
Abstract
We here investigated endothelial nitric oxide synthase (eNOS) expression after 10 minutes of forebrain ischemia. Real-time polymerase chain reaction, immunoblots and immunohistochemical studies revealed up-regulation of eNOS expression in the hippocampal CA1 subfield of gerbil. Immunoreactivity of eNOS significantly increased in endothelium but neither in neurons nor astrocytes after 6 to 168 hours of reperfusion. An increased Akt activity preceded the postischemic eNOS up-regulation. Intracerebroventricular injection (i.c.v.) of wortmannin, an inhibitor of phosphatidylinositol 3-kinase (PI-3K), significantly inhibited the increases in both eNOS mRNA and its protein with concomitant inhibition of Akt activation. The significant increase in the eNOS expression was also evident following preconditioning 2-minute ischemia. Both eNOS up-regulation and acquisition of ischemic tolerance observed at 3 days after preconditioning ischemia were significantly inhibited by pretreatment with wortmannin. Administration (i.c.v.) of NG-nitro-L-arginine methyl ester, but not 7-nitroindazole, 30 minutes prior to lethal 10-minute ischemia, significantly abolished the acquired tolerance. Intraperitoneal injections of aminoguanidine at immediately after, 24, and 48 hours after preconditioning had no effects on the tolerance. These results suggest that eNOS expression is up-regulated in the endothelium via PI-3K pathways after transient forebrain ischemia, and that preconditioning-induced eNOS expression plays an important role in neuroprotection in the ischemic tolerance.
Production of nitric oxide (NO) by nitric oxide synthase (NOS) has important roles in physiological and pathological events in the central nervous system. Accumulating evidence suggests that both neuronal NOS (nNOS) and inducible NOS (iNOS) have detrimental effects on neurons in the ischemic brain, whereas endothelial NOS (eNOS) activity has protective effects (Iadecola, 1997).
Endothelium-derived NO regulates blood pressure, augments regional blood flow, improves cerebral circulation, and inhibits platelet aggregation (Iadecola, 1997; Randomski et al., 1990; Morikawa et al., 1994; Huang et al., 1995; Liao, 1998). Recent experimental and clinical trials have demonstrated that 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins), which had been developed as lipid-lowering drugs, reduce the risk of myocardial infarction and stroke (Marson et al., 2000). Several studies have suggested that treatment with statins up-regulates eNOS expression thereby improving endothelial function (Endres et al., 1998; Laufs et al., 1998; Laufs et al., 2000; Laufs et al., 2002). Beasley et al. (1998) showed eNOS up-regulation via indomethacin-sensitive mechanism in global ischemia model induced by increasing intracranial pressure, whereas it is still controversial whether cerebral ischemia itself causes eNOS induction in vessels (Zhang et al., 1993; Veltkamp et al., 2002; Limbourg et al., 2002). Studies with cultured endothelial cells suggested several protein kinase pathways including kinase insert domain-containing receptor/fetal liver kinase-1 (KDR/Flk-1), protein kinase C (PKC), phosphatidylinositol 3-kinase (PI-3K) γ, Janus kinase 2 (Jak2), extracellular signal-regulated kinase 1/2 (Erk1/2) and Ca2+/calmodulin-dependent protein kinase II (CaMKII) underlie eNOS up-regulation by pharmacological stimuli such as vascular endothelial growth factor, lysophosphatidylcholine (LPC), prostaglandin E2 (PGE2) stimulation (Shen et al., 1999; Cieslik et al., 2001; Cai et al., 2001; Gobeil et al., 2002). However, the mechanisms mediating eNOS expression following brain ischemia are currently unclear.
We recently reported that Akt activation following ischemic preconditioning has a pivotal role in the acquisition of ischemic tolerance (Yano et al., 2001). Because Akt/protein kinase B phosphorylates eNOS on serine1177 in vitro thereby activating the enzyme (Fulton et al., 1999; Gallis et al., 1999), we speculated that phosphorylation of eNOS by Akt accounts for acquisition of ischemic tolerance. In the present study, we found that up-regulation of eNOS but not its phosphorylation by the PI-3K pathway has a pivotal role in the acquisition of ischemic tolerance following ischemic preconditioning.
