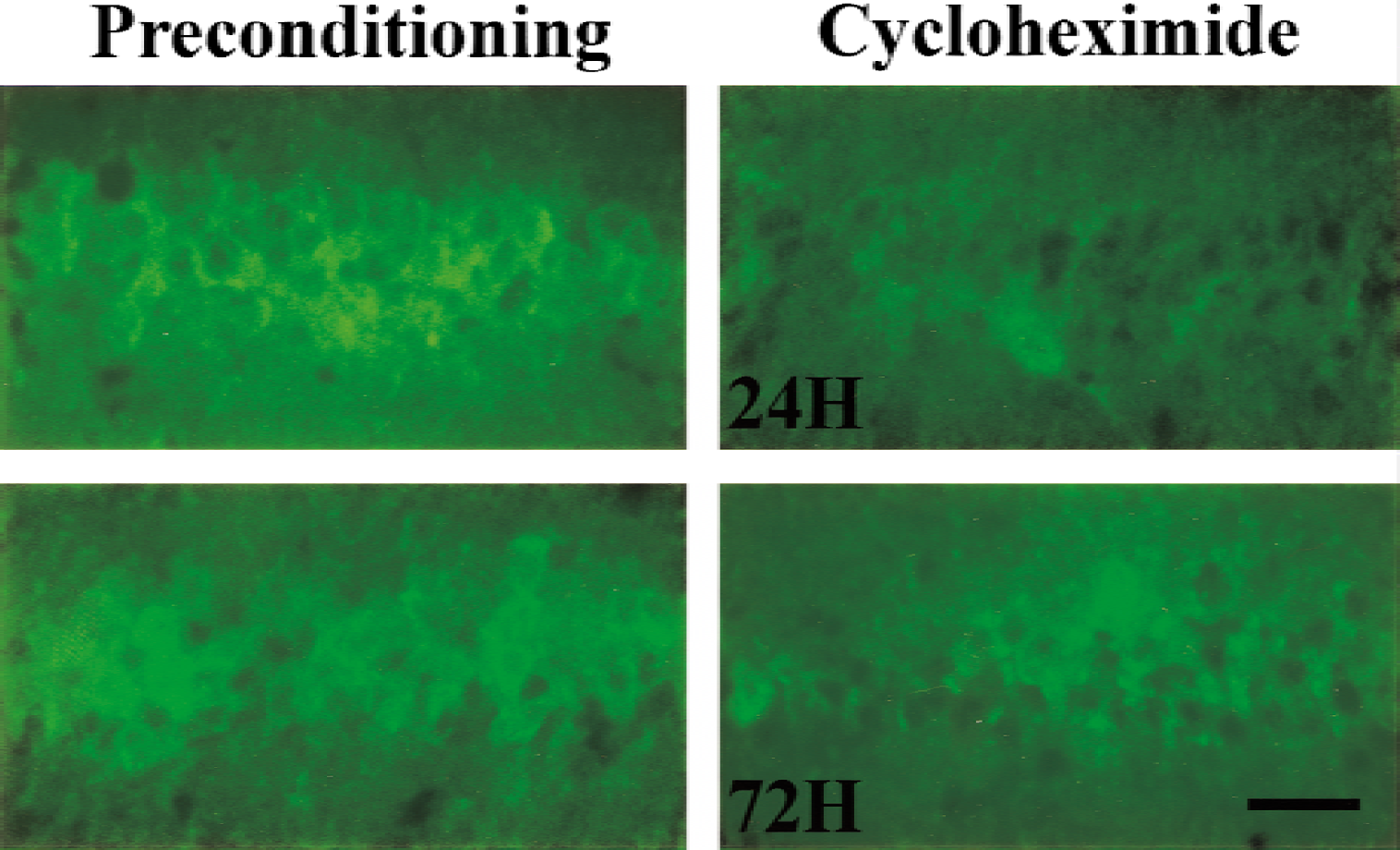
Representative photomicrographs of immunofluorescent staining show that ischemic preconditioning attenuated the early and the late phases of cytochrome c release from mitochondria, and cycloheximide eliminated the late phase of cytochrome c redistribution. Scale bar = 40 μm.
