
Abstract
Release of cytochrome c from mitochondria to cytosol is a critical step in the mitochondrial-dependent signaling pathways of apoptosis. The authors have reported that manganese superoxide dismutase (Mn-SOD) attenuated cytochrome c release and apoptotic cell death after focal cerebral ischemia (FCI). To investigate downstream to the cytochrome c-dependent pathway, the authors examined caspase-9 activation after transient FCI by immunohistochemistry and Western blotting in both wild-type and Sod2 −/+ mice. Mice were subjected to 60 minutes of middle cerebral artery occlusion followed by 1, 2, 4, or 24 hours of reperfusion. Two hours after reperfusion, cytochrome c and caspase-9 were observed in the cytosol and significantly increased in Sod2 −/+ mutants compared with wild-type mice as shown by Western blotting. Immunofluorescent double labeling for cytochrome c and caspase-9 showed cytosolic cytochrome c 1 hour after transient FCI. Cleaved caspase-9 first appeared in the cytosol at 2 hours and colocalized with cytochrome c. Terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick and labeling (TUNEL) showed significant increase of positive cells in Sod2 −/+ mice compared with the wild-type in the cortex, but not in the caudate putamen. The current study revealed Mn-SOD might affect cytochrome c translocation and downstream caspase activation in the mitochondrial-dependent cell death pathway after transient FCI.
Keywords
Caspases, a family of cysteine aspartate-specific proteases have been identified as critical factors in apoptosis. Two major distinct apoptotic pathways may be mediated by caspases. In one pathway, caspase-8 is cleaved after forming the death-inducing signaling complexes of CD95 (Kischkel et al., 1995; Boldin et al., 1996; Muzio et al., 1996; Medema et al., 1997) or the tumor necrosis factor receptor-1 (Hsu et al., 1996). Thereafter, activated caspase-8 cleaves downstream caspases such as caspase-3 or caspase-7 (Boldin et al., 1996; Fernandes-Alnemri et al., 1996; Muzio et al., 1996; Cryns and Yuan, 1998). Another pathway mediated by caspases is known as the cytochrome c–dependent mitochondrial pathway of apoptosis. A number of studies have shown the critical role of cytochrome c in apoptosis in vitro (Liu et al., 1996; Kluck et al., 1997; Yang et al., 1997) and in vivo (Fujimura et al., 1998, 1999, 2000; Sugawara et al., 1999).
Mitochondria are involved in apoptosis by releasing cytochrome c to the cytoplasm where it activates caspases by interacting with cytosolic factors including Apaf-1, a protein homologous to Caenorhabditis elegans CED-4 (Zou et al., 1997; Yoshida et al., 1998). Binding with cytochrome c and deoxy-ATP (dATP), Apaf-1 combines with procaspase-9 in its caspase-recruit domain to cause cleavage of caspase-9 (Li et al., 1997; Zou et al., 1997, 1999; Hakem et al., 1998; Kuida et al., 1998; Kuida, 2000). Caspase-9 is the initial caspase in the cytochrome c–dependent mitochondrial pathway of apoptosis (Zou et al., 1997, 1999; Hakem et al., 1998; Kuida et al., 1998; Kuida, 2000) and becomes activated after cleavage. Activated caspase-9 further cleaves downstream caspases, such as caspase-2, 3, 6, 7, 8, and 10 (Slee et al., 1999). Procaspase-9 resides in the mitocondrial membrane space and is released to the cytosol after opening of the mitochondrial transition pore (MTP) in vitro (Susin et al., 1999). Mitochondrial release of caspase-9 to the cytosol has been observed in neurons after global ischemia (Krajewski et al., 1999). Once in the cytosol, procaspase-9 is cleaved to the active form by a complex containing Apaf-1, dATP, and cytochrome c; thereafter, activated caspase-9 translocates to the nucleus (Krajewski et al., 1999).
Upstream of the cytochrome c–dependent mitochondrial pathway of apoptosis, Bcl-2, a mitochondrial outer membrane protein, inhibits cytochrome c translocation, thereby blocking caspase activation and apoptosis (Kluck et al., 1997; Yang et al., 1997). Overexpression of Bcl-2 prevents superoxide production and attenuates cytochrome c release and apoptosis (Cai and Jones, 1998), suggesting that its antioxidant function contributes to the inhibition of apoptosis.
Antioxidant enzymes are one of the major mechanisms by which cells counteract the deleterious effects of reactive oxygen species (ROS) after cerebral ischemia and reperfusion. Evidence exists that superoxide dismutase (SOD) plays a protective role after focal cerebral ischemia (FCI) (Kinouchi et al., 1991; Chan et al., 1996; Kondo et al., 1997; Murakami et al., 1998; Fujimura et al., 1999, 2000) and global ischemia (Murakami et al., 1997; Chan et al., 1998). Deficiency of mitochondrial manganese superoxide dismutase (Mn-SOD), which prevents mitochondrial ROS production (Murakami et al., 1998), results in a marked increase in the early translocation of mitochondrial cytochrome c and subsequent DNA fragmentation after permanent FCI (Fujimura et al., 1999. This observation suggests the regulatory role of mitochondrial antioxidants in the cytochrome c-dependent apoptotic pathway. However, the effects of ROS on downstream caspase activity remain unclear. In the current study, the authors investigated cytochrome c release and the expression of activated caspase-9 after transient FCI in Sod2 −/+ mutant mice and their wild-type littermates. The authors hypothesized that decreased expression of Mn-SOD would be associated with greater translocation of cytochrome c followed by caspase-9 activation.
MATERIALS AND METHODS
Focal cerebral ischemia
Because homozygous knockouts (Sod2 −/−) showed neonatal lethality because of dilated cardiomyopathy (Li et al., 1995b), only heterozygous knockouts (Sod2 −/+) were used. The Sod2 −/+ mice with a CD1/SV129 background were backcrossed with CD1 mice for more than eight generations. Mn-SOD activity in these mutants is 50% of the activity seen in the brain and other organs of wild-type adult mice (Li et al., 1995b; Chan et al., 1996). These mutants and their wild-type littermates with a genetic background identical to that of the Sod2 −/+ mice (3-month-old males; 35 to 40 g) then were subjected to transient FCI by intraluminal middle cerebral artery (MCA) blockade with a nylon suture as described previously (Yang et al., 1994). There were no differences in the anatomy of the cerebral vasculature and cerebral blood flow after ischemia between the Sod2 −/+ mice and wild-type littermates (Murakami et al., 1998).