MATERIALS AND METHODS
Animal experiments
All animal procedures were approved by the Committee of Animal Experiments at Kumamoto University School of Medicine. All experiments were performed on adult male Mongolian gerbils weighing 60 to 70 g. Anesthesia was achieved with 1% halothane in 70% N2O/30% O2. Gerbils were subjected to transient forebrain ischemia by bilateral occlusion of the common carotid artery with rectal temperature maintained at 37.0°C to 38.0°C as previously reported (Hashiguchi et al., 2003). For western blotting and polymerase chain reaction (PCR) through the manuscript, samples were prepared as previously described (n = 6–8 per group or time point;Hashiguchi et al., 2003). Ischemia was induced as follows: 1) 10 minutes of ischemia, 2) 10 minutes of ischemia following 5 minutes of ischemia with 4 weeks of reperfusion, 3) for experiments of ischemic tolerance, 10 minutes of ischemia following 2 minutes of ischemia with 3 days of reperfusion.
Drug injection
An inhibitor of phosphatidylinositol 3-kinase (PI-3K), wortmannin, a nonselective NOS inhibitor, NG-nitro-L-arginine methyl ester (L-NAME), and a nNOS selective inhibitor, 7-Nitroindazole (7-NI) were purchased from Sigma and dissolved in 2% DMSO in PBS, saline, and 50% DMSO in PBS, respectively. Wortmannin (100 μmol/L) was injected 30 minutes before 2 minutes of ischemia, L-NAME (30 mmol/L) or 7-NI (60 mmol/L) was injected 30 minutes before 10 minutes of ischemia intracerebroventricularly (i.c.v., 2 μL each, bregma; 3.5 mm lateral, 2.0 mm posterior, 2.5 mm deep). To inhibit iNOS activity in vivo, 200 mg/kg aminoguanidine (AG) (Sigma, St. Louis, MO, USA) dissolved in saline was intraperitoneally administered at immediately after, 24, and 48 hours after 2 minutes of ischemia. Each corresponding vehicle was also administered. The doses of wortmannin, L-NAME, and AG used in this study were based on the previous studies (Kohno et al., 1995; Yano et al., 2001; Kapinya et al., 2003).
Immunoblot analysis
Protein extraction from the hippocampal CA1 or CA3 tissues and immunoblotting were performed as previously described (Kawano et al., 2001), with primary monoclonal antibodies against eNOS (1:1000), Akt-1 (1:500) (BD Transduction, Lexington, KY, USA), activated MAP kinase (diphosphorylated Erk-1/2, 1:1000), β-tubulin (1:10000) (Sigma), or polyclonal antibodies against phospho-Ser473 Akt (1:300), phospho-Ser657 PKCα (1:500), phospho-Thr286/287 CaMKIIα (1:2000), or phospho-Ser1177 eNOS (1:200) (Cell Signaling). To quantitate immunoreactive bands, Western blots were scanned and optical densities of bands were analyzed using Scion Image software (Scion Corp., Frederick, MD, USA).
Immunohistochemistry
Immunohistochemistry was performed as previously described (Hashiguchi et al., 2003). The sections were incubated with both primary monoclonal antibody for eNOS (1:300; BD Transduction) and polyclonal antibody for phospho-Ser473 Akt (1:50; Cell Signaling) or glial fibrially acidic protein (GFAP, 1:1000; DAKO, Carpinteria, CA, USA), subsequently labeled with fluorescein-labeled anti-mouse secondary antibodies (1:2000) for the eNOS antibody and with Texas Red-labeled anti-rabbit secondaries (1:2000) for phospho-Ser473 Akt or GFAP antibody, and then analyzed with Olympus (Tokyo, Japan) Fluoview confocal microscope.
Multiplex real-time polymerase chain reaction
Total RNA isolation, reverse transcription, conventional PCR and real-time PCR were performed according to standard techniques. For DNA standards of real-time PCR, products of conventional PCR for mouse eNOS, a 340-bp cDNA (sense: 5′-TTCCGGCTGCCACCTGATCCTAA-3′; antisense: 5′-AA CATATGTCCTTGCTCAAGGCA-3′), and for 18S rRNA, a 362-bp cDNA (sense: 5′-TAGAGGGACAAGTGGCGTTCAGC-3′; antisense: 5′-AAGTTCGACCGTCTTCTCAGCGC-3′) were prepared respectively after confirmation of correct sequences. These PCR products were submitted as target sequences and, designing and ordering LUX™ primers (Invitrogen, Carlsbad, CA, USA) for real-time PCR were done following the step-by-step instruction in the LUX™ Designer Web-based Design Software (Invitrogen LUX™ Web site: http://www.invitrogen.com/LUX). Then, the following primer pairs were selected: eNOS (5′-TCCTAACTTGCCCTGCATCC-3′ forward and 5′-CACGACTAGCCCTTTGATCTCAATGTCGTG-3′ reverse) and 18S rRNA (5′-ATTCCCAGTAAGTGCGGGTCA-3′ forward and 5′-GTACACATAGCGACGGGCGGTGTGTAC-3). Real-time PCR was performed on ABI PRISM 7700 Detection System (Applied Biosystems, Tokyo, Japan). eNOS mRNA was expressed as the ratio of eNOS to 18S rRNA.
