
Abstract
Although protective effects of heat shock protein 70 (HSP70) overproduction after ischemic injury have been shown both in vitro and in vivo in neurons, the mechanisms are not fully understood. The hypothesis of this study is that transgenic mice overexpressing HSP70 (HSP70 Tg) show reduced mitochondrial cytochrome c release into cytosol and diminished apoptotic cell death after permanent focal ischemia in comparison to wild-type (Wt) mice.
Permanent middle cerebral artery occlusion (pMCAO) was produced by intraluminal suture cannulation in HSP70 Tg and Wt mice. DNA fragmentation was evaluated with DNA gel electrophoresis and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) 24 h after pMCAO. Mitochondrial cytochrome c release into cytosol was assessed with Western blotting and immunohistochemistry 4 h after pMCAO. Cytochrome c levels in the cytosolic fraction were significantly reduced and immunoreactivity of cytochrome c in both cortex and striatum was significantly less in HSP70 Tg mice compared with Wt mice after 4-h pMCAO. DNA laddering, which was clearly observed in Wt mice, was markedly attenuated in HSP70 Tg mice 24 h after pMCAO. The number of TUNEL-positive cells was significantly reduced in HSP70 Tg mice compared with Wt mice. Results are consistent with an association between overexpression of HSP70 and reduction of cytochrome c release with subsequent DNA fragmentation. This may contribute to the HSP70-mediated neuroprotective effect observed after cerebral ischemia.
In response to cellular stress, such as cerebral ischemia, the 70-kd heat shock protein (HSP70) is induced in brain and has been established as a stress marker (Massa et al., 1996). Although outcome in neurons expressing HSP70 after various brain injuries is controversial, neuroprotective effects of HSP70 overproduction have been clearly demonstrated after ischemic injury both in vitro (Rordorf et al., 1991; Lowenstein et al., 1991; Papadopoulos et al., 1996; Kelly et al., 2001; Lee et al., 2001) and in vivo (Yenari et al., 1998; Rajdev et al., 2000; Hoehn et al., 2001). The mechanism of neuroprotection has been attributed to the function of HSP70 as a molecular chaperone, assisting in restoration of the structure and function of denatured proteins (Pelham, 1986; Beaucamp et al., 1998). However, further details of HSP70 neuroprotective mechanisms remain to be elucidated (Sharp et al., 2000).
Apoptosis, the programmed destruction of a cell, involves a complex process. Two distinct pathways, namely, the death-receptor pathway and the mitochondrial pathway, are well characterized in various mammalian cell models (Hengartner, 2000; Hsu et al., 1996; Li et al., 1997). In the mitochondrial pathway, mitochondrial cytochrome c release into cytosol triggers caspase cascade activation and subsequent DNA fragmentation after apoptotic stimuli (Liu et al., 1996; Li et al., 1997; Srinivasula et al., 1998). Similarly, the death-receptor pathway triggers the activation of caspases through the binding of ligand to death receptors, such as CD95 (Fas/Apo-1) or tumor necrosis factor receptor-1.
In cerebral ischemia, apoptosis contributes to the spatial and temporal progression of neuronal loss after focal ischemia (Li et al., 1995; Namura et al., 1998; Velier et al., 1999). Several groups have reported that release of mitochondrial cytochrome c into cytosol occurred and resulted in subsequent DNA fragmentation after focal and global ischemia (Fujimura et al., 1998; Sugawara et al., 1999).
In nonneuronal cell lines, recent studies have shown that HSP70 inhibited apoptosis (Beere et al., 2000; Saleh et al., 2000; Creagh et al., 2000; Klein and Brune, 2002). Overproduction of HSP70 suppressed the release of cytochrome c into cytosol after apoptotic stimuli (Creagh et al., 2000; Klein and Brune, 2002). Conversely, heat shock protein synthesis did not reduce apoptosis in Fasmediated apoptosis (Creagh and Cotter, 1999).
Little is known about the antiapoptotic effect of HSP70 in the central nervous system. We hypothesized that overexpression of rat inducible HSP70 transgene would reduce mitochondrial cytochrome c release into cytosol and subsequent DNA fragmentation after focal cerebral ischemia. To test this hypothesis, HSP70 Tg and Wt mice were subjected to permanent middle cerebral artery occlusion (pMCAO). DNA fragmentation was evaluated with DNA gel electrophoresis, and TUNEL staining was performed 24 h after pMCAO. Early mitochondrial cytochrome c release into cytosol was assessed with Western blotting and immunohistochemistry 4 h after pMCAO.
MATERIALS AND METHODS
Animals
This study was conducted in accordance with the animal care guidelines issued by the National Institutes of Health. All protocols were approved by the Animal Care and Use Committee at the Veterans Affairs Medical Center, San Francisco. Transgenic (Tg) mice overexpressing rat HSP70 and transgenicnegative wild-type (Wt) littermates bred on CB6F1 background were obtained from Dr. W. H. Dillmann (University of California, San Diego) and genotyped as previously described (Rajdev et al., 2000). HSP70 Tg mice were produced using a chimeric transgene that consisted of the rat-inducible HSP70 gene inserted into a pCAGGS vector under the control of human cytomegalovirus enhancer and chicken-actin promoter. Adult male mice (28 to 34 g) were used for this experiment. Mice were given free access to food and water before and after surgery. Anesthesia was induced with 2% isoflurane in a closed chamber and maintained with 1.5% isoflurane in 30% O2 and 70% N2O using a face mask. Rectal temperature was monitored and maintained at 37 ± 0.3°C with a thermal blanket throughout the surgical procedure.
