
Abstract
Loss of mitochondrial membrane integrity and the resulting release of apoptogenic factors may play a critical role in mediating hippocampal neurodegeneration after transient global ischemia. In the present study, the authors have cloned and characterized the rat cDNA encoding apoptosis-inducing factor (AIF), an intramitochondrial protein that promotes cell death in a caspase-independent manner upon release into nonmitochondrial compartments. In contrast to the expression patterns of a number of apoptosis-regulatory gene products during brain development, the expression of AIF protein increases gradually with brain maturation and peaks in adulthood. In a rat model of transient global ischemia, AIF was found to translocate from mitochondria to the nucleus in the hippocampal CA1 neurons after ischemia and to manifest a DNA-degrading activity that mimicked the purified AIF protein and was inhibitable by AIF immunodepletion. The temporal profile of AIF translocation after ischemia (24 to 72 hours) coincided with the induction of large-scale DNA fragmentation at the size of 50 kbp, a well-characterized hallmark of AIF-like activity but preceded the formation of internucleosomal DNA fragmentation (72 hours), a DNA degradation associated with the terminal stage of cell death. Further, the nuclear translocation of AIF after ischemia was not blocked by inhibiting caspase-3/-7 activities, but, as shown in neuronal cultures that were challenged with transient oxygen-glucose deprivation, it can be prevented by intracellular delivery of the mitochondria-associated antiapoptotic protein Bcl-xL. The results presented here strongly suggest that mitochondrial release of AIF may be an important factor, in addition to the previously reported cytochrome c and Smac, which could contribute to the selective vulnerability of CA1 neurons to transient global ischemic injury.
Mitochondrial damage and release of apoptogenic factors play a critical role in mediating neuronal cell death after cerebral ischemia (Fujimura et al., 1999;Sugawara et al., 1999). Activation of the mitochondrial-signaling pathway, the so-called intrinsic pathway, is known to trigger caspase-dependent or caspase-independent cell death execution machineries (Graham and Chen, 2001). In the caspase-dependent mechanism, release of mitochondrial cytochrome c activates Apaf-1, which, through the formation of the multimeric caspase-activating complex called apoptosome, recruits and activates caspase-9 and then activates the terminal execution caspases such as caspase-3 and −7 (Li et al., 1997). In the caspase-independent mechanism, loss of mitochondrial integrity leads to the release of apoptogenic factors such as apoptosis-inducing factor (AIF) and endonuclease G, which kill cells without requiring caspase activities (Susin et al., 1999;Widlak et al., 2001).
The activation profile of the caspase-dependent intrinsic pathway has been characterized in several studies (Krajewski et al., 1999;Benchoua et al., 2001;Noshita et al., 2001;Cao et al., 2002a;Sugawara et al., 2002) that showed a correlation of cytochrome c release or caspase-9 activation with selective vulnerability of neuronal cell death after transient ischemia. Furthermore, caspase inhibitors, targeting either the Apaf-1/caspase-9 pathway or its downstream caspases, offered neuroprotection against cerebral infarction after focal ischemia or delayed cell death in the hippocampal CA1 after transient global ischemia (Hara et al., 1997;Chen et al., 1998;Himi et al., 1998;Namura et al., 1998;Gillardon et al., 1999;Cao et al., 2002a). However, whereas these observations point to an important role for mitochondrial damage-mediated caspase activation in ischemic cell death, additional data strongly suggest that caspase-independent mechanisms may be involved as well, and, under certain circumstances, could be a prominent factor in ischemic cell death (Graham and Chen, 2001). For instance, in the rat model of transient global ischemia, inhibition of caspase-3 or caspase-9 activity can delay, but is unable to prevent, delayed cell death in the hippocampal CA1 sector (Chen et al., 1998;Cao et al., 2002a).
The present study aims to investigate the role of AIF, a novel caspase-independent pro-apoptotic molecule, in the rat model of transient global ischemia. AIF is an abundant mitochondrial protein, which is vulnerable to release upon receiving various cell death signals and which potently promotes apoptosis, mainly through its nuclear-degrading activities (Susin et al., 1999;Daugas et al., 2000;Dumont et al., 2000;Ferri and Kroemer, 2000;Vieira et al., 2000). AIF is ubiquitously expressed in the central nervous system and appears to play a critical role in neuronal apoptosis induced by glutamate toxicity or oxidative stress (Yu et al., 2002;Zhang et al., 2002). Accordingly, the authors speculate that the AIF pathway is a legitimate candidate for contributing to the caspase-independent component of cell death processes after ischemic brain injury. To test this hypothesis, the authors cloned the rat AIF cDNA from a brain cDNA library, characterized the temporal profile and cellular/subcellular distribution of AIF expression, and examined the AIF-like activity in the rat four-vessel model of transient global ischemia. The data presented here support the authors' hypothesis.
MATERIALS AND METHODS
The rat model of global ischemia and peptide infusion
Transient global ischemia (15 minutes) was induced in male Sprague-Dawley rats (300–325 g each) using the four-vessel occlusion method, as described previously (Chen et al., 1998). Blood pressure, blood gases, and blood glucose concentration were monitored and maintained in the normal range throughout the experiments. Rectal temperature was continually monitored and kept at 37 to 37.5°C using a heating pad and a temperature-regulated heating lamp. Brain temperature was monitored using a 29 Ga thermocouple implanted in the left caudate-putamen and was kept at 35.8 ± 0.2°C during ischemia and at 37 to 37.5°C thereafter.
Electroencephalogram (EEG) was monitored in all animals to ensure isoelectricity within 10 seconds after the induction of ischemia. Sham operations were performed in additional animals using the same anesthesia and surgical exposure procedures, except that the arteries were not occluded.
In selective experiments, rats were subjected to intracerebral ventricular infusion of the caspase-3/7 inhibitor N-benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoro-methylketone (z-DEVD. fmk) at the dose indicated below (see Results) using procedures described previously (Chen et al., 1998;Cao et al., 2001). The hippocampal tissues were processed for Western blot analysis of AIF nuclear translocation at 3 or 7 days after 15 minutes of transient global ischemia.
cDNA cloning of rat brain AIF
Rat brain AIF cDNA was cloned from a rat brain cDNA library using the Rapid Amplification of cDNA Elongation (RACE) strategy, as described previously (Chen et al., 2000;Cao et al., 2001). Briefly, cerebella were dissected from four 1 week old male Sprague Dawley rats. Polyadenylated RNA was isolated using the polyA-tract-1000 mRNA isolation system (Promega, Madison, WI, U.S.A.) and used as a template for cDNA synthesis. The cDNA library was constructed using a Marathon cDNA amplification kit (Clontech, Palo Alto, CA, U.S.A.) according to the manufacturer's instructions and was subsequently used as a template for RACE. A cDNA fragment encoding the rat brain AIF was first generated by reverse transcription-PCR according to the conserved sequences in human and mouse AIF: 5′-CTTTCTATGTCTCTGCTCAGGACCTGC-3′ (sense) and 5′-GAAGATGACACCTTTGCCGTAGTCCTC −3′ (antisense). Based on the sequence of this cDNA fragment, 5′- and 3′-RACE primers were designed as follows: 5′-CATTTACCCGGAAGCCACCAAAATCGG-3′ and 5′-GAGCTCCAAGCACGTTCTAACATCTGG −3′. The adapter-ligated double-stranded cDNAs served as templates for RACE. The 5′-RACE- and 3′-RACE-amplified fragments were subcloned into pGEM-T easy vector (Promega, Madison, WI, U.S.A.) and then sequenced on both strands (University of Pittsburgh Sequencing Facility). The full-length cDNA of AIF was further obtained using PCR based on the obtained 5′- and 3′-end sequences.
To confirm that the cDNA contained the full open reading frame, the authors performed in vitro transcription/translation to detect its protein product. The Kozak sequence was added to the full-length AIF cDNA before the start code using PCR and then the PCR product was subcloned into the pcDNA3.1 plasmid in forward orientation. Coupled transcription and translation was carried out using the TNT quick-coupled transcription/translation systems (Promega) according to the manufacturer's instructions. In brief, 1 μg of Not I linearized pcDNA-AIF was incubated at 30°C for 2 hours in 40 μL of TNT quick master mix and 20 μCi of 35S-Methionine (ICN Biomedicals, Irvine, CA, U.S.A.). Two microliters of the reaction mixture was electrophoresed on a 12% polyacrylamide gel, transferred to a PVDF membrane, and exposed to x-ray film (Kodak) with intensifying screens. In a separate reaction, 35S-methionine was replaced by cold methionine, and the reaction product was subjected to Western blot confirmation using a rabbit polyclonal anti-AIF antibody raised against the C-terminal sequence of rat AIF.