Neuropathological evaluation
For experiments of ischemic tolerance, we adopted 2 minutes of ischemia for preconditioning, and 10 minutes of ischemia 3 days later as subsequent lethal stimuli. At 7 days after sham operation or 10 minutes of ischemia, histopathological evaluation with propidium iodide (Molecular Probes, Tujunga, CA, USA) was performed (Hashiguchi et al., 2003). Cell viability was expressed as percentages of the averaged number of viable cells from sham-operated gerbils.
Measurement of cerebral blood flow (CBF)
Intra-ischemic CBF changes in the CA1 region were evaluated with a laser bloody flow meter (ALF21, Advance). Gerbils were anesthetized as above, and placed in a stereotaxic apparatus (David Kopf Instruments). A small burr hole was drilled through the skull for stereotaxic insertion of a 1.8 mm diameter laser-Doppler flow probe (Type-E, Advance) into the right hippocampus (2.5 mm posterior and 2 mm lateral to the bregma and 1 mm below brain surface). After a stabilization period, 10-minute ischemia was performed as above. Laser-Doppler flows were continuously recorded throughout the experiments. Proper probe placement was confirmed upon postmortem inspection.
Statistical analysis
Data are expressed as mean ± SD. Statistical evaluation was performed by ANOVA with Scheffé's test, or an unpaired Student t test. P < 0.05 were considered statistically significant.
RESULTS
10-minute ischemia up-regulates eNOS expression in gerbil hippocampus
In the hippcampal CA1 subfield (Fig. 1A), eNOS expression was significantly increased at 6 hours, peaked at 48 hours, and still elevated at 168 hours after 10 minutes of ischemia (180 ± 33%, 289 ± 53%, 259 ± 24% of sham-operated animals, respectively; P < 0.01). Similarly, the eNOS up-regulation was found in the CA3 subfield (data not shown). eNOS immunoreactivity markedly increased in the CA1 subfield after 48 to 168 hours of reperfusion (Fig. 1B). The increased immunoreactivity was predominantly observed in the vascular endothelium around the CA1 subfield. This is in contrast with the previous reports, in which obvious immunoreactivity against eNOS was detected in pyramidal neurons or GFAP-positive astrocytes (Dinerman et al., 1994; Iwase et al., 2000).