Focal ischemia
Permanent focal ischemia was produced using the carotid endovascular suture insertion method (Rajdev et al., 2000). Cortical perfusion was monitored before and for 30 minutes after pMCAO with laser-Doppler flowmetry (Vasamedic, St. Paul, MN, U.S.A.) to confirm appropriate endovascular suture placement in all mice. After mice were anesthetized, a 1.9-mm-diameter laser-Doppler needle probe was placed directly on the skull (1 mm posterior and 5 mm lateral to bregma). Cortical perfusion values after pMCAO were expressed as percentage change relative to baseline. Mice were then placed in the supine position, and a midline skin incision was made. The left common carotid artery and internal carotid artery were surgically isolated from the adjacent vagus nerve. A 5–0 monofilament nylon suture (Dermalon, Davis & Geck, St. Louis, MO, U.S.A.) with heat-rounded tip was advanced approximately 10 mm into the internal carotid artery through the ligated stump of the transected external carotid artery until a faint resistance was felt. After reversal of anesthesia, neurologic deficit was evaluated 45 minutes after pMCAO using a four-point deficit scoring system. While restraining the mouse by holding its tail, motor activity was observed and graded. Neurologic deficit score was defined as follows: 0: normal motor function, 1: flexion of torso and contralateral limbs, 2: leaning or circling to the contralateral side, 3: no spontaneous movement of the contralateral side.
Infarct volume
Mice were deeply anesthetized and killed 24 h after MCAO. Brains were removed, immediately frozen in dry ice, and stored at −80°C until processing. Coronal brain sections (10 μm in thickness) were obtained with a cryostat and stained with cresyl violet. Sections were analyzed with an image scanner (Epson Perfection 1650; Epson America, Inc., Long Beach, CA, U.S.A.). NIH image (version 1.62) was used to measure the infarct area in each section, and infarct volume was calculated. To minimize the effects of brain edema, infarct volume was indirectly measured by subtracting the volume of intact cortex and striatum in the ipsilateral hemisphere from that of the contralateral hemisphere (Swanson et al., 1990).
DNA laddering
Genomic DNA gel electrophoresis was performed as previously described (Velier et al., 1999). Mice were killed 24 h after MCAO. Brains were divided into ipsilateral and contralateral hemispheres. Samples were suspended in lysis buffer (100 mmol/L NaCl, 10 mmol/L Tris, pH 8, 25 mmol/L ethylenediamine tetraacetic acid (EDTA), 0.5% sodium dodecyl sulfate, and 500 μg/mL proteinase K) overnight at 55°C, and then incubated with RNase (5 mg/mL) for 1 h at room temperature. Genomic DNA was extracted with the phenol-chloroform method. DNA sample of 100 mg was loaded and subjected to electrophoresis on a 1% agarose gel containing ethidium bromide (0.5 mg/mL). DNA laddering was visualized by ultraviolet transillumination in each sample.
TUNEL
TUNEL was performed according to the manufacturer's instructions (ApopTag; Serologicals Corp., Norcross, GA, U.S.A.). Frozen sections were fixed with 4% paraformaldehyde in 0.1 mol/L phosphate buffer saline (PBS), pH 7.4 for 5 minutes followed by quenching of endogenous peroxidase activity using 3% hydrogen peroxide in 0.1 mol/L PBS for 5 minutes. Sections were then incubated with equilibration buffer for 30 minutes followed by TdT enzyme for 1 h. Anti-digoxigenin peroxidase conjugate was applied to slides and incubated for 30 minutes. 3,3′-Diaminobenzidine-tetrachloride (DAB; Sigma Chemical Co, St. Louis, MO, U.S.A.) was applied for 6 minutes. Sections were then counterstained with 0.5% methyl green for 10 minutes.
Western blot analysis
Western blotting was performed with brain samples from mice 4 h after pMCAO. Brains were divided into ipsilateral and contralateral hemispheres. Samples were then gently homogenized with a tissue grind pestle (Kontes, Vineland, NJ, U.S.A.) in suspension buffer (20 mmol/L, HEPES-KOH, pH 7.5, 250 mol/L sucrose, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol and proteinase inhibitor cocktail tablets [Boehringer, Mannheim, Germany]). Cytosolic and mitochondrial proteins were isolated as previously described (Klein and Brune, 2002). The homogenates were centrifuged at 14,000g for 30 minutes and the supernatant was used for the cytosolic fraction. The pellets were further sonicated and used for the mitochondrial fraction. Protein concentration was measured by the Bradford method (Bio-Rad, Hercules, CA, U.S.A.). An equal amount of proteins (10 μg for the cytosolic fraction, 5 μg for the mitochondrial fraction) were loaded in each lane. Samples were separated on sodium dodecyl sulfate polyacrylamide gel electrophoresis and electrotransferred onto polyvinyldifluoride membranes (Stratagene, La Jolla, CA, U.S.A.). Membranes were probed with primary antibodies including mouse anti-HSP70 monoclonal (1:4,000; Stressgen, Victoria, BC, Canada), rabbit anti–cytochrome c polyclonal (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), rabbit anti–actin polyclonal (1:1,000, Santa Cruz Biotechnology), and mouse anti–cytochrome oxidase (COX) subunit IV monoclonal (0.2 μg/mL; Molecular Probe, Eugene, OR, U.S.A). After incubation with peroxidaseconjugated secondary antibody, signals were visualized with ECL plus (Amersham, Piscataway, NJ, U.S.A.) and exposed to x-ray films. Films were scanned and optical density was determined with NIH image (version 1.62, NIH). β-Actin and COX subunit IV were used as an internal control for cytosolic and mitochondrial fraction, respectively. Purified cytochrome c from rat heart (Sigma) was used as a positive control.
Immunohistochemistry
Mice were anesthetized and perfused with 0.9% saline followed by 10% formaldehyde in 75 mmol/L PBS 4 h after MCAO. Brains were removed and postfixed overnight. Brains were then transferred to 20% sucrose solution for 48 h. Coronal brain sections 50 μm in thickness were cut with a cryostat. Sections were incubated with 1% hydrogen peroxide for 30 minutes, and then incubated for 2 h in the blocking solution (100 mmol/L PBS, 0.1% Triton-X 100, 3% bovine serum albumin and 3% donkey serum) followed by overnight incubation with primary rabbit polyclonal antibody raised against cytochrome c (Santa Cruz Biotechnology) at 1:100 dilution. Biotinylated donkey anti–rabbit immunoglobulin (1:200; Amersham Life Science, Arlington Heights, IL, U.S.A.) was used as the secondary antibody for 2 h followed by 1 h in avidinbiotin complex solution (Vector Laboratories, Youngstown, OH, U.S.A.) in 100 mmol/L PBS. The sections were then stained with 0.05% diaminobenzidine-tetrachloride (DAB Fast; Sigma).