Generation of rat AIF fusion protein
To determine whether the cloned AIF cDNA encodes a functional protein, AIF fusion proteins were produced using the procedure described previously (Cao et al., 2001). In brief, the rat AIF cDNA was amplified using the primers 5′-ATG TTC CGG TGT GGA GGC CTG GCG G −3′ (sense) or 5′-TTA GGA CTG TCA CCA GAA GAG AAA C −3′ (sense) and 5′-CTA GTT CTT GCC CAC CTC TAA ATC C-3′ (antisense) to generate cDNAs containing the full-length AIF and mature AIF (with the deletion of the first 100 amino acids), respectively. The cDNA was fused into the glutathione S-transferase (GST) gene in PGEX-2T vector according to the manufacturer's instructions (Amersham Pharmacia Biotech, Little Chalfont, Buckinghamshire, England). The GST-AIF fusion proteins were expressed in Escherichia coli BL21 cells and absorbed to glutathione-Sepharose 4B column. The fusion proteins were then cleaved by thrombin for 16 hours at room temperature to remove the GST portion. The elute was collected by centrifugation at 500 × g for 5 minutes at 4°C. The purified AIF proteins were verified by Western blot analysis.
Detection of AIF-like nuclease activity
To determine whether the AIF fusion proteins possess the nuclear degradation activity as anticipated, the AIF protein was added to the in vitro cell-free apoptosis reaction system (Chen et al., 2000;Cao et al., 2001) and evaluated for its ability to induce large-scale DNA fragmentation, as described previously (Susin et al., 1999). The nuclei were isolated and purified from cultured cortical neurons using the procedures previously described (Chen et al., 2000) and were used as the substrate for the detection of nuclease activity of AIF.
To determine whether the AIF-like activity was increased in the brain after ischemia, rats were decapitated at 48 hours after 15 minutes of global ischemia (eight rats per experimental condition). The hippocampi were isolated, and the portion containing the CA1 sector was dissected. Nuclear protein was extracted as described previously (Chen et al., 2000). To perform the assay, protein extracts (200 μg) were incubated with neuronal nuclei (5 × 105 per reaction) overnight at 32°C in the reaction buffer containing 50 mM NaCl, 10 mM Hepes (pH 7.0), 40 mM β-glycerophosphate, 2 mM MgCl2, 5 mM EGTA, 1 mM NAD, 1 mM DTT, and 100 μM z-VAD.fmk (to prevent caspase-related nuclease activity) and supplemented with an ATP-regeneration system containing 2 mM ATP, 10 mM creatine phosphate, and 50 μg/ml creatine kinase. The reaction was terminated by adding the lysis buffer containing 50 mM Tris-HCl (pH 8.0), 10 mM EDTA, protease K (0.5 mg/ml), and RNase (5 μg/ml) and incubated at 50°C for 1 hour. Large-scale DNA fragmentation was examined using pulse-field gel electrophoresis, as previously described (Zhang et al., 2002).
To confirm the specificity of AIF-like nuclease activity, the nuclear extracts were immunoprecipitated using the anti-AIF antibody to deplete the AIF protein before the activity assay.
Northern blot analysis
Total RNA was prepared from rat brains using the total RNA isolation system (Promega) according to the manufacturer's instructions. RNA from the following two sets of tissues was analyzed: set 1, cerebella of rat brains at different ages, including embryonic day 17, postnatal day 1, and weeks 1, 2, 4, and 12 (adult); set 2, hippocampi of adult rat brains subjected to 15 minutes of global ischemia followed by 8, 24, or 72 hours of reperfusion.
Total RNA (30 μg) was electrophoresed on a 1% agarose-formaldehyde gel and blotted onto a zeta-probe GT nylon membrane (Bio-Rad, Hercules, CA, U.S.A.). The membrane was hybridized overnight at 42°C with the AIF cDNA probe (4 × 106 cpm/ml) labeled with 32P using a random primer labeling kit (Boehringer Mannheim, Indianapolis, IN, U.S.A.). To control for sample loading, the membranes were stripped and rehybridized with a 32P-labeled 28S RNA probe. Data analysis was performed based on optical density measurement as previously described (Chen et al., 1998).
In situ hybridization
Rats were anesthetized and decapitated at 4, 8, 24, and 72 hours after 15 minutes of global ischemia or 24 hours after sham operation (n = 4 per time point). Frozen coronal sections (15 μm thick) were cut on a cryostat at −20°C. The 35S-labeled AIF cRNA probe was prepared in both the sense and antisense orientation using the same procedures as previously described (Chen et al., 1998). The sections were hybridized with the labeled probe (1 × 107 cpm/ml) in hybridization cocktail for 18 hours at 55°C. After the washing procedures, the slides were dehydrated, air dried, and exposed to Kodak film for 3 weeks. The relative changes in mRNA expression were quantified by determining the ratio of the optical density of the specified regions in ischemic brains to controls using the MCID system (Chen et al., 1998).
Western blot analysis
Western blotting was performed using the standard method. For the evaluation of AIF protein distribution in various rat organs and in the rat brains of various ages, total protein extracts were prepared from various organ tissues (i.e., heart, liver, kidney, brain, spleen, and lung) of adult rats and from the cerebella of rats at various ages (embryonic day 17, postnatal day 1, and weeks 1, 2, 3, 4, and 12).
For the detection of AIF release and nuclear translocation, mitochondrial and nuclear protein extracts were prepared from the hippocampi at 2, 6, 12, 24, and 72 hours after ischemia or 24 hours after sham operation (n = 3 per group). The tissues were first homogenized in a hypotonic buffer containing 50 mM Tris-HCl (pH 8.0), 25 mM MgCl2, and 0.1 mM phenylmethylsulfonyl fluoride using a Dounce homogenizer and kept on ice for 15 minutes, and the nuclear, cytosolic, and mitochondrial fractions of protein were separately isolated by centrifugation as previously described (Wood and Earnshaw, 1990;Liu et al., 1996;Chen et al., 2000). The protein samples were then immunoreacted with the rabbit polyclonal antibody against AIF at dilution 1:1000, and immunoblotting of COX IV and Histone served as the protein loading controls for the mitochondrial and nuclear fraction, respectively.
Immunohistochemistry
Rats were euthanized in a carbon dioxide chamber for 5 minutes at 2, 6, 24, or 72 hours after ischemia or 24 hours after sham operation (n = 4 per time point). They were perfused with 200 ml heparinized 0.9% saline followed by 500 ml of 4% paraformaldehyde in 0.1 M PBS, pH 7.4. The brains were removed and processed for paraffin embedding and cutting, and coronal sections at the levels of dorsal hippocampus were used for immunohistochemical staining. Immunohistochemical staining for AIF was performed with the same anti-AIF antibody (dilution 1:500) used for the Western blots. The procedures for immunohistochemistry were the same as previously described (Chen et al., 1996). For the assessment of nonspecific staining, alternating sections from each experimental condition were incubated with the primary antibody that had been preabsorbed with the antigen at the antigen/antibody ratio of 10:1 (Chen et al., 1996).
In selective experiments, double-label immunofluorescent staining was performed on freshly frozen sections to co-localize nuclear translocation of AIF and DNA fragmentation. The sections were air dried and then fixed with cold absolute ethanol (−20°C) for 5 minutes. The general procedures for the double labeling were the same as previously described (Cao et al., 2001).
In vitro model of neuronal ischemia
Primary cultures of hippocampal neurons were prepared from 17-day-old Sprague-Dawley rat embryos as previously described (Nagayama et al., 1999). In brief, the hippocampal tissue was cut with a scalpel blade into approximately 1-mm3 pieces and incubated at 37°C for 30 minutes in Ca2+ and Mg2+ free Eagle's balanced salt solution containing 0.01% trypsin (1:250). Ten percent horse serum was then added, and the tissue was triturated with a 5-ml pipette. Cells were centrifuged for 10 minutes at 190 × g and resuspended in Eagle's minimal essential medium prepared without glutamine, with twice the usual concentration of other amino acids, and four times the usual concentration of vitamins (MEM-Pak, UCSF Cell Culture Facility, San Francisco, CA, U.S.A.). The culture was supplemented on the day of plating with 2 mM glutamine, 15 mM HEPES (pH 7.4), and glucose to a final concentration of 30 mM. Cell suspensions were filtered through a 7-μM Falcon nylon cell drainer, supplemented with 10% horse serum and 10% fetal calf serum, and seeded at 3 × 105 cells per well on 24-well Corning cell culture dishes, or at 1.25 × 107 per dish on 10-cm dishes coated with 100 μg/ml of poly-D-lysine. Cytosine arabinoside (AraC, 10 μM) was added on the fifth day in vitro (DIV). At 6 DIV, one half of the medium was replaced with AraC-free medium, and one third of the medium was replaced with fresh medium twice weekly thereafter. Experiments were conducted at 17 DIV, when cultures consisted primarily of neurons (approximately 95% MAP2-immunoreactive cells).