The mice were anesthetized with 1.5% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. The rectal temperature was controlled at 37°C with a homeothermic blanket. Cannulation of the femoral artery allowed the monitoring of blood pressure and arterial blood gases. Samples for analysis were taken immediately after cannulation, 10 minutes after occlusion, and 10 minutes after reperfusion. Blood gases were analyzed by a pH/Blood Gas Analyzer (Chiron Diagnostics, Essex, U.K.). After a midline skin incision, the left external carotid artery was exposed and its branches were electrocoagulated. An 11-mm 5–0 surgical monofilament nylon suture, blunted at the end, was introduced into the left internal carotid artery through the external carotid artery stump. After 60 minutes of MCA occlusion, blood flow was restored by the withdrawal of the nylon suture.
Immunohistochemistry
Anesthetized animals were perfused with 10 U/mL heparin and subsequently with 4% formaldehyde in 0.1 mol/L phosphate-buffered saline (PBS, pH 7.4) 1, 2, 4, and 24 hours (n = 4 each) after transient FCI as previously described (Fujimura et al., 1999). The brains were removed, postfixed for 12 hours, sectioned at 50 μm on a vibratome, and processed for immunohistochemistry. Sections were incubated with blocking solution and reacted with rabbit anti-cytochrome c polyclonal antibody (sc-7159; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) and rabbit anti-caspase-9 polyclonal antibody (sc-8355; Santa Cruz Biotechnology), which recognizes only the p35 subunit of caspase-9, both at a dilution of 1:100.
Immunohistochemistry was performed using the avidin-biotin technique and then the nuclei were counterstained with methyl green solution for 10 minutes. As a negative control, alternative sections were incubated without primary antibodies. For histologic assessment, alternate slices from each brain section also were stained with cresyl violet.
Immunofluorescent double labeling
To clarify the spatial relation between expression in cytosolic cytochrome c and caspase-9, the authors performed double immunofluorescent staining for cytochrome c and caspase-9. To evaluate the subpopulation of cytosolic cytochrome c immunoreactive cells, double staining for cytochrome c and microtube-associated proteins (MAPs), a neuronal specific marker, also was performed. The fixed sections were immunostained with the cytochrome c antibody, the caspase-9 antibody, and rabbit MAPs antiserum (M7273; Sigma, St. Louis, MO, U.S.A.) at a dilution of 1:100. Subsequently, sections were incubated with the secondary antibodies, fluorescein isothiocyanate (FITC)-conjugated Fab fragment goat anti-rabbit IgG polyclonal antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, U.S.A.) or Texas Red conjugated goat anti-rabbit IgG antibody (Vector Laboratories, Burlingame, CA, U.S.A.), at a dilution of 1:100. Sections were mounted on glass slides (Superfrost, Fisher Scientific, Pittsburgh, PA, U.S.A.) and covered with VECTASHIELD Mounting Medium with DAPI (Vector Laboratories). Fluorescence of fluorescein was observed at excitation of 495 nm and emission of >515 nm, and fluorescence of Texas Red was observed at excitation of 510 nm and emission of >580 nm. Fluorescence of DAPI was also observed at excitation of 360 nm and emission of >460 nm.
Western blot analysis
Protein extraction of both the mitochondrial and cytosolic fractions was performed as described (Fujimura et al., 1998). Approximately 50 mg of fresh brain tissue was obtained from the entire MCA territory on the nonischemic and ischemic sides after 1, 2, 4, and 24 hours of reperfusion (n = 4 to 5 each). Tissue was cut into pieces and gently homogenized by douncing 30 times in a glass tissue grinder (Wheaton, Millville, NJ, U.S.A.) in 7 volumes of cold suspension buffer (20 mmol/L HEPES-KOH [pH 7.5], 250 mmol/L sucrose, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA, 0.7% Protease and Phosphatase Inhibitor Cocktails [Sigma]). The homogenates were centrifuged at 750 g at 4°C and then at 8000 g for 20 minutes at 4°C. The 8000 g pellets were used to obtain the mitochondrial fraction. The supernatant was further centrifuged at 100000 g for 60 minutes at 4°C to remove ribosomes of endoplasmic reticulum (ER). Protein concentrations were determined by the Bradford method (Bio-Rad, Hercules, CA, U.S.A.), and approximately 5.7 μg protein from the cytosolic fraction and 1.9 μg from the mitochondrial fraction were loaded per lane. Primary antibodies were either a 1:1000 dilution of rabbit anti-cytochrome c polyclonal (Santa Cruz Biotechnology), 1:1000 dilution of rabbit anti-caspase-9 polyclonal (Santa Cruz Biotechnology), 1:1000 dilution of rabbit anti-Apaf-1 polyclonal (AB16941; Chemicon International, Temecula, CA, U.S.A.), 1 μg/mL of rabbit anti-caspase-9 precursor polyclonal (AF830; R&D Systems, Minneapolis, MN, U.S.A.), or 1 μg/mL of mouse anti-cytochrome oxidase (COX) subunit IV monoclonal (A-6431; Molecular Probes, Eugene, OR, U.S.A.). Western blots were performed with horseradish peroxidase-conjugated anti-rabbit or anti-mouse immunoglobulin G using enhanced chemiluminescence Western blotting detection reagents (Amersham International, Buckinghamshire, England). A densitometric analysis of the cytochrome c and caspase-9 expression was made on the cytosolic fraction of the ischemic brain from both wild-type and Sod2 −/+ mice. The film was scanned with a GS-700 imaging densitometer (Bio-Rad) and the results were quantified using Multi-Analyst software (Bio-Rad). Western blot analysis of β-actin also was performed.
In situ labeling of DNA fragmentation
To confirm the apoptotic cell death, terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL) using both wild-type mice and Sod2 −/+ mutants 24 hours after reperfusion was performed. Sections fixed by formaldehyde were incubated with NeuroPore (Trevigen, Gaithersburg, MD, U.S.A.) for 30 minutes. Slides were placed in 1 × terminal deoxynucleotidyl transferase (TdT) buffer (Life Technologies, Gaithersburg, MD, U.S.A.) for 30 minutes, followed by reaction with TdT enzyme (Life Technologies) and biotinylated 16-dUTP (Boehringer Mannheim, Indianapolis, IN, U.S.A.) at 37°C for 90 minutes. Sections then were washed twice in saline-sodium citrate (SSC, 150 mmol/L sodium chloride, 15 mmol/L sodium citrate, pH 7.4) for 15 minutes, followed by washing in PBS twice for 15 minutes. Avidin-biotin horseradish peroxidase solution (ABC kit, Vector Laboratories) was applied to the sections for 30 minutes, then washed for 15 minutes with 0.175 mol/L sodium acetate. Staining was visualized using 0.025% diaminobenzidine and 0.075% H2O2 in PBS with 0.4 mg/mL nickel sulfate. Sections were rinsed with water and then stained with methyl green for 10 minutes.