Statistical analysis
Student's t-test was used to determine the statistical significance of differences between HSP70 Tg and Wt in infarct volume, the number of TUNEL-positive cells, and densitometric analysis of cytochrome c. The Mann-Whitney U test was used to determine the statistical significance in neurologic deficit score. A P value of less than 0.05 was considered statistically significant. All values are expressed as mean ± SD. All statistical analyses were performed on a personal computer with Stat View (Version 5.0.1, SAS Institute Inc, Cary, NC, U.S.A.).
RESULTS
Neuroprotection in HSP70 Tg mice 24 hours after permanent middle cerebral artery occlusion
Infarct volume was significantly reduced in HSP70 Tg mice compared with Wt mice 24 h after pMCAO (P = 0.013). (Figs. 1A and 1B). Infarct volumes 24 h after pMCAO in HSP70 Tg (n = 7) and Wt mice (n = 8) were 32.22 ± 2.29 mm3 and 46.18 ± 2.38 mm3, respectively (Fig. 1D). Cortical perfusion was reduced to approximately 10% of baseline immediately after suture advancement and reduction persisted at approximately 20% of baseline value for 30 minutes during occlusion (Fig. 1C). Although the SD was greater in the HSP70 Tg group, there were no significant differences in cortical perfusion values between HSP70 Tg and Wt mice at any given time point, suggesting that appropriate suture insertion was accomplished in both groups. Neurologic deficit was reduced significantly in HSP70 Tg compared with Wt mice. Scores were 1.86 ± 0.69 and 2.63 ± 0.52, respectively (P = 0.037).
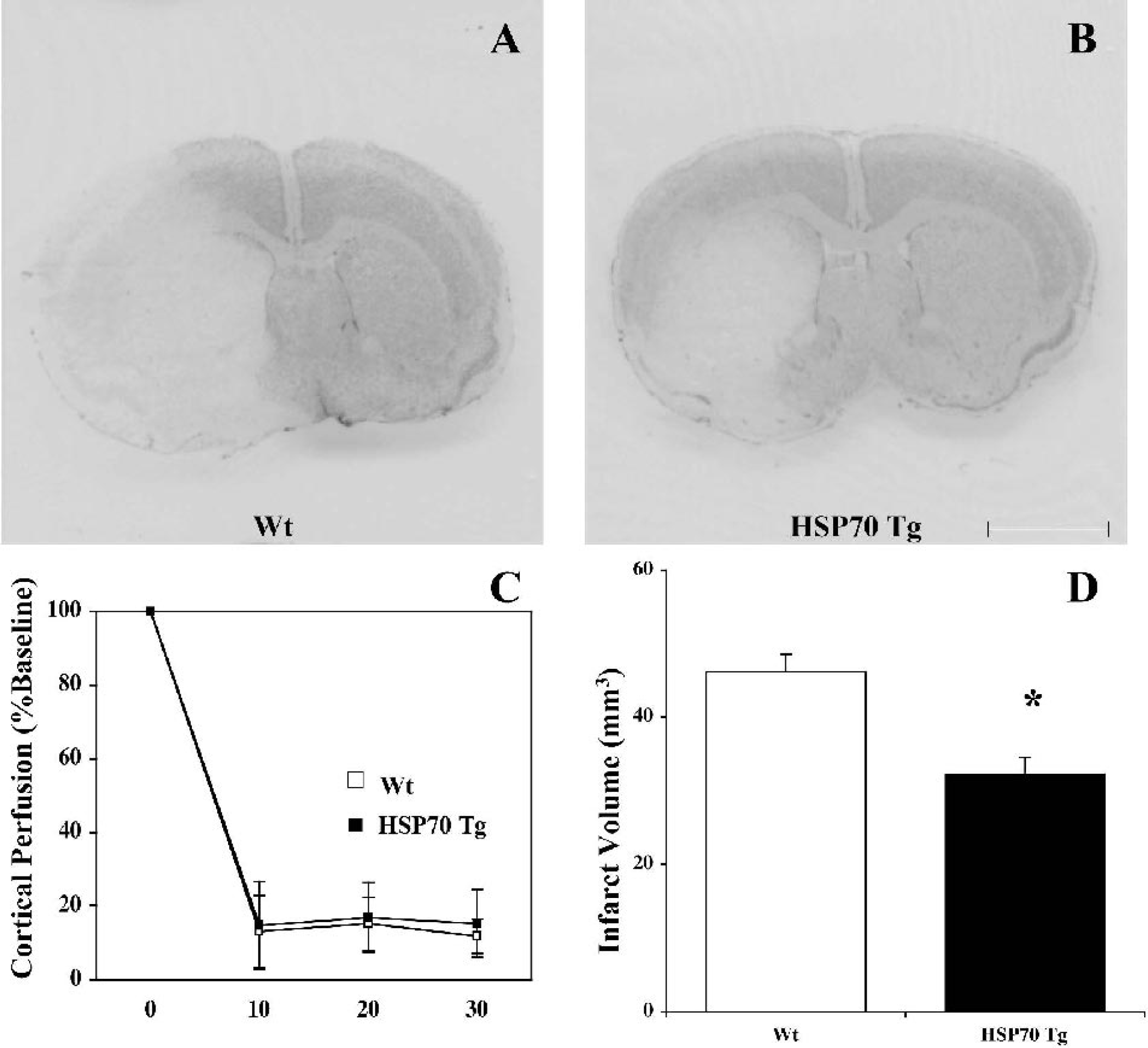
Reduced infarct size in the middle cerebral artery (MCA) territory of heat shock protein (HSP)70 transgenic (Tg) mice after permanent MCA occlusion (MCAO) without increased cortical perfusion. Representative photographs of coronal sections with cresyl violet staining 24 h after permanent MCAO in wild-type (Wt)
Attenuation of DNA laddering in HSP70 Tg mice
The presence of DNA fragments with multiple integers of approximately 180 base pairs in length was used as a marker for apoptotic cell death. In Wt mice, a laddering pattern was clearly observed in the ischemic hemisphere samples. DNA laddering was attenuated in the samples from HSP70 mice, suggesting that apoptotic cell death in HSP70 Tg mice was reduced in comparison with Wt mice (Fig. 2A). In nonischemic mice, the laddering pattern was not observed in either HSP70 Tg or Wt mice (data not shown).