To model ischemia-like conditions in vitro, primary cultures were exposed to combined oxygen and glucose deprivation (OGD) (Nagayama et al., 1999;Cao et al., 2001). In brief, two thirds of the culture medium was replaced four times with serum- and glucose-free medium, resulting in a final glucose concentration of less than 1 mM. The glucose-deprived cultures were then placed in a Billups-Rothenberg modular incubator chamber (Del Mar, San Diego, CA, U.S.A.), which was flushed for 5 minutes with 95% Argon and 5% CO2 and then sealed. The chamber was placed in a water-jacketed incubator (Thermo, Cambridge, UK) at 37°C for 60 minutes and then returned to 95% air, 5% CO2, and glucose-containing medium for the period of time indicated in each experiment. Control glucose-containing cultures were incubated for the same periods of time at 37°C in humidified 95% air and 5% CO2.
Measurement of cell viability
Fluorescence of Alamar Blue (Accumed International, Westlake, OH, U.S.A.), an indicator that changes from blue to red and fluoresces when reduced by cellular metabolic activity, was used to measure the viability of the cultured neuron (Nagayama et al., 1999). One half of the culture medium was replaced with MEM-Pak containing 10% (v/v) Alamar Blue, and cultures were incubated for 2 hours at 37°C in humidified 95% air and 5% CO2. Fluorescence was determined in a Millipore CytoFluor 2300 automated plate-reading fluorometer (Bedford, MA, U.S.A.), with excitation at 530 nm and emission at 590 nm. As reported previously, Alamar Blue fluorescence in these cultures varies linearly with cell number, decreases with exposure to hypoxia or OGD, and correlates with the extent of cellular injury determined by lactate dehydrogenase (White et al., 1996).
Data analysis
All data are presented as mean ± SD. Comparisons of AIF release or the viability of cultured neurons were made between experimental groups using analysis of variance (ANOVA) and post hoc Scheffe's tests. A level of P < 0.05 was considered statistically significant.
RESULTS
cDNA cloning of rat brain AIF
To characterize the expression pattern and determine the role of AIF in the brain after ischemia, the authors cloned cDNA containing the entire open reading frame of AIF (GenBank accession numbers: AF375656) from a cDNA library constructed from 1-week-old rat cerebellum. Sequence analysis revealed that this cDNA encodes 612 amino acids (Fig. 1A). The deduced amino acid sequence showed 92.6% and 98.2% identity to the published sequences of human and mouse AIF (Susin et al., 1999), respectively (Fig. 1B). The rat AIF contains two stretches of 24 and 6 amino acids at positions aa 277 to 300 and aa 445 to 450, respectively, consisting of repetitive glutamine, proline, arginine, and lysine, strongly suggestive of nuclear localization segments. In addition, the 100-amino acid N-terminus of the rat AIF is almost identical to that of its mouse counterpart, which has been identified as the mitochondrial localization signal of AIF (Susin et al., 1999).
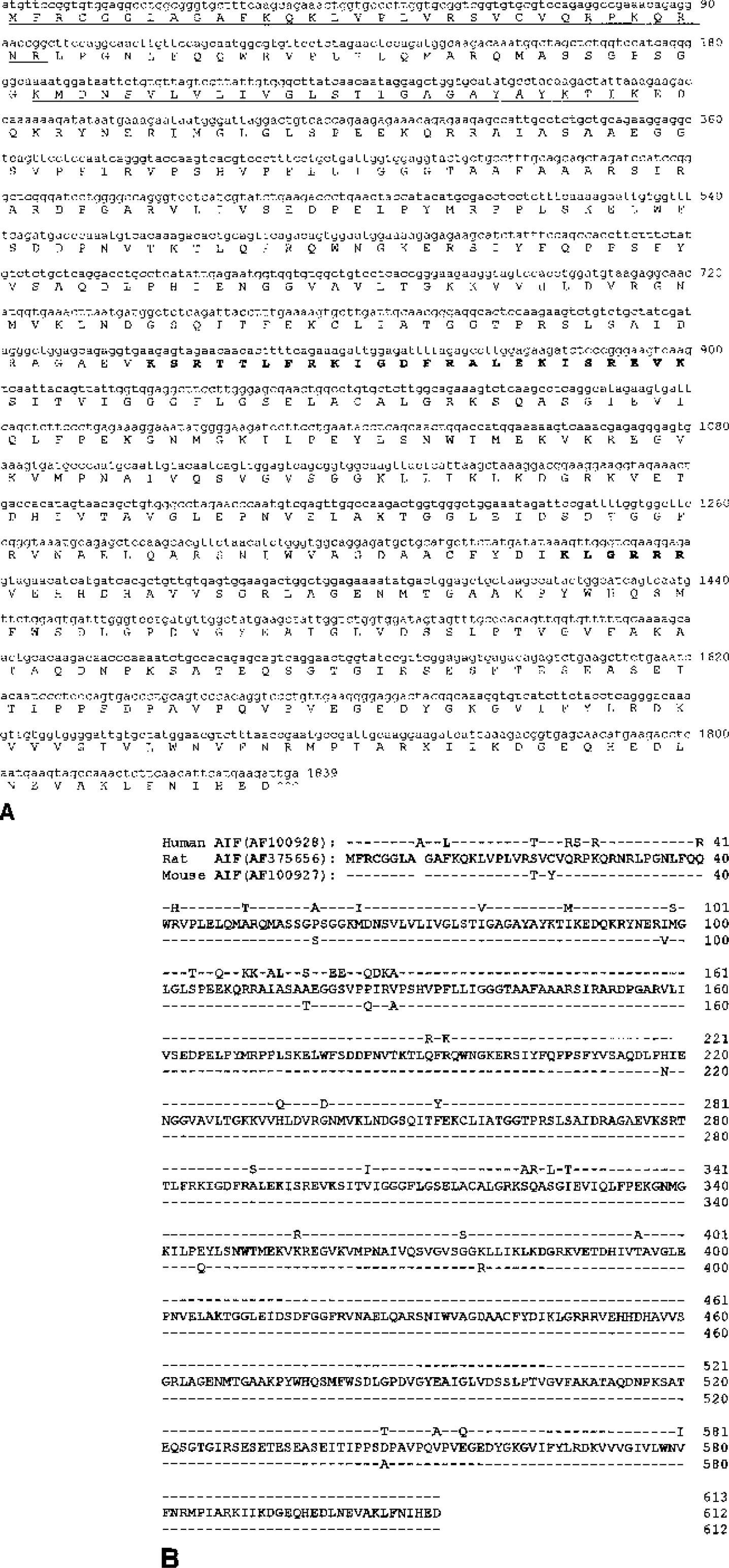
Cloning of rat AIF.
Using the cloned cDNA as a template, the in vitro transcription/translation assay produced a radiolabeled protein product at approximately 67 kDa, which was also recognized by the anti-AIF immunoblotting (data not shown). This result confirms the validity of the sequence of rat AIF cDNA.
Expression of AIF in the normal brain
Northern blot analysis was performed using the random primer labeled cRNA probe to confirm the expression of AIF mRNA in rat brain tissues. Because the expression of a number of apoptosis-regulatory genes, such as caspase-9, caspase-3, Apaf-1, and CAD, has shown marked changes during brain development (Cao et al., 2001, 2002a;Yakovlev et al., 2001), the authors examined AIF mRNA expression in rat brains of various ages. As shown (Fig. 2A), Northern blots detected single mRNA transcript at the size of approximately 2.4 kb in all samples tested. As determined by semiquantitatively based optical density measurements, AIF mRNA expression showed slight to moderate decreases (up to 20%) in the brain as age increased (Fig. 2B).