Statistical analysis
Optical density (OD) was used as the unit to analyze the amount of the cytochrome c and caspase-9. The number of the TUNEL-positive cells was counted as previously described (Fujimura et al., 1998). Data are expressed as mean ± SD. Comparisons between two groups were achieved using the Student's t-test. P < 0.05 was considered statistically significant.
RESULTS
Physiologic data and cerebral infarction
Physiologic parameters showed no significant differences in mean arterial blood pressure (MABP) and arterial blood gas analysis between each group. The preischemic physiologic values for wild-type and Sod2 −/+ mice were as follows: wild-type: MABP, 97 ± 4.5 mm Hg; Pao2 130 ± 11 mm Hg; Paco2, 32 ± 3.5 mm Hg; pH, 7.3 ± 0.07; and Sod2 −/+: MABP, 93 ± 12 mm Hg; Pao2 120 ± 18 mm Hg; Paco2, 29 ± 2.5 mm Hg; pH, 7.3 ± 0.09 (all values are mean ± SD, n = 4). There was no deviation from these values over the period of assessment. An ischemic lesion of the core of the caudate putamen was visible as a pale, slightly stained area in the ischemic hemisphere as early as 1 hour after reperfusion and extended to the entire MCA territory at 4 hours by cresyl violet staining (data not shown). The time-dependent increase of infarction in mouse brain using the intraluminal suture blockade was consistent with previous reports that used the same focal stroke model in mice (Murakami et al., 1998).
Cytosolic expression of cytochrome c and translocation of cleaved caspase-9 from cytosol to nuclei were detected by immunohistochemistry in ischemic brain after transient FCI
Cytochrome c protein expression before and after transient FCI was analyzed by immunohistochemistry in both the wild-type (Fig. 1A to 1I and 1K) and Sod2 −/+ mice (Fig. 1J and 1L).
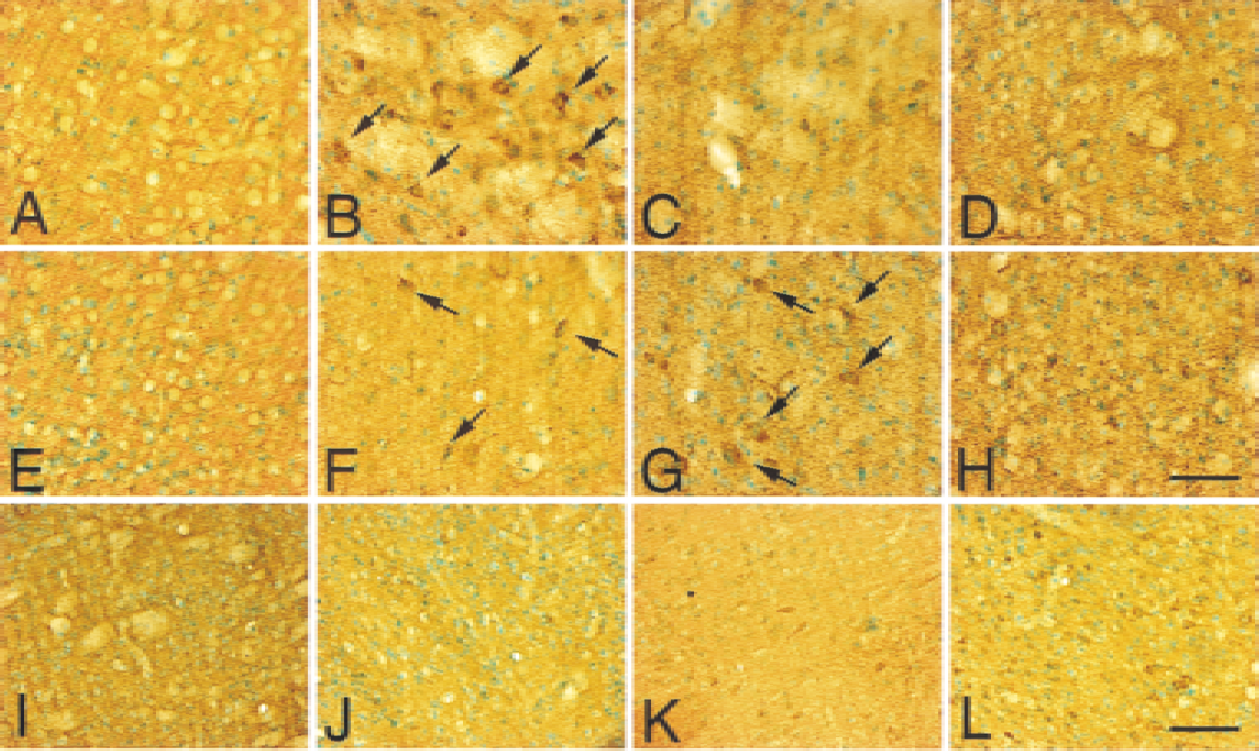
Cytochrome c immunostaining with methyl green counterstaining in coronal cortical brain sections from wild-type (
In the caudate putamen of wild-type mice, homogeneous cytoplasmic immunoreactivity of cytochrome c was visible 1 hour after transient FCI (Fig. 1B, arrows), whereas it became faint at 2 hours (Fig. 1I) and 4 hours (Fig. 1C). Although the cytoplasmic immunoreactivity was faint in the caudate putamen at 24 hours, the immunoreactivity also was observed in the intercellular space (Fig. 1D). In the cortex of wild-type mice, the cytoplasmic immunoreactivity was slightly visible 1 hour after reperfusion (Fig. 1F, arrows). By 4 hours after reperfusion, the cytoplasmic immunoreactivity became prominent in the cortex (Fig. 1G, arrows). After 24 hours of reperfusion, the immunoreactivity became faint in the cortex, however, the extracellular stain was also observed (Fig. 1H). No immunoreactivity was observed in the nonischemic control in either the caudate putamen (Fig. 1A) or the cortex (Fig. 1E). In Sod2 −/+ mutants, the number of cytochrome c–positive cells in the cortex was significantly increased (Fig. 1L) as compared with wild-type mice (Fig. 1K) 2 hours after FCI. In the caudate putamen, no obvious differences were seen between the wild-type (Fig. 1I) and Sod2 −/+ mutants (Fig. 1J) at 2 hours.