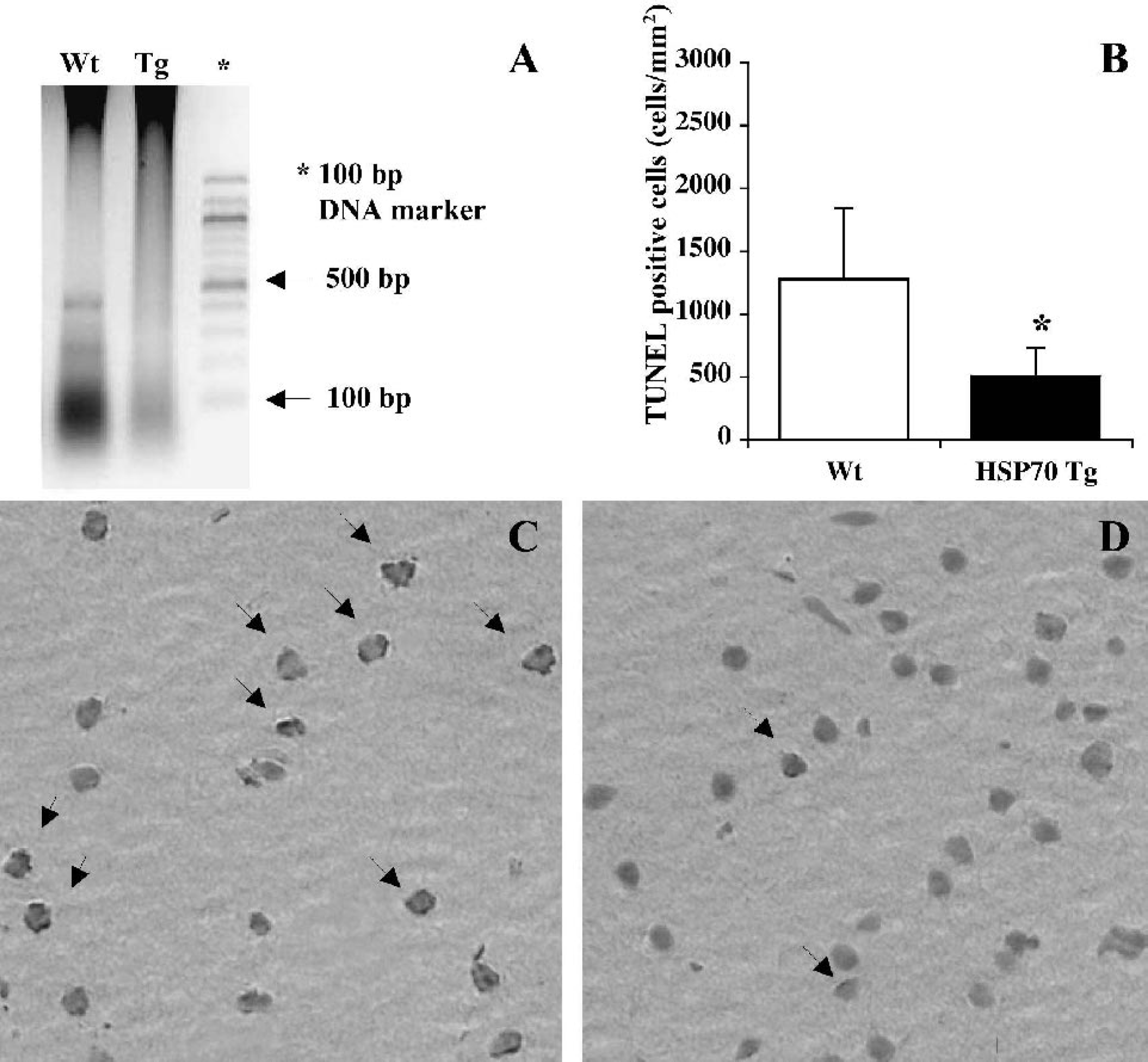
DNA laddering and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)-positive cells are significantly reduced in heat shock protein (HSP)70 transgenic (Tg) mice.
Reduction of TUNEL-positive cells in HSP70 Tg mice
TUNEL staining was used for analysis of spatial propagation of apoptotic cell death 24 h after pMCAO (Figs. 2B–D). For comparison, TUNEL-positive cells showing morphologically apoptotic features such as cell shrinkage, chromatin condensation in nuclei, and apoptotic bodies were counted (Figs. 2C and 2D, arrows). Cells exhibiting weakly stained, large and swollen nuclei were considered necrotic and were eliminated from counting. In the ischemic cortex, the number of TUNEL-positive cells was significantly reduced in HSP70 Tg (n = 5) compared with Wt mice (n = 6), as indicated by the cell counts of 507 ± 223 and 1,278 ± 564, respectively (P = 0.019) (Figs. 2B–D). In the lateral striatum, the number of TUNEL-positive cells in HSP70 Tg (n = 5) and Wt mice (n = 6) was 1,487 ± 773 and 1,922 ± 669, respectively (P > 0.05). Although there was a trend toward reduction of TUNEL-positive cells in lateral striatum of HSP70 Tg mice, it was not significant. The lack of statistical significance suggests that reduction of apoptosis may have occurred in HSP70 Tg mice due to the severity of ischemia and lack of collateral blood flow.
Overexpression of HSP70 in HSP70 Tg mice
To confirm HSP70 overexpression in HSP70 Tg mice, Western blots from nonischemic brain samples in HSP70 Tg and Wt mice (n = 3 each) were compared. Strong signals were detected in HSP70 Tg mice, documenting overexpression of HSP70. In Wt mice, a faint or absent HSP70 band was observed (Fig. 3).