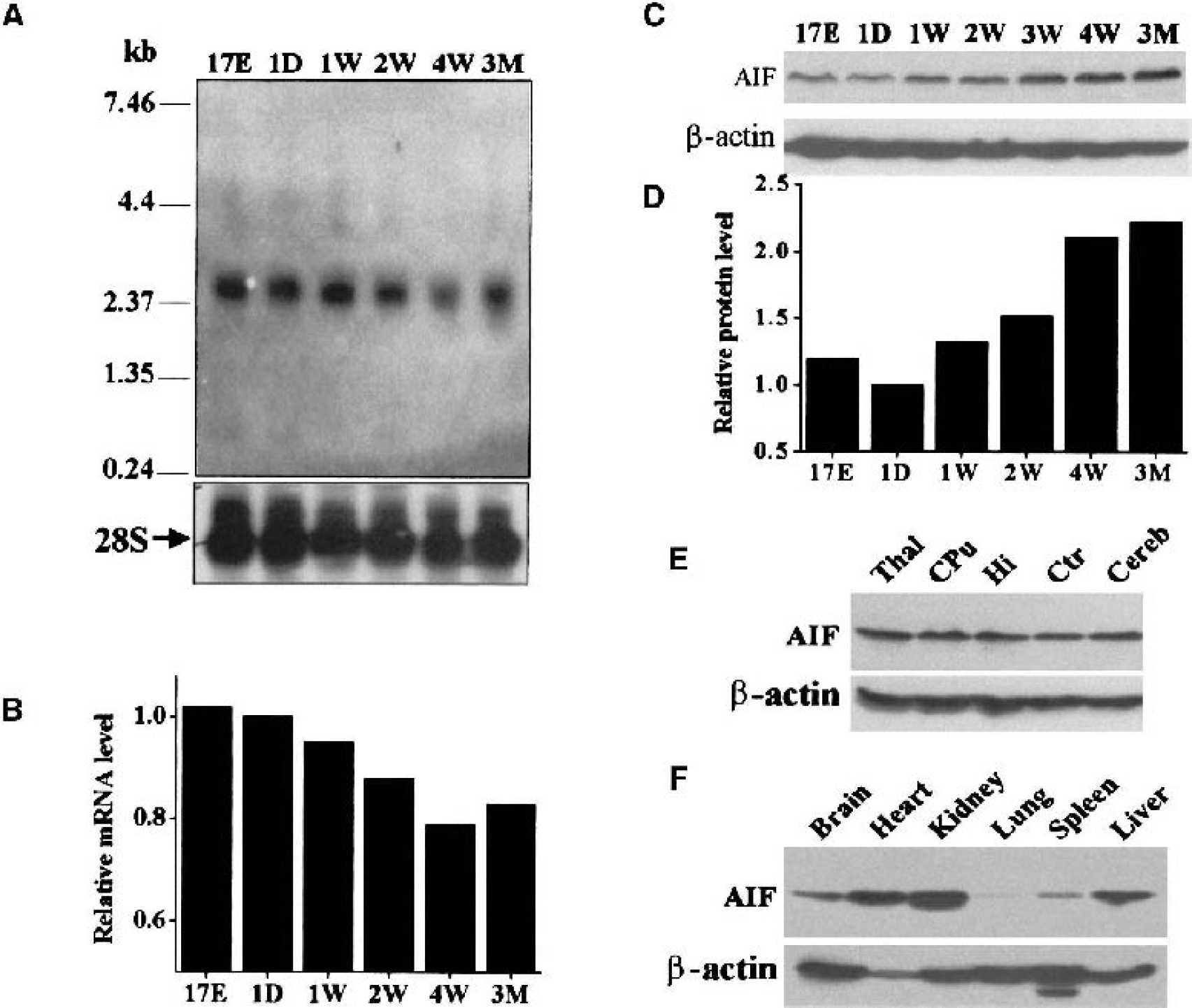
Characterization of AIF expression in the rat.
Using an antibody raised against the C-terminal amino acid sequence of rat AIF, Western blot analysis was performed to examine the expression of AIF protein in the rat brain cerebellum of various ages (Fig. 2C), various regions of adult brains (Fig. 2E), and various organ tissues of adult rats (Fig. 2F). Surprisingly, AIF immunoreactivity showed marked increases (greater than twofold) in the cerebellum as age increased (Fig. 2D), especially after postnatal 3 weeks. This pattern of AIF protein expression is distinctive from that of its mRNA expression.
AIF protein was readily detectable in adult rats with comparable expression levels among different brain regions (Fig. 2E). In contrast, the levels of AIF immunoreactivity were variable among different organ tissues, with relatively higher levels in the kidney, liver, and heart, followed by brain and spleen, with the lowest level in the lungs.
Expression of AIF mRNA in the brain after ischemia
To investigate the potential role of AIF in ischemic neuronal cell death, the authors examined the expression of AIF at both mRNA and protein levels in normal and ischemic brains, focusing on the hippocampal CA1 sector, where neurons are particularly vulnerable to transient global ischemia. Using Northern blot analysis, the authors failed to detect increased expression of AIF mRNA throughout the time course studied (Fig. 3A). In fact, a moderate decrease (approximately 15–30%) in the signal was seen at 24 and 72 hours after ischemia. In situ hybridization with the antisense probe showed that AIF mRNA expression was detectable in the hippocampal pyramidal and granular cell layers in the sham-control brains, and the levels of expression were not obviously changed at 4, 8, or 24 hours after ischemia (Fig. 3B). However, at 72 hours after ischemia, a decrease in the signal was seen in the CA1 pyramidal layer, consistent with the maturation of cell degeneration in this region (Fig. 3B, panel E). In situ hybridization with the sense probe on sections obtained at 24 hours after ischemia showed homogenous background signals (Fig. 3B, panel F), thus confirming the specificity of AIF mRNA signals as detected using the antisense probe.
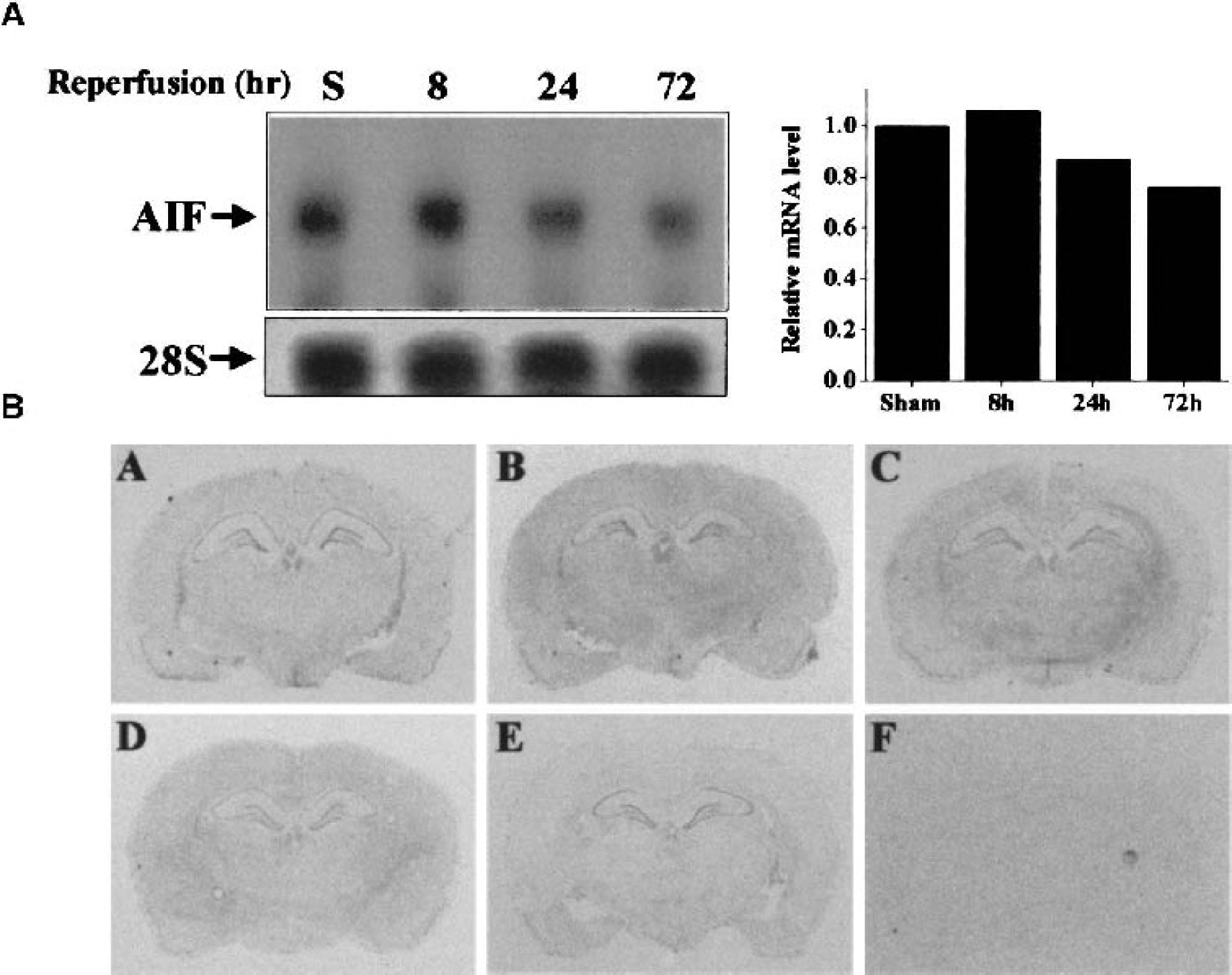
Characterization of AIF mRNA expression after transient global ischemia.
Nuclear translocation of AIF after ischemia
The expression of AIF protein in hippocampal whole-cell extracts was examined using Western blotting in sham controls and brains at 2, 6, 12, 24, and 72 hours after ischemia. Results from optical density measurement indicated that the total AIF protein level was not significantly changed after ischemia (Fig. 4B).