In the caudate putamen of wild-type mice, the immunoreactivity of cleaved caspase-9 was barely detected 1 hour after reperfusion and in nonischemic control animals (Fig. 2B and 2A, respectively). The immunoreactivity of caspase-9 was detected in cytosolic compartment 2 hours (Fig. 2I) and 4 hours (Fig. 2C, arrows) after reperfusion, whereas it became unclear at 24 hours (Fig. 2D). In the cortex of wild-type mice, the immunoreactivity of caspase-9 was visible 2 hours (Fig. 2K) and 4 hours (Fig. 2G, arrows) after reperfusion, whereas the immunoreactivity was barely detected at 1 hour (Fig. 2F) and in nonischemic controls (Fig. 2E). Twenty-four hours after reperfusion, the immunoreactivity of cleaved caspase-9 accumulated inside and around the nuclei in the cortex (Fig. 2H, arrowheads). The number of caspase-9 immunopositive cells and the intensity of the immunoreactivity were significantly increased in Sod2 −/+ mutants compared with the wild-type littermates 2 hours after FCI, both in the caudate putamen (Fig. 2I, wild-type; Fig. 2J, Sod2 −/+) and the cortex (Fig. 2K, wild-type; Fig. 2L, Sod2 −/+). No immunoreactivity was present without primary antibody (data not shown). To analyze the spatial relation between cytosolic cytochrome c expression and cleaved caspase-9, immunofluorescent double staining for cytochrome c and cleaved caspase-9 was performed on wild-type mice (Fig. 3). One hour after transient FCI, a small number of cytochrome c–positive cells were observed, which did not colocalize for cleaved caspase-9 (Fig. 3A to 3C). Two hours after FCI, all cytochrome c–immunopositive cells colocalized with cleaved caspase-9 (Fig. 3D to 3F). At 24 hours reperfusion, cleaved caspase-9 accumulated inside and around nuclei (Fig. 3G to 3I), whereas cytochrome c stain was unclear. To determine the cell population of immunoreactive for cytochrome c, fluorescent double-labeling for cytochrome c and MAPs, as a neuronal marker, was performed (Fig. 3J to 3L) 4 hours after transient FCI. Cytochrome c-immunopositive cells (Fig. 3K) colocalized with MAPs-positive cells (Fig. 3L), indicating the cell population of cytochrome c–positive cells to be mainly neuronal.
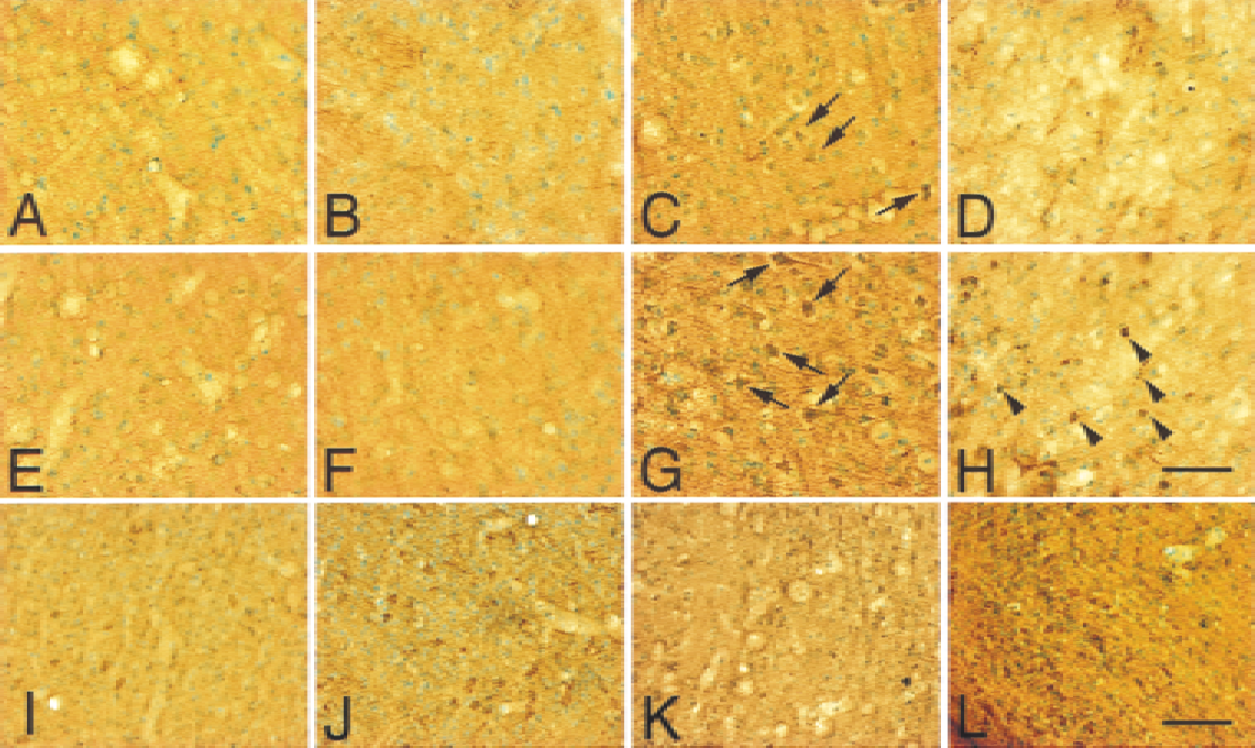
Cleaved caspase-9 (p35) immunostaining with methyl green counterstaining in coronal cortical brain sections from wild-type (
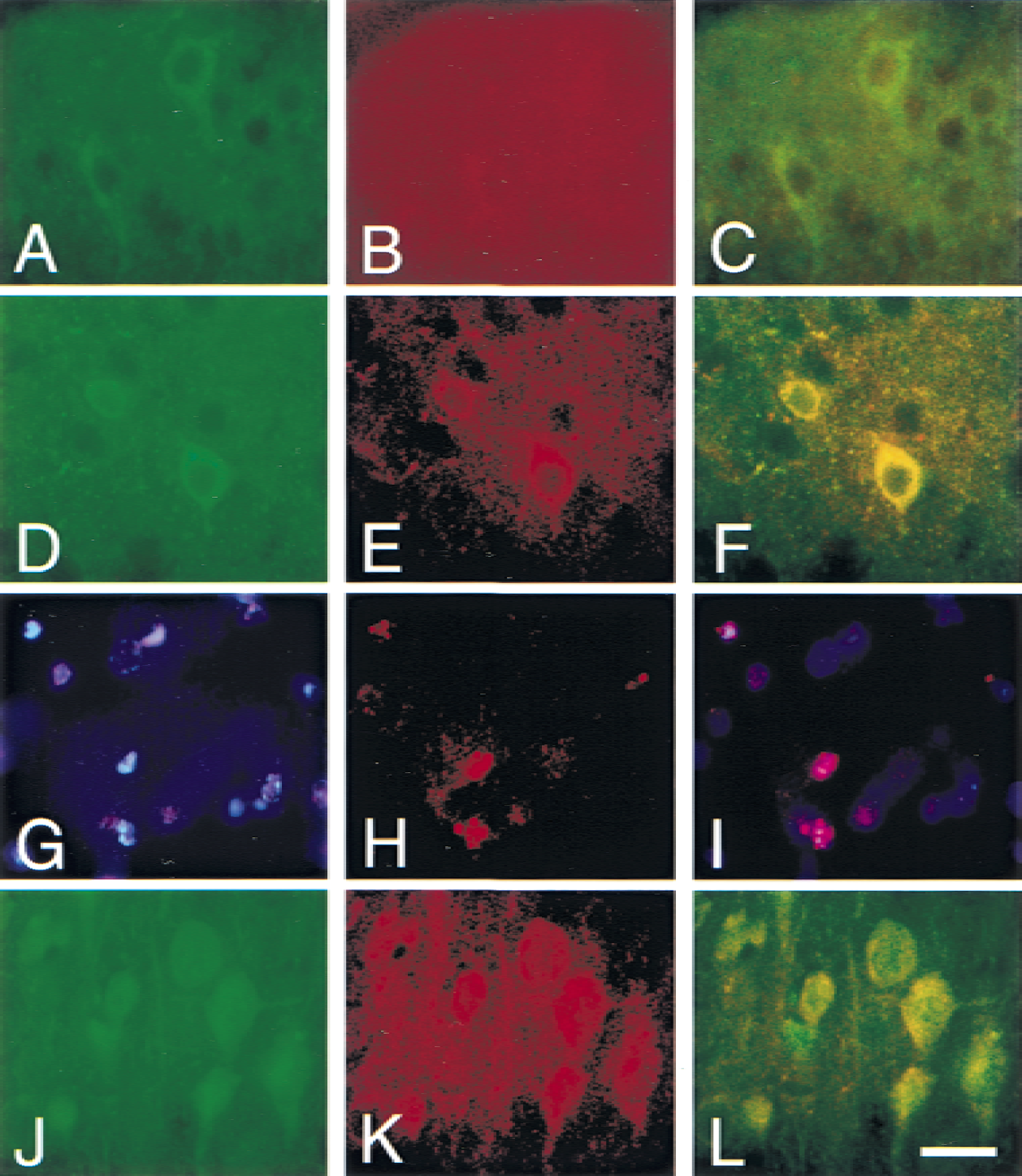
Representative photomicrographs show immunofluorescent staining for cytochrome c, caspase-9, and MAPs in wild-type mice after transient FCI. Double immunofluorescent staining with cytochrome c and cleaved caspase-9 one hour