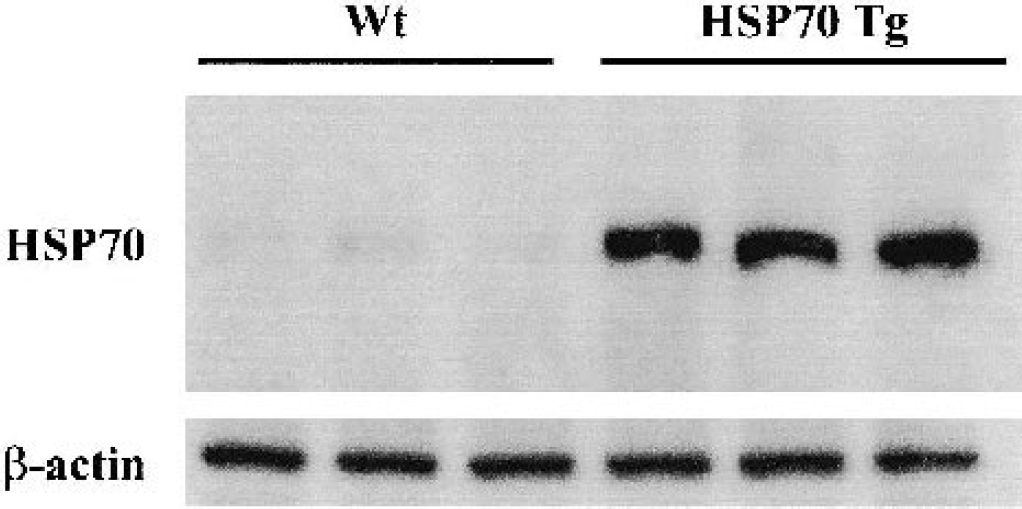
Strong constitutive heat shock protein (HSP)70 expression in the HSP70 transgenic (Tg) mice. Western blot analysis of HSP70 and β-actin from nonischemic mice brain samples. HSP70 is strongly expressed in HSP70 Tg mice, whereas little or no expression of HSP70 was seen in wild-type (Wt) mice.
Total amount of cytochrome c expression unaltered in HSP70 Tg mice
Western blots were performed to confirm the total amount of cytochrome c expression in nonischemic HSP70 Tg and Wt mice. Cytochrome c immunoreactivity was clearly observed as a single band at 15 kd (Fig. 4A). In the cytosolic fraction, little or no signal was detected in either HSP70 Tg or Wt mice, indicating successful separation of fractions. In the mitochondrial fraction, strong signal was observed in both HSP70 Tg and Wt mice (Fig. 4B). Comparing HSP70 Tg and Wt groups, there was no difference in the expression of cytochrome c in the mitochondrial fraction, suggesting that cytochrome c expression was not altered in nonischemic HSP70 Tg mice.
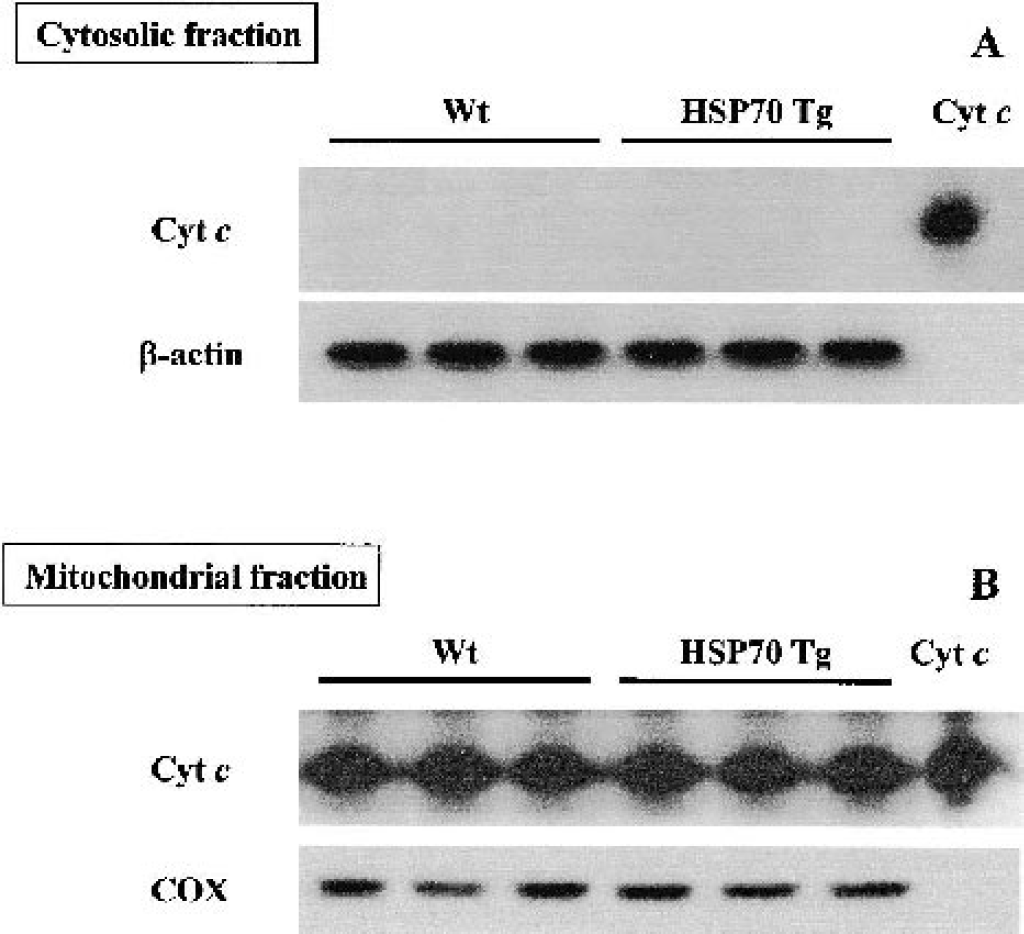
Lack of cytochrome c expression in the cytosolic fraction of both normal wild-type (Wt) and heat shock protein (HSP)70 transgenic (Tg) mice. Western blot analysis of cytochrome (Cyt) c of the cytosolic
Reduction of cytochrome c release in HSP70 Tg mice 4 hours after permanent middle cerebral artery occlusion
Mitochondrial cytochrome c release into cytosol was less in HSP70 Tg (n = 5) than in Wt mice (n = 6) 4 h after pMCAO (Fig. 5). In the cytosolic fraction, a strong signal was detected by Western blotting in the samples from Wt mice, whereas only a weak signal was seen in HSP70 Tg samples (Fig. 5A). Samples obtained from the nonischemic hemisphere showed few or no bands. The relative optical density of cytosolic cytochrome c in Wt mice (0.92 ± 0.06) (p < 0.01) was significantly greater than that in HSP70 Tg mice (0.71 ± 0.03). In the mitochondrial fraction, however, there is no apparent difference in cytochrome c expression between HSP70 Tg and Wt mice (Fig. 5C).