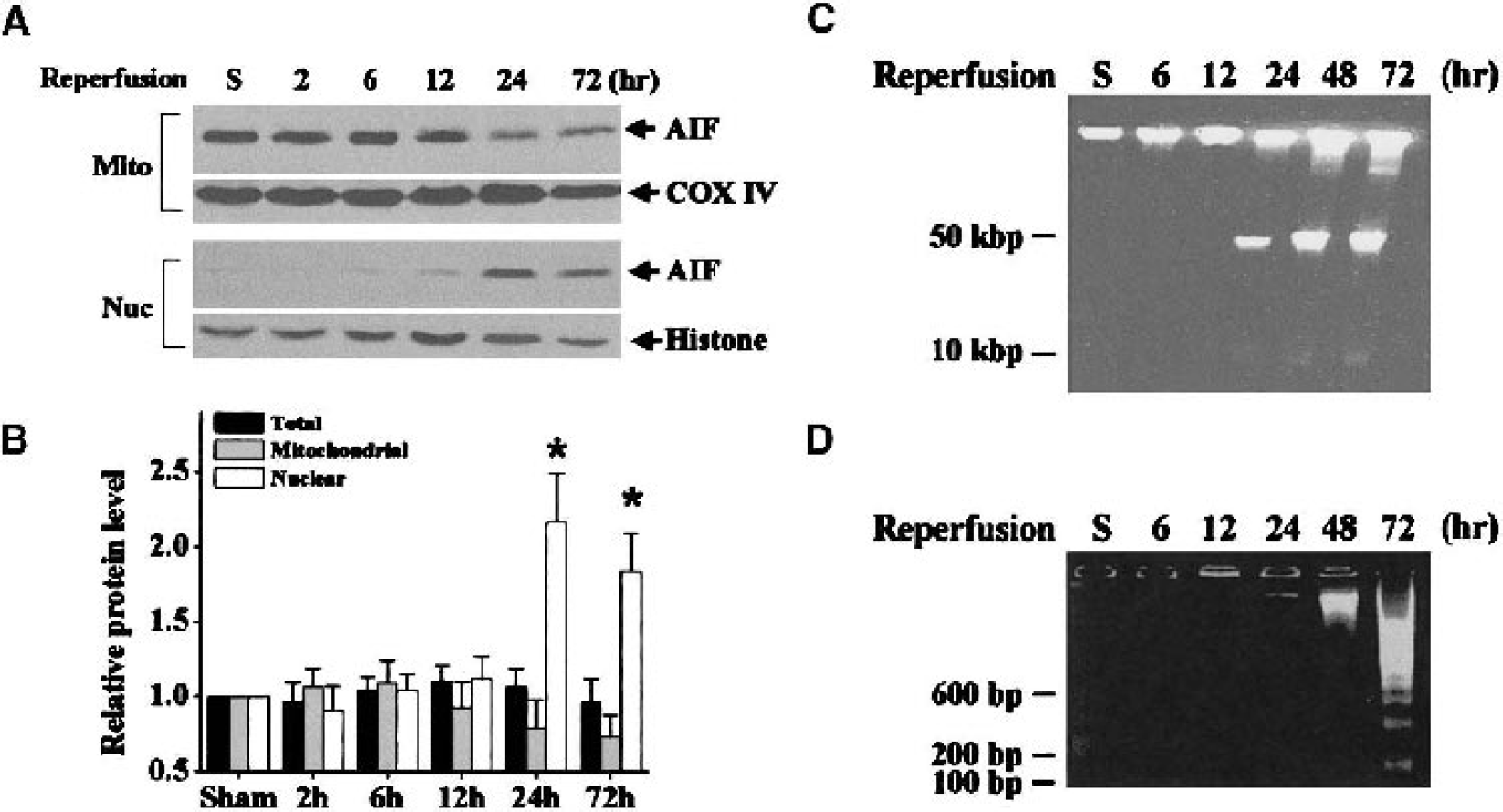
AIF translocation and DNA fragmentation in the hippocampus after transient global ischemia.
Western blots performed following subcellular protein fractionation demonstrated that AIF was subjected to translocation from mitochondria to nucleus in the hippocampus after ischemia (Fig. 4A and Fig. 4B). In the nuclear fraction, little AIF immunoreactivity was detectable in the sham controls, or at 2, 6, and 12 hours after ischemia. However, AIF immunoreactivity was significantly increased in the nuclear fraction at 24 and 72 hours after ischemia. Consistent with its nuclear translocation, the immunoreactivity of AIF was decreased in the mitochondrial fraction at 24 and 72 hours after ischemia. This time course of AIF translocation coincided with that of the formation of large-scale DNA fragmentation (Fig. 4C), a hallmark of AIF activity (Susin et al., 1999), detected in CA1 sector after ischemia, but preceded that of intranucleosomal DNA fragmentation (Fig. 4D), a terminal DNA degradation that is likely caused by increased CAD activity (Cao et al., 2001).
To characterize the cellular distribution of AIF after ischemia, the authors performed immunohistochemical staining in PFA-fixed/paraffin-embedded brain sections obtained at 24 and 72 hours after ischemia or 24 hours after sham operation (n = 4 per time point). Similar to what has been seen in cytochrome c immunohistochemistry experiments (Sugawara et al., 1999), AIF immunostaining in control brain sections resulted in no specific staining in the hippocampal neurons (Fig. 5A), probably because of the incapability of the antibody to access the intramitochondrial antigen in PFA-fixed/paraffin-embedded cells. In contrast, a markedly increased AIF immunoreactivity with nuclear localization was detected in many hippocampal CA1 neurons at both the 24- and 72-hour time points after ischemia. The increased AIF immunoreactivity was not detected in CA3 neurons or in the dentate granule cell layer, which are resistant to ischemic injury in the global ischemia model, suggesting that nuclear translocation of AIF occurs selectively in degenerating neurons.
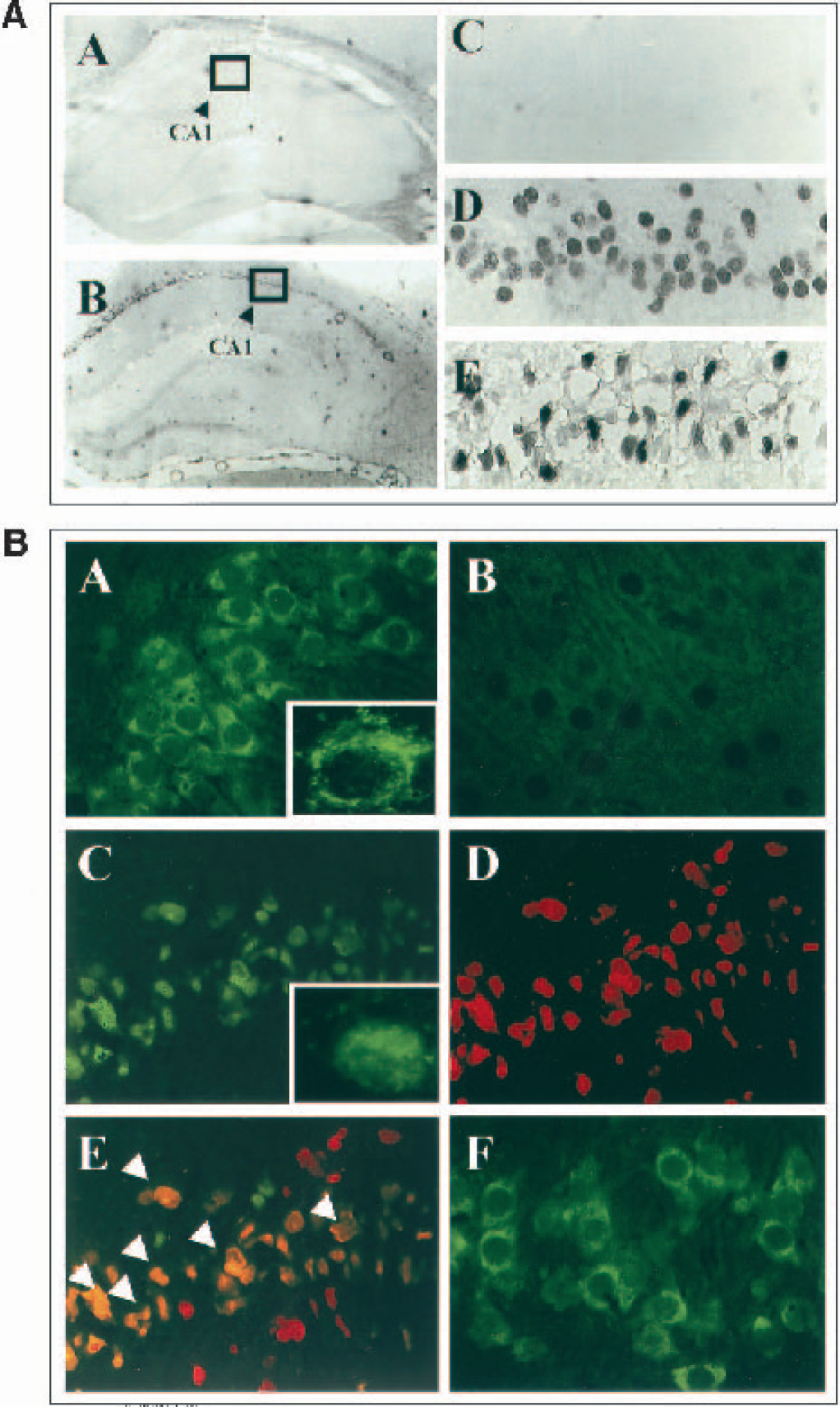
Immunohistochemical detection of AIF translocation in CA1 neurons after ischemia.