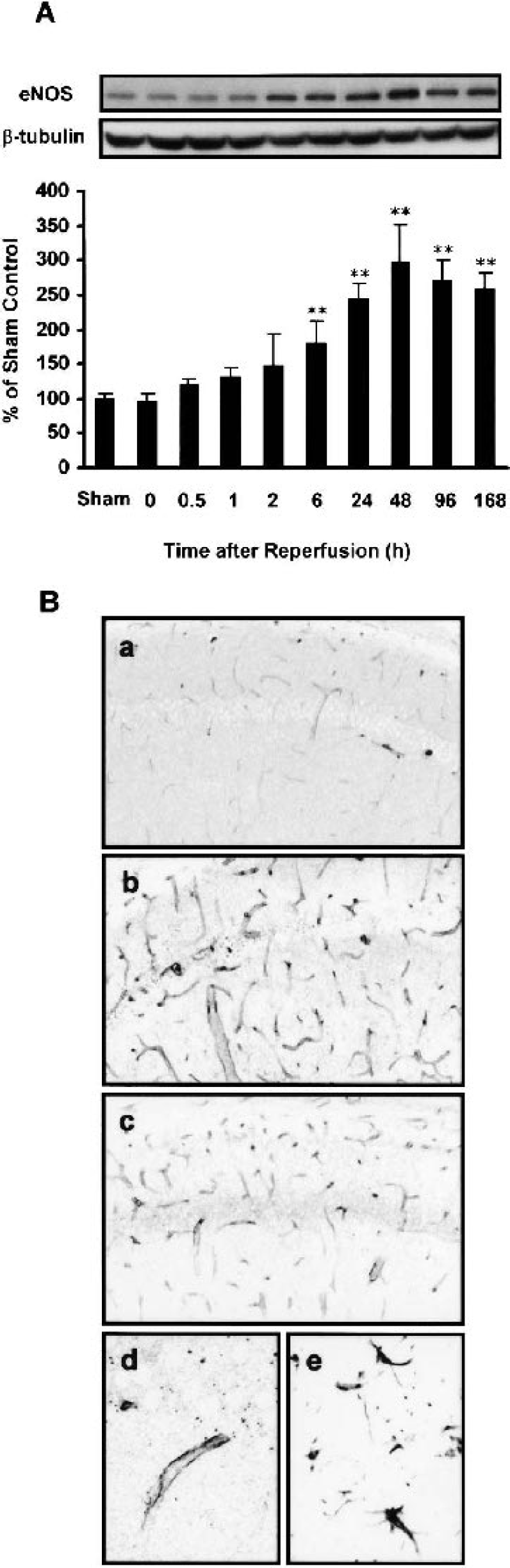
Time course of eNOS expression in the CA1 subfield after 10 minutes of ischemia. A, Representative image of immunoblot with anti-eNOS antibody and its quantitative analyses by densitometric scanning of immunoreactive bands (mean ± SD). β-tubulin was used to normalize amount of proteins applied in each lane. (n = 6 per time point). ∗∗ P < 0.01 versus sham-operated animals. B, Hippocampal CA1 sections from sham-operated (a), or at 48 hours (b and d), 168 hours (c) after reperfusion (n = 6) were stained with monoclonal anti-eNOS antibody. Panels (a) through (c) indicate the CA1 pyramidal cell layer. At higher magnificationin in panels (d) and (e) after double staining with anti-eNOS and anti-GFAP antibodies, eNOS (d) was not colocalized with GFAP-positive cells (e).
10-minute ischemia enhances phosphorylation level of Akt, neither of CaMKIIα, nor Erk1/2, nor PKCα in gerbil hippocampus
To determine potential mediators for the eNOS up-regulation, we first examined phosphorylation levels of four major kinases, Akt, Erk1/2, CaMKIIα and PKCα, since these kinases were reported to mediate eNOS up-regulation in the many types of cells (Fig. 2A; Shen et al., 1999; Cieslik et al., 2001; Cai et al., 2001; Gobeil et al., 2002). Akt-Ser473 phosphorylation significantly decreased to 25 ± 14% of sham-operated animals immediately after 10 minutes of ischemia, followed by persistently increased to 198 ± 101% and 216 ± 65% at 0.5 and 6 hours after reperfusion, respectively. Akt phosphorylation returned to near basal level of 158 ± 69% at 48 hours. However, phospho-Ser1177 in eNOS was unchanged over time course after ischemia. The levels of Erk1/2 and CaMKIIα phosphorylation significantly and persistently decreased throughout the experimental periods. No significant changes were observed in PKCα phosphorylation.
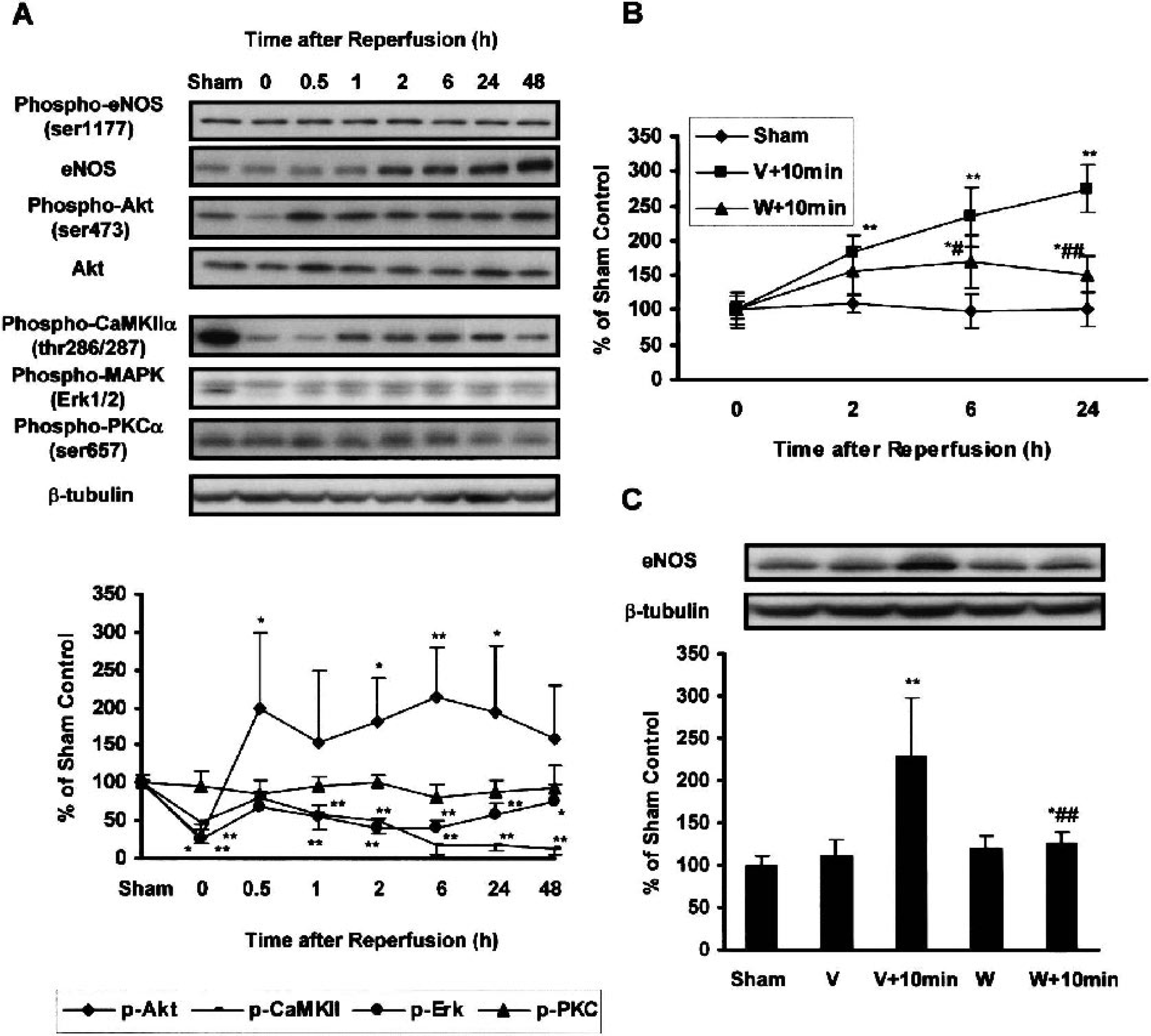