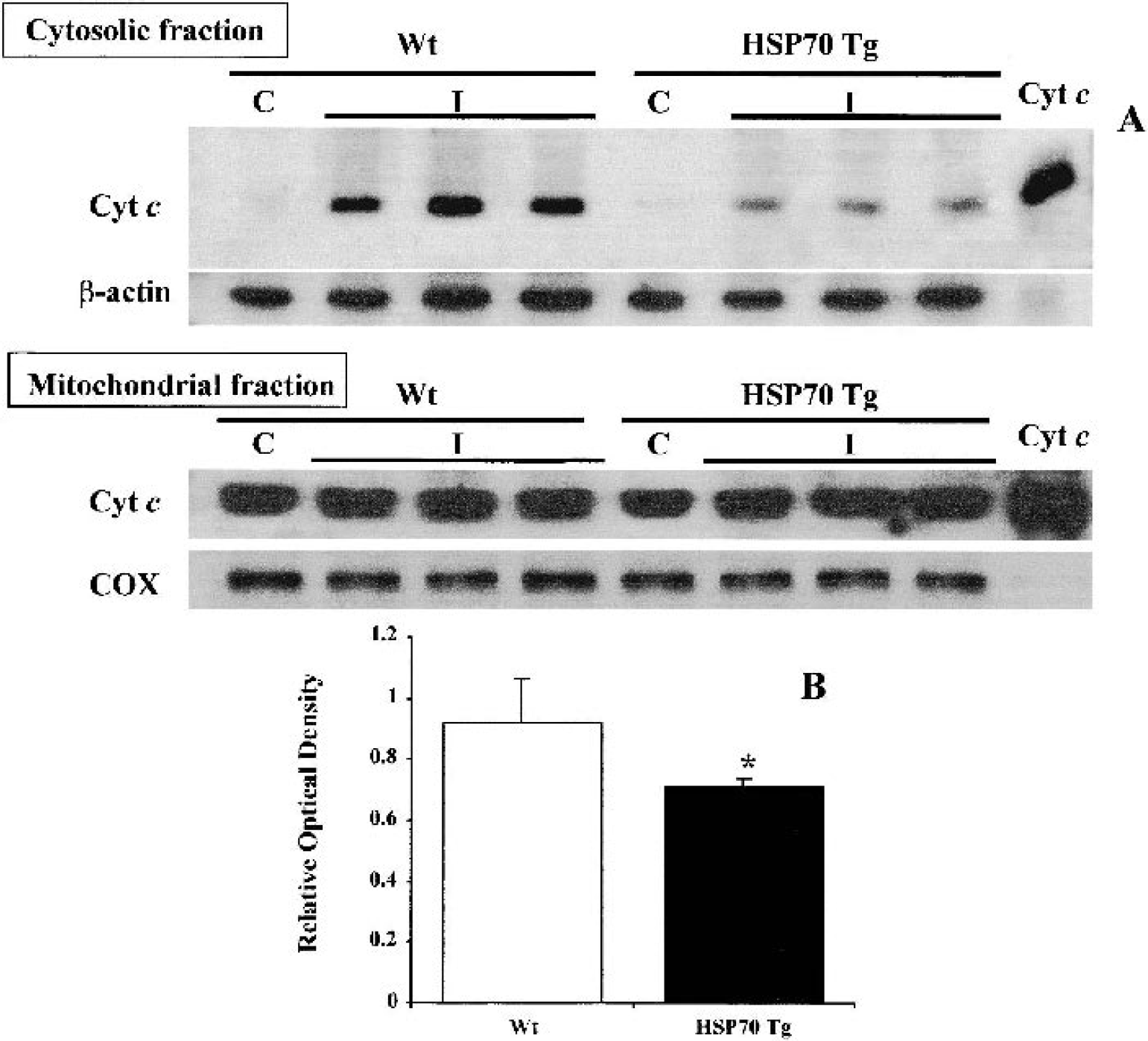
Significantly less cytochrome (Cyt) c is translocated into the cytosol of heat shock protein (HSP)70 transgenic (Tg) mice 4 h after permanent middle cerebral artery occlusion (pMCAO). Western blot analysis of cytochrome c of the cytosolic
Reduction of cytochrome c immunoreactivity in HSP70 Tg mice 4 hours after permanent middle cerebral artery occlusion
Cytochrome c immunohistochemistry staining was performed 4 h after pMCAO. Strong cytosolic cytochrome c immunoreactivity in neuronal cells was observed in the ischemic cortex and lateral striatum of Wt mice, whereas only weak immunostaining was observed in HSP70Tg mice (Figs. 6B, 6C, 6E, and 6F). Little or no immunoreactivity in either cortex or lateral striatum of nonischemic brain (Figs. 6A and 6D) was observed. This was possibly because of increased intensity of the tissue fixation, since antibody reacted with cytosolic but not with mitochondrial-bound protein (Fujimura et al., 1999, 2000; Noshita et al., 2001). Nissl and TUNEL staining performed on adjacent sections of HSP70 Tg mice 4 h after pMCAO showed that neither ischemic neuronal damage nor TUNEL-positive cells had developed (data not shown).