Western blot analysis demonstrating the early release of mitochondrial cytochrome c to the cytosol and activation of caspase-9 after transient FCI
As shown in Fig. 4A, cytochrome c immunoreactivity was evident as a 15 kDa band (Fujimura et al., 1999) in the cytosolic fraction from the ischemic brain of wild-type mice as early as 1 hour after reperfusion, whereas it was barely detected in the control brain. This result is in complete accordance with the previous studies (Fujimura et al., 2000). Cytochrome c immunoblotting in the ischemic sample was sustained through 24 hours of reperfusion (Fig. 4A, top panel). These data not only confirm the specificity of the polyclonal antibody for cytochrome c used in the current study, but also show that cytosolic localization of cytochrome c was significantly increased after transient FCI. Cleaved caspase-9 immunoblots (35 kDa) were prominent 2 hours after reperfusion in the cytosolic fraction from the ischemic brain compared with the nonischemic brain. This band persisted at 4 hours after reperfusion; however, it was faint in the cytosolic fraction 24 hours after transient FCI (Fig. 4A, second panel). A band characteristic for Apaf-1 was evident in the cytosolic fraction in the control brains. No significant modifications of Apaf-1 expression were evident after 1 to 24 hours of transient FCI (Fig. 4A, third panel). A consistent amount of β-actin was observed in control and ischemic animals. The mitochondrial fraction of cytochrome c and procaspase-9 also were examined in the wild-type animals after transient FCI (Fig. 4B). A significant amount of mitochondrial cytochrome c was detected in the nonischemic brain; however, the decrease of cytochrome c was unclear (Fig. 4B, top panel). A band characteristic for procaspase-9 did not show significant changes before and after transient FCI (Fig. 4B, middle panel). Cytochrome oxidase was used as an internal control for the mitochondrial fraction, and no difference was observed between the samples (Fig. 4B, bottom panel).
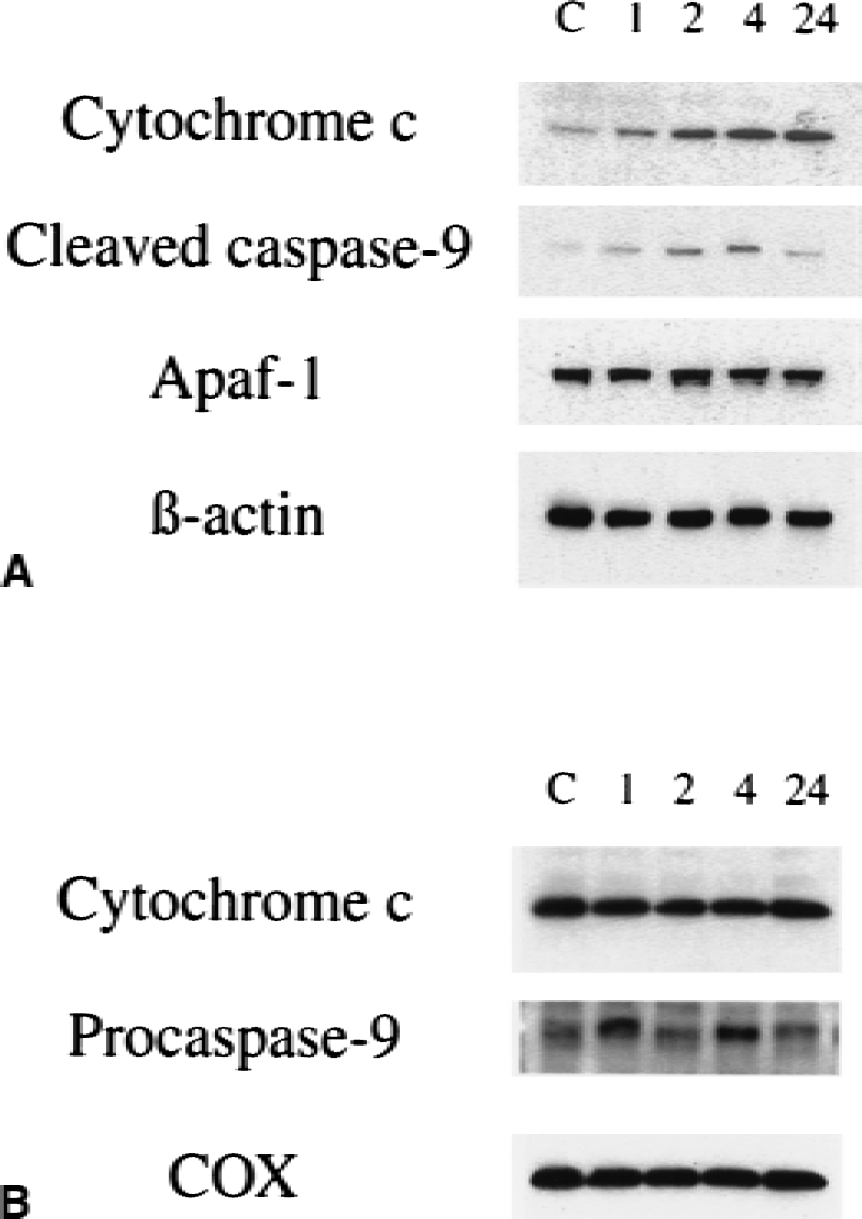
Western blot analysis of cytosolic
Early release of mitochondrial cytochrome c to cytosol and expression of active caspase-9 are significantly increased in Sod2 −/+ mice compared with wild-type mice after transient FCI