Phosphorylation levels of eNOS and protein kinases, and up-regulation of eNOS expression following 10-minutes ischemia. A, Upper panel, Representative images of immunoblots with anti-phospho-Ser1177 eNOS, eNOS, phopho-Ser473 Akt, phospho-Thr286/287 CaMKIIα, diphosphorylated Erk-1/2, or phospho-Ser657 PKCα antibody. Immunoblot image with anti-Akt or β-tubulin antibody revealed that the same amount of proteins was loaded in each lane. Lower panel, Densitometric analyses of Akt, CaMKIIα, Erk-1/2, or PKCα phosphorylation in the hippocampal CA1 subfields following ischemia (mean ± SD, n = 6 per time point). ∗∗ P < 0.01, ∗ P < 0.05 versus corresponding phosphorylation levels in sham-operated animals. B, Changes in real-time PCR product of eNOS mRNA after reperfusion. The levels of eNOS mRNA normalized to 18S rRNA mRNA in sham-operated, or vehicle-treated, wortmannin-treated ischemic animals. (mean ± SD, n = 6 per time point). ∗∗ P < 0.01, ∗ P < 0.05 versus sham-operated, ##P < 0.01, #P < 0.05 versus vehicle-treated ischemic animals. C, Changes in eNOS protein following brain ischemia and effect of wortmannin treatment. Upper panel, representative image of immunoblots with anti-eNOS or anti-β-tubulin antibody at 48 hours after 10 minutes of ischemia. Lower panel, densitometric analyses of eNOS expression (mean ± SD, n = 6). ∗∗ P < 0.01, ∗ P < 0.05 versus sham-operated, ##P < 0.01 versus vehicle-treated ischemic animals. V, vehicle; W, wortmannin; 10min, 10 minutes of ischemia.
Wortmannin prevents ischemia-induced eNOS up-regulation
Because PI-3K/Akt pathways were activated after forebrain ischemia, we then determined whether wortmannin, a PI-3K inhibitor, affected the eNOS up-regulation in both mRNA (Fig. 2B) and protein levels (Fig. 2C) after 10 minutes of ischemia. The increased Akt-Ser473 phosphorylation was blocked by treatment with wortmannin as previously reported (data not shown;Yano et al., 2001). Real-time PCR analyses showed that eNOS mRNA levels significantly increased to 183 ± 26%, 234 ± 42% and 274 ± 34% of sham-operated animals at 2, 6 and 24 hours after reperfusion, respectively (P < 0.01; Fig. 2B). Intracerebroventricular injection of 2 μl of 100 μmol/L wortmannin significantly but partly suppressed the increased eNOS mRNA by 169 ± 38%, 151 ± 27% of sham-operated animals at 6, 24 hours, respectively. Similarly, the up-regulation in eNOS protein level was markedly inhibited by wortmannin injection (Fig. 2C). Vehicle or wortmannin alone did not affect eNOS expression.