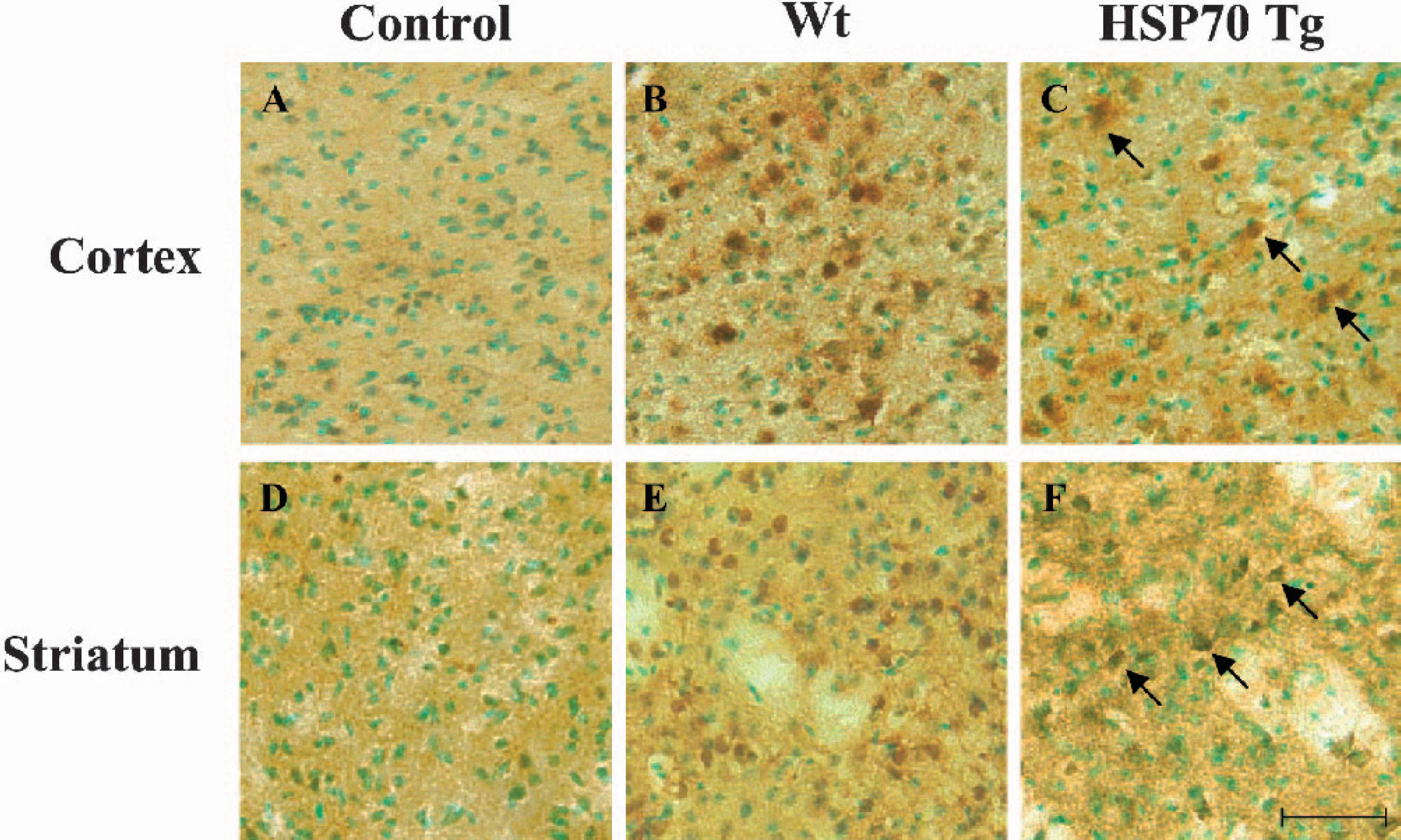
Reduced cytochrome c immunoreactivity in both cortex and striatum of heat shock protein (HSP)70 Tg mice 4 h after permanent middle cerebral artery occlusion (pMCAO). Cytochrome c immunohistochemistry with methyl green counterstaining was performed in wild-type (Wt) and HSP70 Tg mice. Representative photomicrographs of cytochrome c immunohistochemistry are shown from the cortex and striatum of nonischemic control Wt
DISCUSSION
These results show that overexpression of HSP70 reduces mitochondrial cytochrome c release into cytosol 4 h after pMCAO and subsequent DNA fragmentation 24 h after pMCAO. These results suggest that overproduction of HSP70 is associated with inhibition of apoptosis, possibly due to reduction of mitochondrial cytochrome c release. This reduced release was correlated with neuroprotection manifested as reduced infarct volume and neurologic deficits after permanent focal ischemia.
Apoptosis and signal transduction pathways
Apoptosis is defined by morphologically distinct criteria, such as membrane blebbing, chromatin condensation, and fragmentation of the nucleus (Kerr et al., 1972). Most of the morphologic and biochemical changes are caused by the activation of the caspase cascade (Hengartner, 2000). Caspases are categorized as either the executioner type, such as caspase-3, or initiators, such as caspase-8 or −9. Two major pathways for transmission of the apoptotic stimuli have been established (Hengartner, 2000; Yuan and Yanker, 2000). The death-receptor pathway is triggered by binding of the ligand to death receptors such as CD95 (Fas/Apo-1) or tumor necrosis factor receptor-1, forming a death-inducing signal complex (Kischkel et al., 1995; Hsu et al., 1996). This complex cleaves caspase-8 followed by cleavage of caspase-3. The mitochondrial pathway is initiated by the release of mitochondrial cytochrome c into cytosol. Although the mechanism of cytochrome c release across the mitochondrial outer membrane is still under debate, the BCL-2 family of proteins plays a crucial role in regulating this process (Kroemer and Reed, 2000). After being released into cytosol, cytochrome c binds to apoptotic protease, activating factor-1 (Apaf-1) in the presence of dATP, promoting the oligomerization of Apaf-1 itself. Concurrently or subsequently, this complex recruits procaspase-9, forming the complex called “apoptosome.” The apoptosome assembly allows procaspase-9 to be autoactivated, and this is followed by the recruitment and activation of procaspase-3 (Li et al., 1997; Srinivasula et al., 1998). Caspase-3 cleaves the inhibitor of caspaseactivated deoxyribonuclease and activates DNase, leading to DNA fragmentation (Hengartner, 2000).