To further confirm that AIF translocation occurs in degenerating neurons after ischemia, double-label immunofluorescent staining of AIF and TUNEL was performed using freshly frozen brain sections obtained at 72 hours after ischemia (n = 6) or sham operation (n = 3). In ethanol-fixed frozen sections, the authors observed AIF immunofluorescence in normal hippocampal neurons, which showed predominantly cytoplasmic localization (Fig. 5B, panel A). Under high-power microscopic fields, AIF immunofluorescence showed a punctate pattern (insert in panel A), which is consistent with the notion that AIF is an intramitochondrial protein. In sections where the primary antibody had been preabsorbed with the AIF antigen, AIF immunofluorescence was not seen (Fig. 5B, panel B), thus confirming the specificity of the immunofluorescent signals resulting from the anti-AIF antibody. In sections obtained at 72 hours after ischemia, a co-localization of nuclear AIF immunofluorescence and TUNEL staining was found in the CA1 sector, in which many, but not all, TUNEL-positive neurons also showed nuclear AIF staining (Fig. 5B, panels C–E). In contrast, in CA3 neurons that do not degenerate in this model, AIF immunofluorescence remained in a cytoplasmic pattern after ischemia (Fig. 5B, panel F).
Effect of caspase inhibition on AIF translocation
Previous studies have shown that caspase inhibition can delay but is unable to prevent completely CA1 cell death after global ischemia, because a portion of the CA1 neurons rescued by caspase inhibitors for 3 days continued to degenerate when the duration of reperfusion was extended to 7 days (Chen et al., 1998;Cao et al., 2002a). Therefore, the authors speculated that AIF translocation might contribute to this continuing cell death in a caspase-independent manner. To test this hypothesis, additional rats were treated with z-DEVD. fmk (4.5 μg, intracerebroventricular (ICV) infusion) or vehicle 30 minutes before and 1 and 24 hours after 15 minutes of transient global ischemia (n = 6 per condition), and their hippocampi were processed for nuclear protein extraction followed by Western blot analysis of AIF at 3 or 7 days of reperfusion. At neither time point did infusion of z-DEVD. fmk have any effect on nuclear accumulation of AIF (Fig. 6), despite the fact that at the dose studied (3 × 4.5 μg), z-DEVD. fmk provided optimal protection in this ischemia model (Chen et al., 1998;Cao et al., 2002a). In the present study, the effect of caspase inhibition on AIF release was not determined at earlier time point (24 hours) after ischemia; thus the possibility that z-DEVD. fmk might delay AIF release could not be excluded. Nevertheless, the data presented here strongly suggest that the caspase inhibitor was incapable of preventing ischemia-induced AIF release.
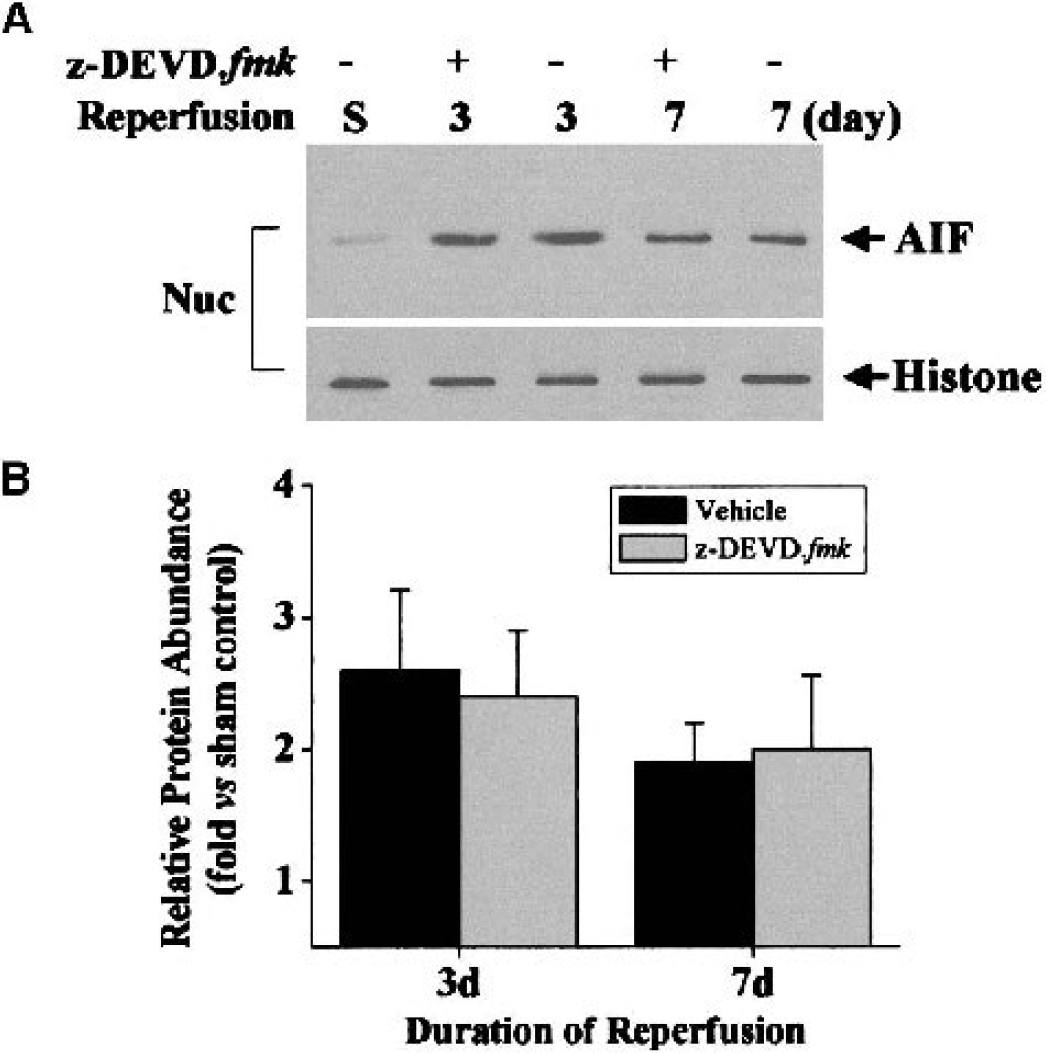
Inhibition of caspase activity does not prevent AIF translocation after ischemia.
Detection of induced AIF-like activity after ischemia
Nuclear translocation of AIF in CA1 neurons strongly suggests that AIF is activated in dying neurons after ischemia. To directly detect the AIF-like nuclease activity, the authors isolated nuclear protein from the hippocampal CA1 sector of control non-ischemic brains or brains at 48 hours after ischemia (8 rats per condition, 2 brains per sample) and performed the in vitro cell-free apoptosis assay using purified nuclei from neurons as the nuclease substrate (Cao et al., 2001;Luo et al., 2002). The assay was carried out in the presence of z-VAD. fmk (100 μM) to prevent any caspase-related nuclease activities. Nuclear extracts from ischemic brains, but not from the non-ischemic control brains, induced large-scale DNA fragmentation in genomic DNA of purified nuclei, as determined using pulse field gel electrophoresis (Fig. 7A). The induced DNA fragmentation mainly occurred at the size of 50 kbp, which was consistent with the size of DNA fragments induced by the purified AIF fusion proteins in isolated nuclei (Fig. 7B). Moreover, this ischemia-induced DNA-degrading activity was completely prevented by immunodepletion of AIF from the nuclear extracts before the assay, but was not affected by the addition of the ICAD fusion protein (2 μg/ml) that is capable of selectively inhibiting caspase-dependent DNA fragmentation (Chen et al., 2000;Cao et al., 2001).