Cycloheximide eliminates the late but not the early phase of cytochrome redistribution
To confirm the role of cytochrome c redistribution in ischemic hippocampal CA1 neuronal death, the authors then examined the effect of cycloheximide on ischemic neuronal death and cytochrome c release. Previous in vivo studies (Goto et al., 1990; Tortosa et al., 1994) have shown that cycloheximide reduces ischemic hippocampal CA1 neuronal damage. Recent studies showed that cycloheximide blocks the release of cytochrome c from mitochondria to the cytosol in response to apoptotic stimuli (Deshmukh and Johnson, 1998; Neame et al., 1998; Putcha et al., 1999). Because cycloheximide at doses more than 0.6 mg/kg is known to reduce protein synthesis (Pavlik and Teisinger, 1980), a dose of 1 mg/kg was chosen for the current study.
Although immunofluorescence revealed that cycloheximide was not able to attenuate the loss of mitochondrial cytochrome c 24 hours after reperfusion, it blocked the late phase of cytochrome c redistribution that occurs 48 to 72 hours after reperfusion in the hippocampal CA1 region (Fig. 8), indicating that cycloheximide acted to inhibit the late but not the early phase of cytochrome c release. In contrast to the effect of ischemic preconditioning, mitochondrial cytochrome c was not regained in most of the hippocampal CA1 neurons in cycloheximide-treated animals. Although cycloheximide rendered strong protection 4 days after reperfusion, the protection diminished within 4 to 7 days after reperfusion (Fig. 9).
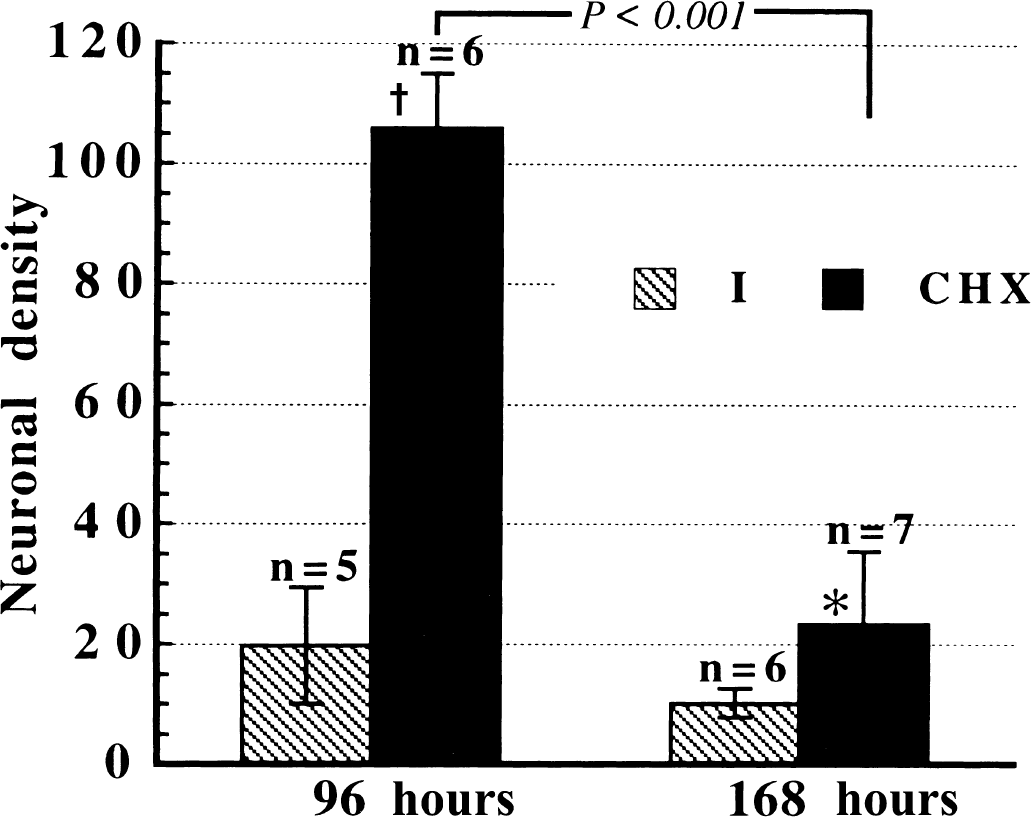
Effect of cycloheximide (CHX) on ischemic hippocampal CA1 neuronal damage on days 4 (96 hours) and 7 (168 hours) after reperfusion. The very strong protection of cycloheximide on day 4 diminished within 4 to 7 days after reperfusion. * P < 0.05 and †P < 0.001 versus single 10-minute ischemia (I).