The amount of cytosolic cytochrome c and cleaved caspase-9 were compared between wild-type mice and Sod2 −/+ mutants 2 hours after transient FCI (Fig. 5). Western blots were performed, using four of the nonischemic control samples and five of the ischemic samples from different animals in each group in both the wild-type littermates and Sod2 −/+ mutants. There was no difference in the β-actin level between the wild-type and Sod2 −/+ mutants. The OD of cytochrome c in Sod2 −/+ mutants was significantly greater (P < 0.05) than that from wild-type mice (wild-type, 1.31 ± 0.64;Sod2 −/+, 2.35 ± 0.75) 2 hours after transient FCI. The OD of caspase-9 in Sod2 −/+ mutants also was significantly increased (P < 0.001) compared with that from the wild-type (wild-type, 0.77 ± 0.18;Sod2 −/+, 1.60 ± 0.21) 2 hours after FCI. These results suggested that cytosolic localization of cytochrome c was significantly increased in the Sod2 −/+ mutants as compared with the wild-type mice after transient FCI, as shown after permanent FCI (Fujimura et al., 1999). Caspase-9 activation also was greater in Sod2 −/+ mutants than in wild-type mice. There was no significant difference in Apaf-1 expression between wild-type mice and Sod2 −/+ mutants 2 hours after transient FCI (data not shown). To evaluate apoptotic cell death, TUNEL staining was performed 24 hours after transient FCI. TUNEL-positive cells were observed in the entire MCA territory in both wild-type mice (Fig. 6A) and Sod2 −/+ mutants (Fig. 6B), with shrunken, darkly stained nuclei and apoptotic bodies. Quantitative analyses (n = 4) showed a significant increase of TUNEL-positive cells in Sod2 −/+ mutants as compared with the wild-type in the cortex (Fig. 6D, P < 0.05; wild-type, 0.832 ± 0.500;Sod2 −/+, 2.332 ± 1.032), but not in the caudate putamen (Fig. 6C; wild-type, 1.856 ± 0.926;Sod2 −/+, 2.100 ± 0.526).
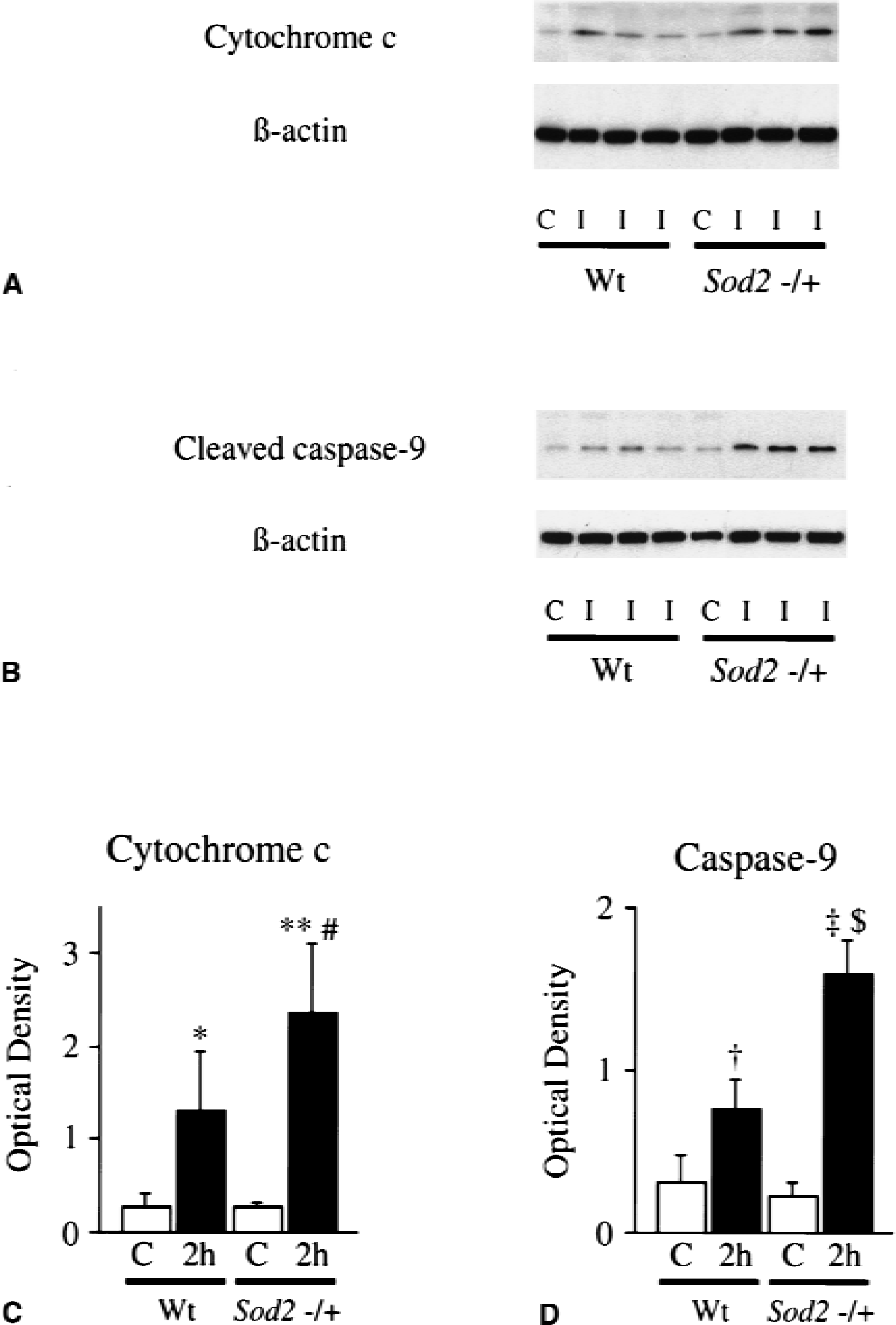
Western blot analyzes of cytochrome c
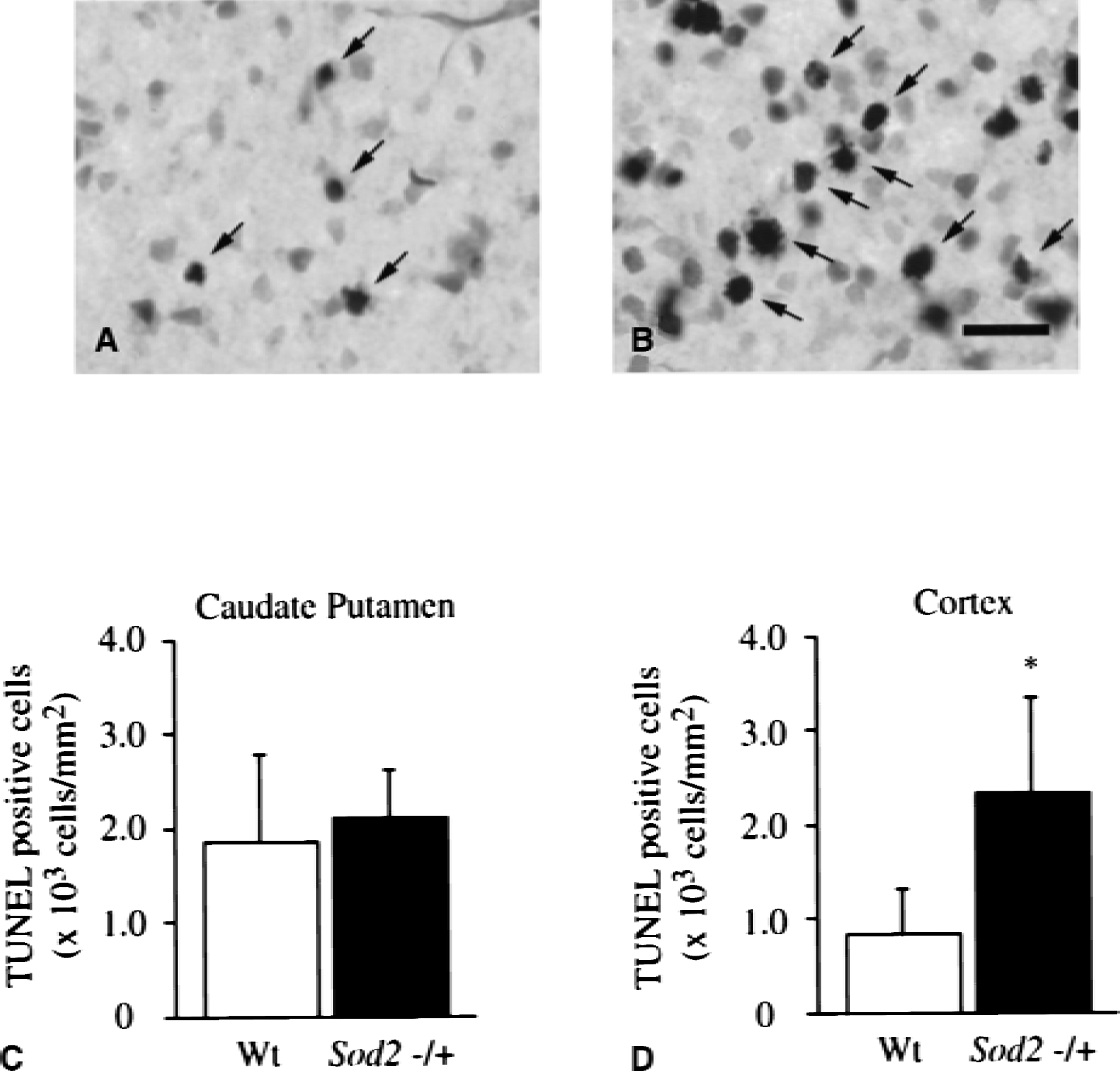
Representative photomicrographs show TUNEL-positive cells in the cortex of wild-type (Wt) mice
DISCUSSION
Caspase-9 activation after mitochondrial cytochrome c release to the cytosol
The cytochrome c–dependent mitochondrial pathway plays a critical role in apoptosis. The authors have shown the early release of mitochondrial cytochrome c to the cytosol and subsequent DNA fragmentation after focal ischemia (Fujimura et al., 1998) and global ischemia (Sugawara et al., 1999). The current study provides the first evidence that caspase-9 is activated in the cytoplasm after the cytosolic translocation of cytochrome c after focal ischemia and reperfusion (Figs. 3 and 4). The activation of