Apoptosis and focal cerebral ischemia
Apoptosis and necrosis have been identified as a mechanism of ischemic neuronal cell death that occurs in the penumbra after focal vascular occlusion. The mechanism of induction of apoptosis by focal ischemia is complex, including activation of several genes after the onset of ischemia. Among them are the apoptosis-related genes that produce caspases (Sharp et al., 2000). In cerebral ischemia, apoptosis has been considered to be important for the propagation of ischemic injury, especially in the penumbra (Yuan and Yankner, 2000). After onset of focal cerebral ischemia, the temporal profile of DNA fragmentation has been described (Li et al., 1995). Since DNA fragmentation resulting from apoptosis is not always correlated with TUNEL-positive staining, colocalization of cleaved caspase-3 immunoactivity and TUNEL-positive cells has been used to describe apoptosis induced after focal ischemia (Namura et al., 1998). Induction of caspase-8 and caspase-9 has also been documented after focal ischemia (Velier et al., 1999; Noshita et al., 2001). A recent report indicates that activity of caspase-1 and caspase-8 was increased 30 minutes after pMCAO and that increase of caspase-3 activity was observed 1 h after pMCAO in the ischemic core, confirming that apoptotic cell death contributes to the acute neuronal cell death in the core as well as the penumbra after focal ischemia (Benchoua et al., 2001). Caspase inhibitors have been shown to significantly reduce infarct volume, which further strengthens the case for a role of apoptosis in ischemic injury (Hara et al., 1997)
It has been reported that cytochrome c in mitochondria is translocated to the cytosol after focal ischemia (Noshita et al., 2001), preceding the activation of caspase-9. The spatial and temporal pattern of cytochrome c translocation has also been repeatedly shown in both rats and mice (Fujimura et al., 1999, 2000; Noshita et al., 2001; Namura et al., 2001). Cytochrome c in the cytosolic fraction was observed by Western blotting at early time points, such as 1 or 2 h after MCAO and sustained up to 24 h after MCAO (Fujimura et al., 1999, 2000; Noshita et al., 2001). Consistent with these reports, our pilot studies in Wt mice indicated that cytochrome c release was observed in the cytosolic fraction both 2 h and 24 h after pMCAO. Other focal ischemia studies indicate that cytochrome c translocation was reduced in the copper/zinc-superoxide dismutase transgenic mice (Fujimura et al., 2000) and after antioxidant drug administration in normal ICR mice (Namura et al., 2001). Mild hypothermia reduced cytochrome c release temporarily after focal ischemia (Yenari et al., 2002). Furthermore, manganese–superoxide dismutase knockout mice showed increased cytochrome c release after focal ischemia, which correlated with greater ischemic injury observed in these transgenic mice compared with their Wt counterparts (Fujimura et al., 1999).
Mechanism of neuroprotective effect of HSP70
A neuroprotective effect of HSP70 overproduction has been shown with in vitro studies after a variety of ischemic insults (Rordorf et al., 1991; Lowenstein et al., 1991; Papadopoulos et al., 1996; Kelly et al., 2001; Lee et al., 2001). Both pre- and postischemic gene therapy inducing overproduction of HSP70 in vivo showed neuroprotective effect against focal ischemic injury (Yenari et al., 1998; Hoehn et al., 2001). In our previous study, rat-inducible HSP70 Tg mice provided a potent neuroprotective effect 6 and 24 h after permanent focal ischemia (Rajdev et al., 2000). However, two other studies reported no evidence of protection after pMCAO using different strains of HSP70 overexpressing transgenic mice (Plumier et al., 1997; Lee et al., 2001). These conflicting observations may be caused by differences in the level of HSP70 expression. As previously reported, HSP70 protein level was 5- to 10-fold higher in our HSP70 Tg mice compared with Wt mice (Rajdev et al., 2000), in contrast to a 2- to 2.5-fold difference between the Tg and Wt mice used by Lee et al. (2001). Thus, higher levels of HSP70 expression may be required to confer protection against focal ischemic injury. In contrast, increased infarct volume in HSP70 knockout mice compared with Wt littermates was also reported after transient MCAO, further suggesting a possible neuroprotective role for HSP70 against focal ischemic injury (Lee et al., 2001). Studies of rats pretreated with intraventricular administration of geldanamycin to induce HSP70 in brain also show significant reduction of infarction after focal ischemia (Lu et al., 2002).
Alternative mechanisms for the neuroprotective effects of HSP70 have been investigated. HSP70 is thought to function as a molecular chaperone, providing protection to neurons against various insults (Pelham, 1986; Beaucamp et al., 1998). Recent studies in nonneuronal cell lines indicate that HSP70 inhibited the apoptotic pathway by reducing caspase activation (Beere et al., 2000). Relevant to the mitochondrial pathway, studies with human acute lymphoblastic leukemia T-cell line, and ME-180 cervix carcinoma cells as well as WEHI-S fibrosarcoma cells, showed that overproduction of HSP70 inhibited caspase-3 activation (Mosser et al., 1997; Jaattela et al., 1998). Recently, two other studies in preheated HSP70 expressing nonneuronal cells found that HSP70 interacted with Apaf-1, interfering with the formation of cytochrome c/Apf-1/procaspase-9 complex. (Beere et al., 2000; Saleh et al., 2000). HSP70 overexpression in nonneuronal cells is also associated with suppression of cytochrome c release from mitochondria after exposure to apoptotic stimuli (Creagh et al., 2000; Mosser et al., 2000; Klein and Brune, 2002). However, a contradictory finding by Li et al. (2000) showed that cytochrome c release did occur in nonneuronal cells, such as rat cardiomyocytes, myogenic cells, primary human fibroblast, human lymphoblastic leukemia T-cell line, and tumor cell line, showing overexpression of HSP70 after heat treatment. As Klein and Brune mentioned in their discussion, it may turn out that higher HSP70 concentrations are needed for blocking apoptosome assembly, whereas lower levels of cellular HSP70 are associated with alternative pathways, such as cytochrome c release, to attenuate cell death. HSP70 was also found to prevent apoptosis by interfering with apoptosis-inducing factor, a caspase-independent apoptosis effector (Ravagnan et al., 2001). Conversely, the death-receptor pathway is not known to be affected by the overexpression of HSP70 (Liossis et al., 1997; Creagh and Cotter, 1999).
In conclusion, our results provide the first in vivo evidence that HSP70 overexpression is associated with early reduction of mitochondrial cytochrome c release and subsequent DNA fragmentation, possibly contributing to a significant neuroprotective effect observed after permanent focal ischemia. Since cytochrome c is highly regulated by the BCL-2 gene family (Yuan and Yankner, 2000; Shimizu et al., 1999), future studies of proapoptotic and antiapoptotic proteins in the BCL-2 family may further clarify the relation between HSP70 and cytochrome c release.
Footnotes
Acknowledgments:
The authors thank Angelo Zegna and Theresa Marsh for the technical assistance provided.