Both ischemic preconditioning and cycloheximide reduce ischemia-induced DNA fragmentation
Although TUNEL staining alone is not specific for apoptotic cell death, cell death with or without DNA fragmentation may implicate different underlying mechanisms. There were no TUNEL-positive cells in the sections prepared from sham animals (Fig. 10A). Also, no detectable TUNEL-positive cells were found in the hippocampal CA1 region 48 hours after reperfusion (data not shown). Extensive TUNEL-positive cells appeared 96 hours after reperfusion in the CA1 region (Fig. 10B) but not in the dentate gyrus and CA3 regions. Among TUNEL-positive cells, few showed chromatin condensation or chromatin aggregation in the perinuclear regions, which are generally accepted as hallmarks of apoptotic cell death. Ischemic preconditioning largely reduced and cycloheximide moderately reduced TUNEL-positive cells (Figs. 10C, 10D, and 11).
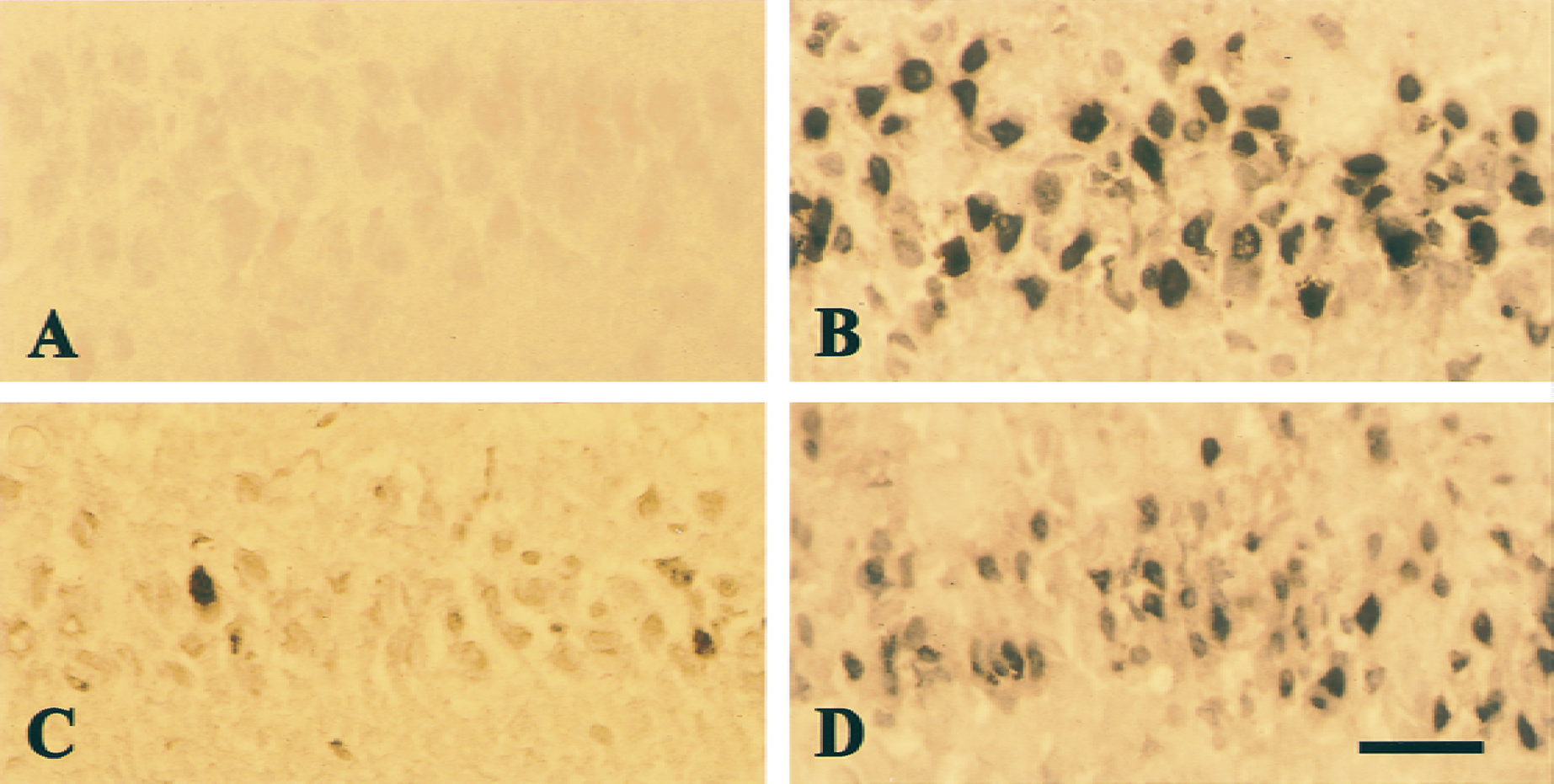
Representative photomicrographs of TUNEL staining in the hippocampal CA1 region in sham-operated
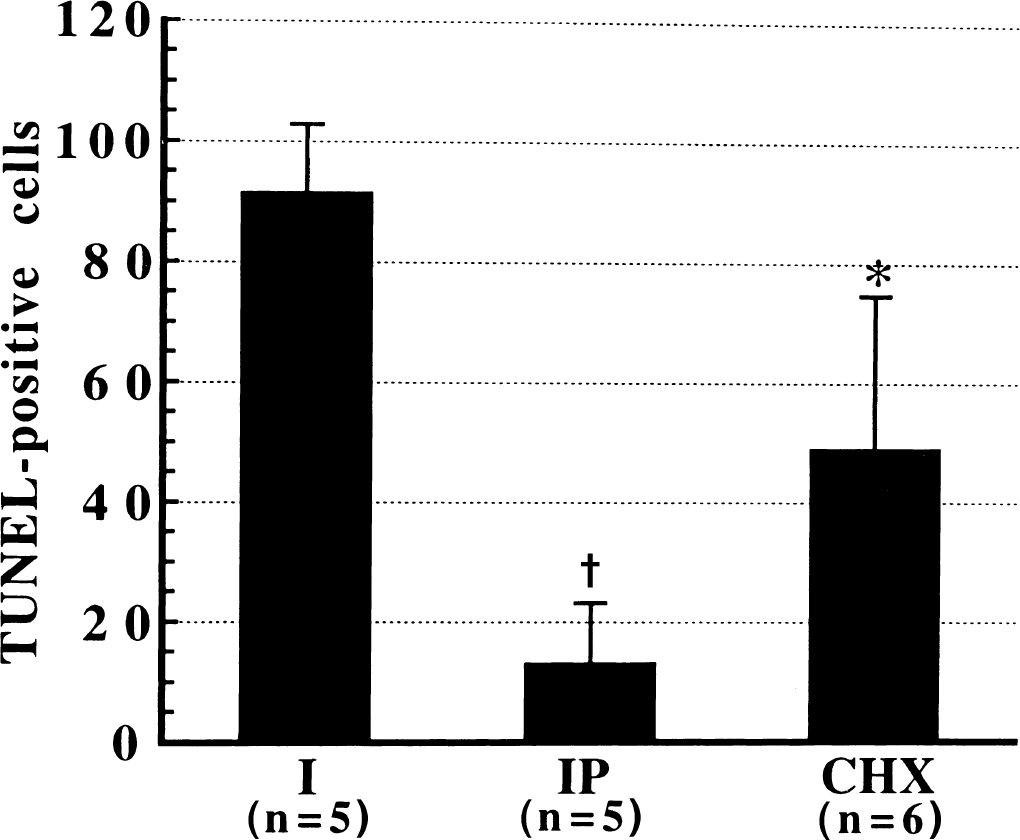
Comparison of the number of TUNEL-positive cells in the hippocampal CA1 region in single ischemic (I), ischemic-preconditioned (IP), and cycloheximide-treated rats (CHX) at 96 hours after reperfusion. * P <0.05 and †P < 0.001 versus single 10-minute ischemia.