An increased Akt phosphorylation is also observed in endothelium as well as neurons
As previously reported (Yano et al., 2001; Kawano et al., 2001), increased Akt phosphorylation was predominantly observed in cytoplasm and apical dendrites, and weakly in nuclei of CA1 pyramidal neurons in sham-operated gerbils. Little eNOS immunoreactivity was observed in the neurons. In the endothelium, intensive eNOS immunoreactivity and moderate immunoreactivity against phospho-Ser473 Akt antibody was found (Fig. 3A and Fig. 3B). At 6 hours after reperfusion when the maximal Akt phosphorylation occurred (Fig. 2A), immunoreactivity for phospho-Ser473 Akt markedly increased in the endothelium as well as neurons (Fig. 3C and Fig. 3D), whereas wortmannin injection inhibited the increased immunoreactivity (Fig. 3G and Fig. 3H). Importantly, Akt phosphorylation was still evident in the endothelium even at 48 hours after reperfusion, when it was totally lost in the neurons (Fig. 3E and Fig. 3F).
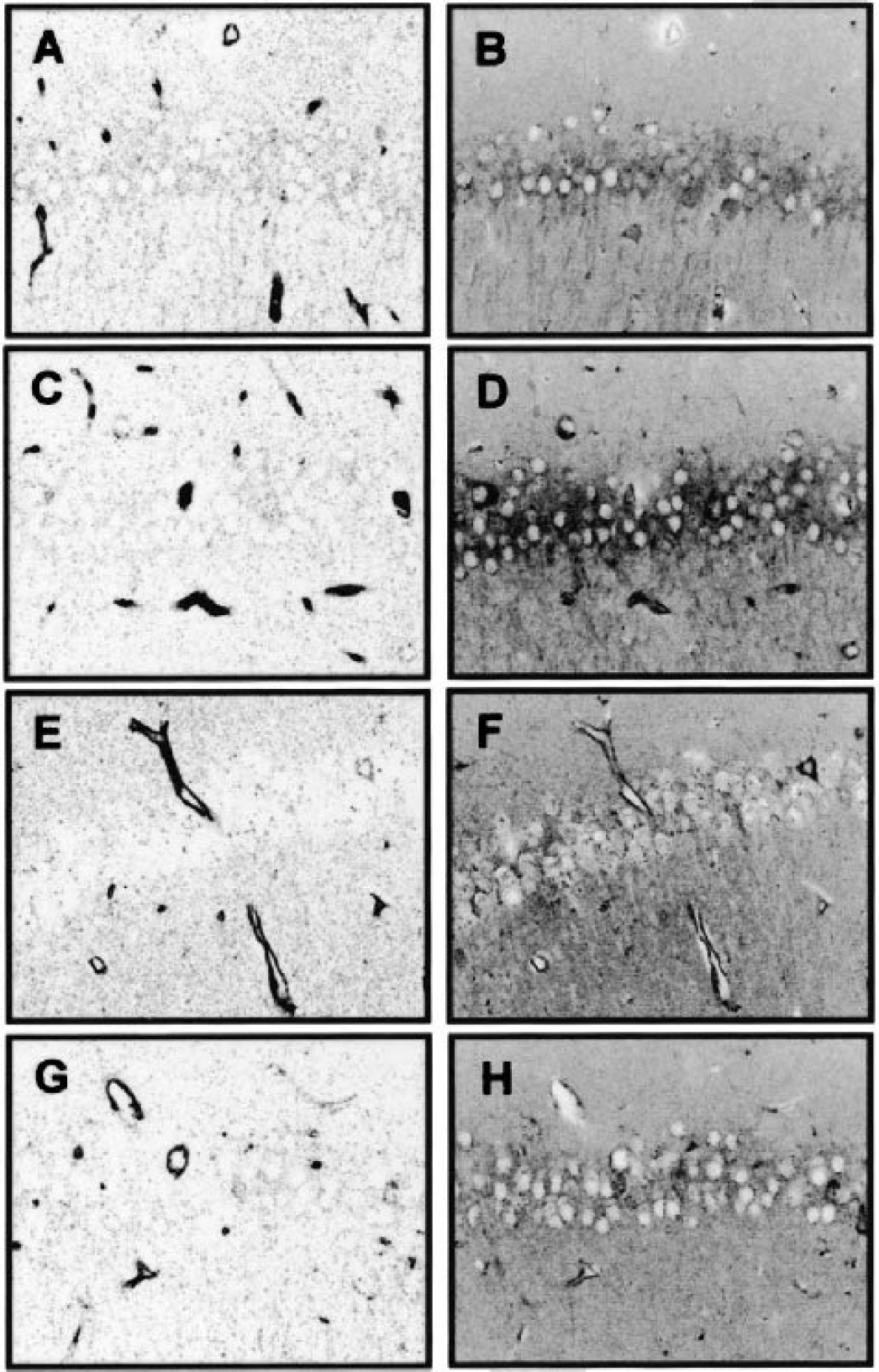
Immunohistochemical localization of eNOS and phospho-Ser473 Akt following 10-minutes ischemia. Hippocampal sections were double stained with anti-eNOS (A, C, E and G) and anti-phospho-Ser473 Akt antibodies (B, D, F and H). Representative images of CA1 areas in sham-operated gerbils (A and B), in vehicle-treated gerbils subjected to 10 minutes of ischemia followed by 6 hours (C and D) or 48 hours of reperfusion (E and F), or in wortmannin-treated gerbils subjected to 10 minutes of ischemia followed by 6 hours of reperfusion (G and H) (n = 6).
10-minute ischemia up-regulates eNOS expression independently of neuronal injury