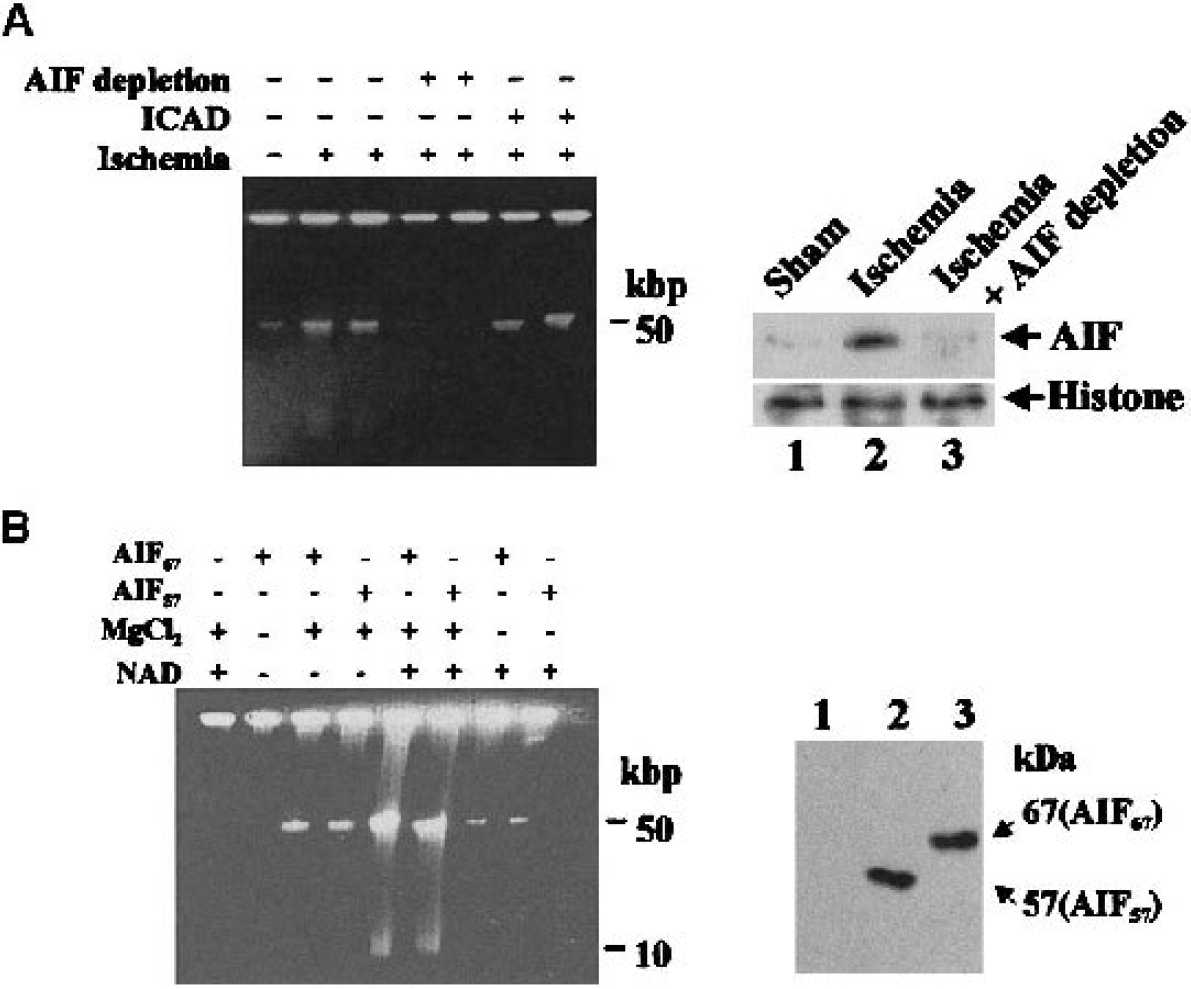
Induction of AIF-like nuclease activity in the hippocampus after ischemia.
Nuclear translocation of AIF after in vitro ischemia and inhibition by Bcl-xL
To determine whether the nuclear translocation of AIF is a unique phenomenon in global ischemia or represents a common mechanism for nuclear degradation in other ischemia-relevant models, the authors examined cellular distribution of AIF in the in vitro model of ischemia in primary neuronal cultures induced by oxygen-glucose deprivation (OGD) (Nagayama et al., 1999;Cao et al., 2001;Plesnila et al., 2001). In the present study, the authors subjected neurons to 60 minutes of OGD (which causes cell death in approximately 60% neurons at 24 hours). Under this injury paradigm, neurons showed marked cell body swelling during early phase of reperfusion (0–2 hours) but cell body shrinkage and destruction of neuronal processes during the later phases. It is likely that cell death in this model involves a mixture of necrosis (excitotoxic) and apoptosis, that is, the caspase inhibitor z-DEVD. fmk (100 μM) reduced cell death by 25% to 30% at 24 hours, whereas the NMDA receptor antagonist MK801 (1 μM) reduced cell death by 50% to 70% (data not shown). As determined at 2, 6, and 16 hours after OGD, Western blot analysis following subcellular fractionation revealed that AIF immunoreactivity was increased in the nuclear fractions at all time points after OGD (Fig. 8A). A corresponding decrease in mitochondrial AIF was also observed at 6 and 16 hours after OGD.
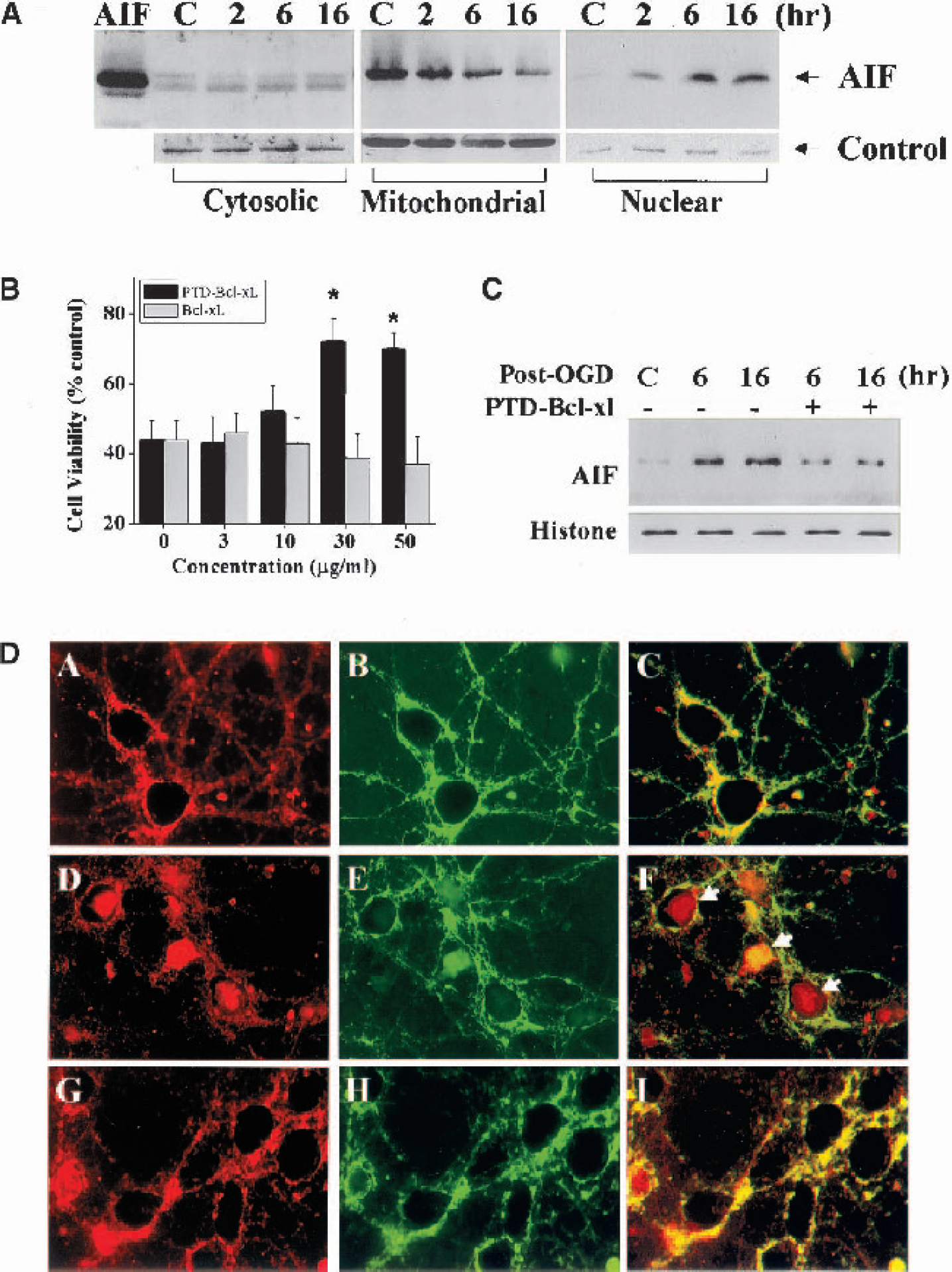
Translocation of AIF in neurons after oxygen-glucose deprivation and inhibition by Bcl-xL.
To determine whether the anti-apoptotic members of the Bcl-2 family can regulate OGD-induced release of AIF from mitochondria, the authors examined the effect of Bcl-xL on AIF translocation at 6 and 16 hours after 60 minutes of OGD. The Bcl-xL fusion protein was delivered into neurons (30 minutes before OGD) with the assistance of the 11-amino-acid HIV/TAT protein transduction domain (PTD), which was fused into the N-terminus of Bcl-xL (Cao et al., 2002b). The PTD-Bcl-xL fusion protein at the concentration of 30 to 50 μg/ml provided significant protection against OGD-induced cell death (Fig. 8B). Furthermore, Western blot and immunofluorescent-staining experiments demonstrated that PTD-Bcl-xL attenuated nuclear accumulation of AIF at both the 6- and 16-hour time points after OGD (Fig. 8C and Fig. 8D). In contrast, the addition of z-DEVD. fmk (30 to 100 μM) had no effect on AIF release (data not shown).