DISCUSSION
Even with examination by MAP-2 immunohistochemistry, a more sensitive method for detecting ischemic neuronal damage, most of the hippocampal CA1 neurons were found to die in a delayed process. Although the question has been raised regarding whether the delayed hippocampal CA1 neuronal death is morphologically apoptotic or not (Colbourne et al., 1999; Deshpande et al., 1992; Martin et al., 2000; Nitatori et al.,1995), there should be no doubt that both apoptotic and nonapoptotic mechanisms are involved (Fiskum et al., 1999; Lipton, 1999).
The current results show that cytochrome c release from mitochondria is responsible for hippocampal CA1 neuronal death after transient cerebral ischemia, as evidenced by the observations that persistent loss of cytochrome c selectively occurs in the hippocampal CA1 region and inhibition of cytochrome c release from mitochondria to the cytosol is associated with attenuation of neuronal damage and DNA fragmentation.
Characteristics of cytochrome c release after transient forebrain ischemia
Although previous studies have shown that cytochrome c release from mitochondria occurs after transient ischemia, the characteristics of cytochrome c redistribution are not well illustrated. As reported by Ouyang et al. (1999), cytochrome c release occurred as late as 36 hours after ischemia, whereas another study revealed that the denatured form of cytochrome c was detectable in the cytosol as early as 2 hours after reperfusion in the same ischemic model. A combination of immunofluorescence and Western blotting analysis revealed that two phases of cytochrome c redistribution occur in the hippocampal CA1 region after reperfusion. Although the behavior of cytochrome c release from mitochondria after cerebral ischemia may not be the same as what has been obtained during apoptosis (Goldstein et al., 2000), the results obtained from immunofluorescence and Western blotting analysis clearly indicate that a major part of cytochrome c was released soon after reperfusion. The difference obtained by Western blotting analysis and immunofluorescence may reflect that the kinetic natures of cytochrome c release from mitochondria and its degradation in the cytosol are different. The early phase of cytochrome c release was attenuated by ischemic preconditioning but not by cycloheximide. Unlike the early phase, the late phase occurred in a pattern similar to that observed in the cell-free system (Bossy-Wetzel et al., 1998; Shimizu et al., 1999); cytochrome c diffused to the nuclear space in some neurons, and this was eliminated either by ischemic preconditioning or by cycloheximide.
Although the precise mechanisms by which the two phases of cytochrome c redistribution are initiated and executed are not understood, it seems that different mechanisms were involved. It is known that antiapoptotic members of the Bcl-2 family block cytochrome c release, whereas the proapoptotic member Bax promotes cytochrome c release (Jürgensmeier et al., 1998; Kluck et al., 1997; Shimizu et al., 1999; Yang et al., 1997). Studying the expression of Bcl-2 family proteins after cerebral ischemia has revealed that Bax, a proapoptotic protein, is expressed in the hippocampal CA1 region (Krajewski et al., 1995), whereas neither Bcl-2 nor Bcl-xl proteins are (Chen et al., 1997). Based on the fact that new protein synthesis is necessary for the induction of ischemic preconditioning (Barone et al., 1998) and ischemic preconditioning could induce Bcl-2 expression (Shimazaki et al., 1994), the attenuation of cytochrome c release by ischemic preconditioning may be in part attributable to the up-regulation of Bcl-2 expression. However, cycloheximide may inhibit the synthesis of proapoptotic proteins responsible for cytochrome c release and stimulate Bcl-2 expression (Furakawa et al., 1997).