To confirm that ischemia-induced eNOS up-regulation in the endothelium is independent of neuronal injury, which activates neuronal Akt (Fig. 3), we first killed neurons by 5 minutes of ischemia and 4 weeks later, examined eNOS expression in the endothelium by 10 minutes of ischemia using the same animals. In sham-operated gerbils (Fig. 4A and Fig. 4B), viable CA1 pyramidal neurons and moderate eNOS immunoreactivity were observed. Although the pyramidal neurons were totally lost 4 weeks after a 5-minute ischemia (Fig. 4C and Fig. 4E), eNOS up-regulation after 10 minutes of ischemia (Fig. 4F) was still observed compared with sham-operated gerbils (Fig. 4D). The eNOS up-regulation at 48 hours after reperfusion was significantly inhibited by wortmannin treatment (188 ± 17% and 142 ± 15% of sham-operated animals without or with wortmannin, respectively; Fig. 4G).
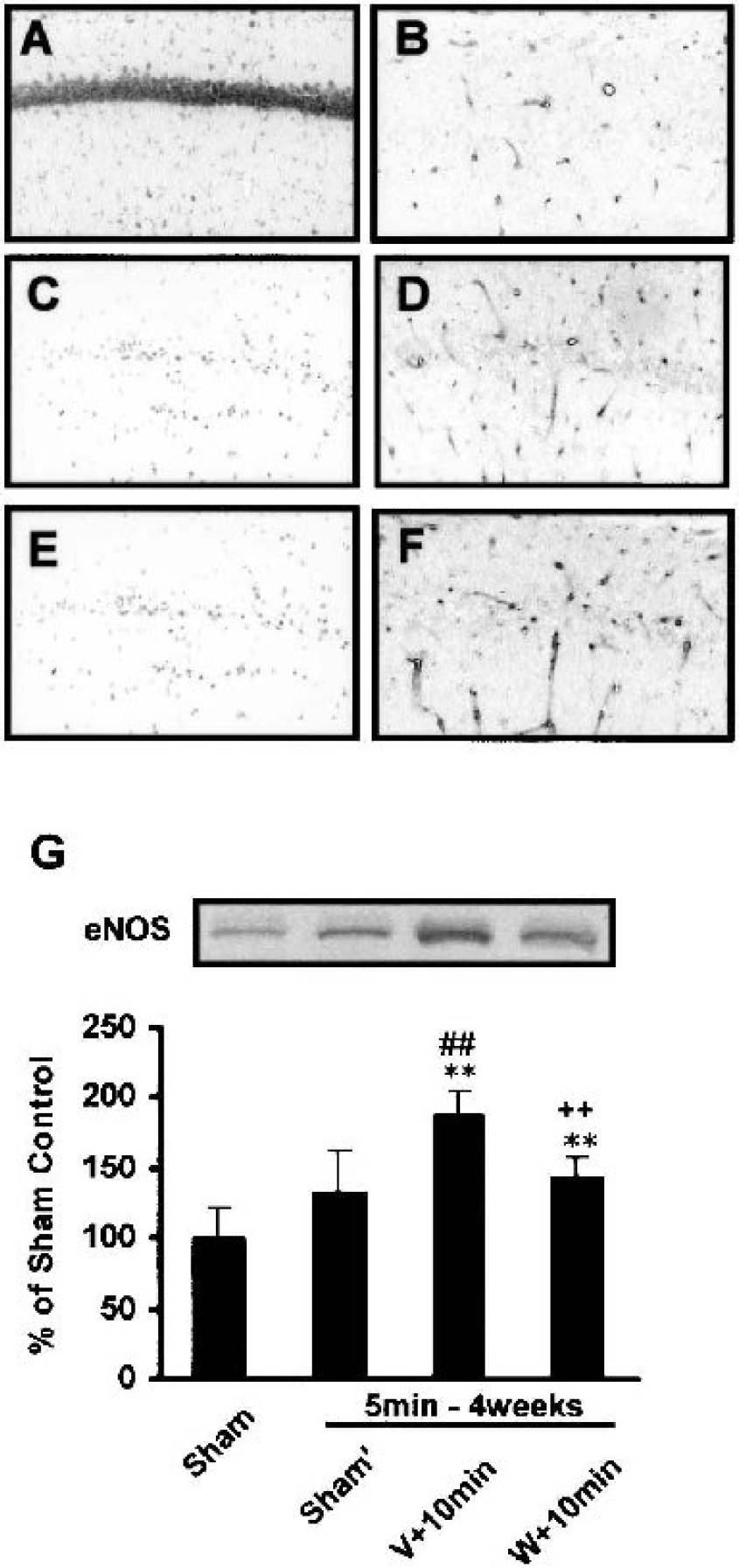
Up-regulation of eNOS in the hippocampal CA1 subfield having neuronal loss by 5-minute ischemia. Gerbils were processed to 5 minutes of ischemia followed by 4 weeks of reperfusion, which caused neuronal death in the CA1 subfield and then subjected to the second 10 minutes of ischemia. Hippocampal sections were double stained with propidium iodide (A, C and E) and anti-eNOS antibody (B, D and F). Representative images of CA1 areas in sham-operated gerbils (A and B), in gerbils subjected to 5 minutes of ischemia followed by 4 weeks of reperfusion (C and D), or in gerbils subjected to the second 10 minutes of ischemia followed by 48 hours of reperfusion (E and F) (n = 4). As is the case with 10 minutes of ischemia (Fig. 2C), up-regulation of eNOS was still observed in the CA1 subfied 48 hours after the second ischemia. Densitometric analyses of the immunoreactive bands