caspase-9, which occurs downstream to cytochrome c release and upstream to activation of executor caspases such as caspase-3 or −7, is thought to begin after mitochondrial release of procaspase-9 to the cytosol followed by its cleavage to an active form by binding with Apaf-1 complex containing cytochrome c and dATP (Li et al., 1997; Krajewski et al., 1999). Although decreases of cytochrome c and procaspase-9 were not evident in mitochondria after FCI, it is conceivable that only relatively small levels of cytochrome c and procaspase-9 were released in contrast to their abundance in the mitochondria. Concerning the activity of caspase-9, a recent study indicated the importance of Apaf-1 (Rodriguez and Lazebnik, 1999). The authors of the study demonstrated that the proteolytic activity of caspase-9 in a complex with APAF-1 was much greater than that of free caspase-9, suggesting the complex functioning as an active holoenzyme. Once activated, caspase-9 cleaves procaspase-3 to activated caspase-3 and subsequently other executor caspases (Slee et al., 1999).
In the current study, the authors could show that the cytochrome c was released in the cytosol followed by cleaved caspase-9 expression after transient FCI, using immunofluorescent double labeling (Fig. 3) and Western blotting (Fig. 4). Moreover, immunofluorescent double labeling revealed the spatial coincidence of these two proteins (Fig. 3). With regard to cell type, cytochrome c–positive cells were thought to be neurons because the size of their nuclei was obviously larger than that of other cell populations, and because immunofluorescent double staining showed them to colocalize with MAPs (Fig. 3J to 3L). That cytochrome c translocation is seen in neurons has been reported in prior studies (Morita-Fujimura et al., 1999; Sugawara et al., 1999) through the use of neuron-specific nuclear protein (NeuN) immunohistochemistry, which reacts with neuronal nuclei and their cell bodies throughout the adult nervous system (Mullen et al., 1992). The absence of mitochondrial immunoreactivity of cytochrome c in the nonischemic brain, which is consistent with previous studies (Fujimura et al., 1998, 1999, 2000), is thought to occur because of thorough fixation of the brain with formaldehyde that prevents the antibody from reaching the mitochondrial intermembrane space but not the cytosol. In fact, immunohistochemistry with frozen sections results in dotted cytoplasmic immunoreactivity of cytochrome c in control brain (Fujimura et al., 1998, 1999, 2000).