In contrast to the late phase of cytochrome c redistribution, the early phase occurred rapidly after reperfusion and could be attenuated by ischemic preconditioning but not cycloheximide. This phase may be a result of the mitochondrial swelling that follows cerebral ischemia within several hours after reperfusion (Lipton, 1999). In support of this, a study has shown that ischemic preconditioning enables attenuation of mitochondrial swelling (Kaeffer et al., 1996).
How cytochrome c release may cause ischemic hippocampal CA1 neurons to die
Although cytochrome c redistribution has been shown to occur in the hippocampal CA1 region after transient forebrain ischemia (Ouyang et al., 1999; Sugawara et al., 1999), issues regarding the necessity of cytochrome c redistribution for hippocampal CA1 neuronal death and the shape of neuronal death remain unclear. The fact of inhibition of both early and late phases of cytochrome c release by ischemic preconditioning in association with drastic protection against ischemic neuronal damage and DNA fragmentation indicates that cytochrome c release is responsible for ischemic hippocampal CA1 neuronal death.
One explanation for the role of cytochrome c release in ischemic neuronal death has been that once released, cytochrome c can initiate a caspase-dependent cascade that results in apoptotic neuronal death (Ouyang et al., 1999; Sugawara et al., 1999). However, it seems that the early phase of cytochrome c release might not cause apoptotic neuronal death, because DNA fragmentation was not detectable within 48 hours after reperfusion. Rather, the authors' results indicate that the late phase of cytochrome c redistribution may relate to apoptotic neuronal death based on the fact that the late-phase cytochrome c redistribution was similar to that obtained in the cell-free system after application of apoptotic stimuli (Bossy-Wetzel et al., 1998; Shimizu et al., 1999), and the late phase was followed by intensive DNA fragmentation. Considering that only a smaller part of neurons showed the late phase of cytochrome c redistribution, the results also imply that apoptotic cell death may be a small percentage of the total cell death. This notion is further supported by the fact that inhibition of caspase-3 activity with Z-DEVD-FMK provided only a weaker protection against ischemic neuronal damage. Indeed, the effectiveness of caspase-3 inhibitors on hippocampal CA1 neuronal damage in forebrain ischemia models is under debate. In the 4-VO rat ischemia model, conflicting results exist regarding whether the caspase-3 inhibitor Z-DEVD-FMK is effective (Chen et al., 1998; Li et al., 2000). Two recent studies provided insights to explain the difference. As shown in rat models of hypoxia–ischemia (Hu et al., 2000) and trauma (Bittigau et al., 1999), both caspase-3 activation and caspase-3 activation-related neuronal death were found to depend tightly on the age of animals, with an insignificant increase of caspase-3 activation in mature animals (Hu et al., 2000).
In the absence of properly working apoptotic machinery or before completing apoptosis, the persistent loss of mitochondrial cytochrome c may cause cell death through other pathways, such as failure of oxidative phosphorylation, an increase in superoxide production, or both.