DISCUSSION
Neuronal cell death resulting from cerebral ischemia may involve the activation of a host of death-execution gene products (Lipton, 1999;Graham and Chen, 2001). Among the signaling cascades leading to the activation of execution molecules in ischemic neurons, the mitochondrial death pathway appears to play a central role (Fujimura et al., 1999;Sugawara et al., 1999). After ischemic injury, a number of damaging stimuli such as oxidative stress and the pro-apoptotic molecules Bid, Bax, and Bad may activate the mitochondrial death pathway by triggering the release of soluble intermembrane proteins, some of which are strong apoptogenic factors. Factors such as cytochrome c and Smac are known to activate terminal caspases, but others may cause cell death via caspase-independent mechanisms. In the present study, the authors demonstrate that AIF, a caspase-independent death execution molecule, is released from mitochondria in the hippocampal neurons after transient global ischemia and in cortical cultures following oxygen-glucose deprivation. Furthermore, AIF is found to translocate into nuclei and manifest a DNA-degrading activity characterized by the induction of large-scale DNA fragmentation in purified nuclei. The mitochondrial release and nuclear translocation of AIF after ischemia are not blocked by inhibiting caspase activities but, as shown in neuronal cultures, can be prevented by intracellular delivery of the mitochondria-associated anti-apoptotic protein Bcl-xL. Because the mitochondrial release of AIF occurs predominantly in the hippocampal CA1 neurons that are selectively vulnerable to ischemic injury, the authors suggest that AIF may contribute to the caspase-independent mechanism of neuronal cell death after transient cerebral ischemia and in neuronal cultures under ischemia-like conditions.
The role of AIF as a potent caspase-independent cell death executor is well established in a variety of cell types (Susin et al., 1999;Daugas et al., 2000). AIF is an ubiquitously expressed protein that has been detected in virtually every type of tissue or mammalian cell tested thus far (Daugas et al., 2000). The AIF gene is highly conserved across species; it has been identified in the human (Susin et al., 1999), mouse (Susin et al., 1999), and rat (present study). Sequence analysis revealed that the deduced amino acid sequence of AIF shares high homology (greater than 90% identity) among the human, mouse, and rat (Fig. 1). Moreover, the compositions of several functional sequences are nearly identical among the three species, including the N-terminal mitochondrial localization sequence, which is essential for AIF to be imported to the mitochondria, two nuclear localization sequences, which are required for AIF to translocate to the nucleus, and the C-terminal oxidoreductase domain, which contains the sequence for the cell-killing activity of AIF (Susin et al., 1999). The pattern of AIF protein expression in the brain during development (age-dependent increases; see Fig. 2) shows strong similarity to thoat of several mitochondrial proteins such as cytochrome c and cytochrome c oxidase (Chen et al., 2002) but is different from that of caspase-3, caspase-9, Apaf-1, and CAD (Cao et al., 2001;Yakovlev et al., 2001;Cao et al., 2002a). The pathologic significance of the marked increases in AIF protein expression in the adult brain compared with that in neonates is unclear. However, previous studies have shown that the caspase dependence of ischemic neuronal death markedly declines with brain maturation (Benedict et al., 2000), which is strongly correlated with the age-dependent decreases in brain expression of caspase-9, caspase-3, Apaf-1, and CAD. Accordingly, the authors suggest that it is possible that the involvement of AIF in ischemic neuronal death may be increased with brain maturation and therefore could be an important factor for cell death in the adult brain under pathologic conditions.
The data presented here demonstrate that AIF is released from mitochondria and redistributed to the nucleus in CA1 neurons after transient global ischemia (Fig. 4 and Fig. 5). Consistent with the nuclear translocation of AIF is the detection of AIF-like DNA-degrading activity in nuclear extracts prepared from the ischemic hippocampal CA1 sector (Fig. 7). Of note is the fact that the translocation and activation of AIF in ischemic neurons occur without the alterations in overall AIF gene expression at either mRNA or protein levels (Fig. 3 and Fig. 4). Furthermore, the hippocampal activation of AIF occurs with a temporal profile (24–72 hours after ischemia) closely correlated with the endogenous induction of large-scale DNA fragmentation, mainly the 50-kbp fragments, which is a well characterized hallmark of AIF-like activity (Susin et al., 1999), but preceding CA1 cell death and terminal DNA fragmentation (72 hours after ischemia). These results suggest that AIF, upon nuclear translocation after ischemia, may result in the degradation of genomic DNA and may contribute to a set of molecular events that ultimately lead to CA1 cell death.
The precise mechanism by which AIF executes cell death is not fully understood; however, it likely involves both nuclear-dependent and nuclear-independent processes. Like cytochrome c, AIF is confined exclusively to the mitochondria in healthy cells and gains its pro-apoptotic activity only when it is released into the nonmitochondrial compartments. Confirming this notion, the authors have observed that enforced expression of the extramitochondrial form (the mature form) of AIF lacking the mitochondrial-localization sequence, but not the full-length AIF, induced apoptosis in cultured mammalian cells (unpublished data). As determined in the microinjection experiments (Susin et al., 1999) or in the cell-free assays (present study), AIF can induce the condensation of nuclear chromatin and large-scale DNA fragmentation, which are believed to be the main cell-killing effects of AIF. The nuclear-degrading effect of AIF has been attributed to its direct interaction with genomic DNA through a recently defined DNA-binding sequence (Cande et al., 2002a;Cande et al., 2002b;Ye et al., 2002). In addition, extramitochondrial AIF can cause the dissipation of mitochondrial transmembrane potential, which may further exacerbate mitochondrial dysfunction and release of apoptogenic factors (Susin et al., 1999;Vieira et al., 2000). However, the deduced nuclear-independent effect of endogenous AIF remains to be confirmed in live cells.
In the present study, the authors have shown that the release and nuclear translocation of AIF after ischemic injury is independent of caspase activities. Brain infusion of the caspase inhibitor z-DEVD-fmk, which showed significant protection in this model, failed to prevent the release of AIF in CA1 neurons (Fig. 6). Similarly, OGD-induced AIF release in cultured neurons was not blocked by z-DEVD-fmk. These results are consistent with the recent observations in neuronal cultures where AIF release was induced by NMDA toxicity, oxidative stress, or DNA damage in a caspase-independent manner (Cregan et al., 2002;Yu et al., 2002;Zhang et al., 2002). Also proposed to mediate AIF release are a number of factors that are potent cytochrome c release inducers, such as the pro-apoptotic Bcl-2 family proteins Bid, Bax, and Bak, and reactive oxygen or nitrogen species (Cande et al., 2002a;Cande et al., 2002b). However, detailed biochemical analyses of these factors and their role in inducing AIF and cytochrome c release have produced various results thus far, depending upon the specific model systems tested. For instance, the truncated Bid, representing the active form of Bid, is a potent inducer of release of AIF and cytochrome c from isolated liver mitochondria (van Loo et al., 2002), but it is incapable of inducing AIF release in HeLa cells (Arnoult et al., 2002). Moreover, Bax, which appears to be incapable of inducing AIF release from isolated liver mitochondria (Arnoult et al., 2002), has been shown to be an important mediator of AIF release in neurons in response to DNA damage (Cregan et al., 2002). These contradicting results emphasize the importance of choosing appropriate cell phenotypes and injury stimuli for modeling the pathologic processes in relevance to the diseases to be studied. Therefore, future studies are warranted to further elucidate the precise mechanisms underlying AIF release in ischemic neurons in the brain.
The authors have tested the efficacy of Bcl-xL in inhibiting AIF release in cultured neurons receiving OGD challenges. The results demonstrate that Bcl-xL is a potent inhibitor of AIF release (Fig. 8). As demonstrated previously, Bcl-xL is capable of inhibiting cytochrome c release and caspase activation induced by a variety of apoptosis stimuli in neurons and in many other cell types (Frankowski et al., 1995;Gonzalez-Garcia et al., 1995;Kharbanda et al., 1997;Sastry and Rao, 2000;Shinoura et al., 2000). Accordingly, Bcl-xL potentially can prevent the activation of both caspase-dependent and AIF-dependent (but caspase-independent) cell death pathways through its protective actions on the mitochondria. Therefore, the results reinforce the notion that Bcl-xL may be a potentially important therapeutic target for ischemic brain injury and neuronal apoptosis.
Collectively, the data presented here suggest that AIF is a contributing factor in delayed cell death in the hippocampus after cerebral ischemia. Although the gene expression levels of AIF are not altered in the ischemic brain, the release and translocation of AIF may cause chromatin condensation and large-scale DNA fragmentation, and may ultimately contribute to the execution of cell death. The results support the notion that the loss of mitochondrial integrity may play a critical role in mediating ischemic neuronal death, perhaps by triggering a diversity of death pathways involving both caspase-dependent and caspase-independent mechanisms. Hence, it is likely that therapeutic interventions that target the mitochondrion or its upstream signaling pathways to prevent the release of apoptogenic factors may offer more favorable protective effects against ischemic injury than those that aim at an individual downstream cell death pathway such as caspase-3.
Footnotes
Acknowledgment
The authors thank Juan Xin for technical support, Carol Culver for editorial assistance, and Pat Strickler for secretarial support.