Caspase-9 translocates from the cytosol to nuclei after transient FCI
Immunohistochemistry revealed that cleaved caspase-9 translocated from the cytosol to nuclei (Figs. 2 and 3G to 3I). Western blotting demonstrated decreased caspase-9 expression in the cytosol 24 hours after reperfusion (Fig. 4A)—a result which suggests that cleaved caspase-9 translocates from the cytosol to the nucleus. These results are in accordance with a prior study of immunoelectron microscopy (Krajewski et al., 1999) using in vitro and global ischemia models. These authors suggest the possibility that nuclear targeting sequences, contained in procaspase-9, become exposed in the active processed protease and that they account for the translocation of caspase-9 to the nucleus (Krajewski et al., 1999).
Cytochrome c release and caspase-9 activation are accelerated in Sod2 −/+ mice compared with wild-type mice
The authors have reported that ROS, especially superoxide, are involved in neuronal cell death, including apoptosis and necrosis, and in the pathogenesis of brain edema after FCI (Kinouchi et al., 1991; Chan, 1996; Kondo et al., 1997; Murakami et al., 1998). Evidence exists that mitochondria are the sites at which ROS are produced after cerebral ischemia (Piantadosi and Zhang, 1996). Furthermore, studies have demonstrated that superoxide production and cytochrome c release from mitochondria after permanent FCI are exacerbated in Sod2 −/+ mice (Fujimura et al., 1999). Because the mechanism by which cytochrome c exits the mitochondria is thought to be the formation of the MTP, a process inhibited by the mitochondrial antioxidant Bcl-2 (Marzo et al., 1998; Reed et al., 1998), it is conceivable that Mn-SOD may prevent cytochrome c release by blocking the formation of the MTP. This hypothesis is supported by a previous study in which the mitochondrial transmembrane potential, measured by rhodamine 123, was significantly decreased in Sod2 −/+ mice after permanent FCI (Murakami et al., 1998). Consequently, overproduction of ROS in mitochondria could cause MTP opening and cytochrome c release from mitochondria leading to subsequent DNA damage. In the current study, the authors confirmed that Mn-SOD deficiency accelerated caspase-9 activation by showing a significant increase of active caspase-9 2 hours after transient FCI (Fig. 5D), followed by the increase of apoptotic cell death 24 hours after reperfusion (Fig. 6D), in Sod2 −/+ mutants compared with wild-type mice. It is possible that the increased cytochrome c in the cytosol might accelerate caspase-9 activation in Sod2 −/+ mutants.
Alternatively, the increased active form of caspase-9 in Sod2 −/+ mutants might be a result of increased release of procaspase-9 from mitochondria because the activation of caspase-9 begins with the release of procaspase-9 from mitochondria to cytosol (Susin et al., 1999; Krajewski et al., 1999). The mechanism responsible for procaspase-9 release from mitochondria is still unclear, and so far the authors have been unable to demonstrate evidence of procaspase-9 release from mitochondria in the ischemic brain. However, considering the possible effect of Mn-SOD on MTP reduction, it is conceivable that mitochondrial procaspase-9 release to the cytosol may be regulated by superoxide dismutase, or Bcl-2, or both (Susin et al., 1999). Further studies are now being undertaken in the authors' laboratory to address this issue.
The current study demonstrated that Apaf-1 was constitutively expressed in adult mouse brain and did not change after 1 to 24 hours of transient FCI (Fig. 4A). Also, no difference in Apaf-1 expression was observed before and after FCI between the wild-type littermates and Sod2 −/+ mutants 2 hours after ischemia/reperfusion (data not shown). These results suggested that differences in caspase activation were not caused by modifications of the Apaf-1 level after transient FCI.
Caspase-9 in the cytochrome c–dependent apoptosis pathway
Increasing evidence suggests that an active process similar to programed cell death contributes to the death of neurons (Linnik et al., 1993; Tominaga et al., 1993; Li et al., 1995a; Gillardon et al., 1996; Asahi et al., 1997; Hara et al., 1997; Namura et al., 1998) and to the expansion of the lesion after FCI (Du et al., 1996). The authors have shown that cytochrome c is released from mitochondria to cytoplasm after FCI (Fujimura et al., 1998, 1999, 2000) and global ischemia (Sugawara et al., 1999). Furthermore, it has also been reported that the activated form of caspase-3 is detected at the early stage of ischemia and reperfusion injury in the brain (Namura et al., 1998), and that inhibition of the caspase family of protease may reduce infarct volume and apoptosis after transient ischemia (Hara et al., 1997; Endres et al., 1998; Chen et al., 1998). These studies suggest that a biochemical cascade, including caspase activation and subsequent DNA fragmentation, plays a major role in neuronal cell death after ischemia and reperfusion. In the current study, the authors have demonstrated the relationship and colocalization between cytochrome c and caspase-9 (Fig. 3), which is known as the apical caspase in the cytochrome c-dependent apoptotic pathway.
Caspase-3 has a critical role in apoptosis because it directly activates DNase and leads to DNA fragmentation and cell death (Enari et al., 1998). Regarding the relationship between caspase-3 and DNA damage in focal ischemia, one study has demonstrated the spatial colocalization between caspase-3 activation and DNA fragmentation (Namura et al., 1998). Caspase-3 activation is preceded by caspase-9 activation in the cytochrome c-dependent apoptotic pathway. However, caspase-9 is not the only initiator of caspase-3 activation. Caspase-3 also may be activated through another pathway mediated by caspase-8, in which the cleaved form of caspase-8 activates executor caspases like caspase-3 (Boldin et al., 1996; Fernandes-Alnemri et al., 1996; Muzio et al., 1996; Cryns and Yuan, 1998). To clarify the main pathway of apoptosis in this model, further investigations including the interrelationship among the activations of caspase-9, caspase-3, and caspase-8 are necessary. Moreover, recent evidence has shown that the early release of mitochondrial cytochrome c also is mediated by caspase-8 (Li et al., 1998). Simultaneous examination of cytochrome c, caspase-9, caspase-3, and caspase-8 will form the basis of future studies to elucidate the mechanism of apoptosis in focal ischemia.
Conclusion
The current results have demonstrated that activated caspase-9 is evident after mitochondrial cytochrome c release to the cytosol after transient FCI, and that Mn-SOD may attenuate caspase-9 activation and cytochrome c release. These results reveal the spatial and temporal relation between cytochrome c and caspase-9 and suggest that ROS affects caspase activation in apoptosis.
Footnotes
Acknowledgments:
The authors thank Dr. Paul Matz for his reviewing this manuscript, and Liza Reola, Bernard Calagui, and Jane O. Kim for their technical assistance.