Several experiments have shown that mitochondrial dysfunction follows cerebral ischemia, but the mechanisms for mitochondrial dysfunction are unknown (Fiskum et al., 1999; Lipton, 1999). Logically, the failure of oxidative phosphorylation may follow the persistent loss of cytochrome c, resulting in CA1 neuronal death after the glycolytic capacity of cells is insufficient to maintain ATP production. Considering that small reductions in ATP production (20% to 30%) lasting for several hours can lead to delayed cell death (Pang and Geddes, 1997; Schulz et al., 1997), the decrease in mitochondrial ATP synthesis resulting from the loss of mitochondrial cytochrome c may contribute to delayed hippocampal CA1 neuronal death. The result obtained from cycloheximide-treated animals supports this possibility. In cycloheximide-treated animals, most of the hippocampal CA1 neurons survived until 96 hours after reperfusion. However, without cytochrome c regain, cycloheximide protection was almost lost by 7 days after reperfusion. By inference, approaches aimed at enhancing the capacity of glycolysis after reperfusion should afford neuroprotection. Indeed, recent studies have demonstrated that high glucose effectively reduces neuronal death induced by N-methyl-d-aspartate, free radicals, and in vitro and in vivo ischemia (Schurr et al., 1999; Seo et al., 1999).
In addition to failure of oxidative phosphorylation, loss of cytochrome c from mitochondria may contribute to mitochondrial superoxide formation (Cai and Jones, 1998). Cytochrome c transports electrons between mitochondrial complexes III and IV. A disruption of the mitochondrial electron flow caused by a significant loss of cytochrome c maintains complex I and the ubiquinone at complex II in the reduced state (Luetjens et al., 2000). This condition has previously been shown to favor one-electron reduction of molecular oxygen, presumably because of an autooxidation of complex I and ubiquinone (Boveris et al., 1976). Given the importance of superoxide production in mediating ischemic neuronal death (Chan et al., 1998), facilitation of superoxide production may also be a pathway for ischemic neuronal death resulting from the loss of cytochrome c in mitochondria.
Taken together, the loss of cytochrome c from mitochondria may cause ischemic hippocampal CA1 neurons to die by both caspase-dependent and -independent mechanisms. The shape of neuronal death may be dependent on intracellular ATP levels. If the caspase-dependent pathway works foremost and intracellular ATP levels are maintained, cell death would more likely be apoptotic rather than necrotic. Further, if both caspase-dependent and -independent mechanisms work together, ischemic hippocampal CA1 neuronal death may share features of necrosis and apoptosis, implying that typical apoptotic cells may rarely exist; this is in agreement with recent morphologic findings (Colbourne et al., 1999; Martin et al., 2000).
Persistent loss of mitochondrial cytochrome c in the hippocampal CA1 region may be attributable to protein synthesis inhibition
Like most mitochondrial proteins, cytochrome c is encoded by a nuclear gene and synthesized as a cytoplasmic precursor molecule, apocytochrome c, which becomes selectively imported into the mitochondrial intermembrane space (Bossy-Wetzel et al., 1998). The persistent disappearance of cytochrome c in mitochondria after ischemia is consistent with the previous observation that total protein is severely reduced as early as 6 hours after reperfusion and is reduced persistently in hippocampal CA1 neurons, with most hippocampal CA1 neurons never regaining their biosynthesis activity (Araki et al., 1990; Furuta et al., 1993; Johansen and Diemer, 1990; Thilmann et al., 1986). In support of this possibility, ischemic preconditioning protects neurons against ischemic damage that has been reported to be associated with rapid restoration of protein synthesis in the gerbil (Furuta et al., 1993; Kato et al., 1995; Nakagomi et al., 1993).
The authors conclude that loss of cytochrome c from mitochondria correlates with hippocampal CA1 neuronal death after transient cerebral ischemia in relation to both caspase-dependent and -independent mechanisms. The amount of mitochondrial cytochrome c regained may determine whether ischemic hippocampal CA1 neurons survive or succumb to a late-phase death. Further works are needed to elucidate the factors responsible for the early phase of cytochrome c release and to search for effective approaches to improve the regain of mitochondrial cytochrome c.
Footnotes
Acknowledgment:
The authors thank Dr. J. Yao, Department of Cellular Physiology, Institute of Nephrology, Niigata Univertsity School of Medicine, for his assistance in performing Western blotting analysis.