for anti-eNOS antibody were performed (Fig G) (n = 6). ∗∗ P < 0.01 versus sham-operated, ##P < 0.01 versus sham'-operated, ++P < 0.01 versus vehicle-treated ischemic animals. Sham'; only 5 minutes of ischemia without second 10 minutes of ischemia; V, vehicle; W, wortmannin; 10min, 10 minutes of ischemia.
Ischemic tolerance needs eNOS up-regulation
Preconditioning ischemia increased eNOS expression at 3 days after reperfusion in the present study, concomitant with increased Akt phosphorylation as reported previously (Yano et al., 2001). The increased eNOS expression was to lesser extent than that by 10 minutes of ischemia (Fig. 5A; 174 ± 34% compared with 265 ± 16% in 10-minute ischemia). The increased eNOS expression by preconditioning was partly inhibited by wortmannin injection (131 ± 13% of sham-operated animals; Fig. 5A). Further eNOS up-regulation was not potentiated at 48 hours after additional 10 minutes of ischemia (188 ± 45%). By histopathological analysis, compared with sham-operated animals, 10 minutes of lethal ischemia induced cell death to 4.5 ± 2.1% and the preconditioning rescued neurons by 82 ± 22% of sham-operated animals (Fig. 5B). Wortmannin injection prior to preconditioning and L-NAME treatment prior to lethal ischemia blocked the neuroprotection by preconditioning, whereas neither 7-NI treatment prior to lethal ischemia nor daily injections of AG after preconditioning had any effects on the preconditioning-induced neuroprotection. Treatment with vehicle or drug alone had no effects on the neuronal viability (data not shown). Although preconditioning ischemia resulted in an increased eNOS expression, the preconditioning did not affect hippocampal CBF during ischemia (Fig. 5C).
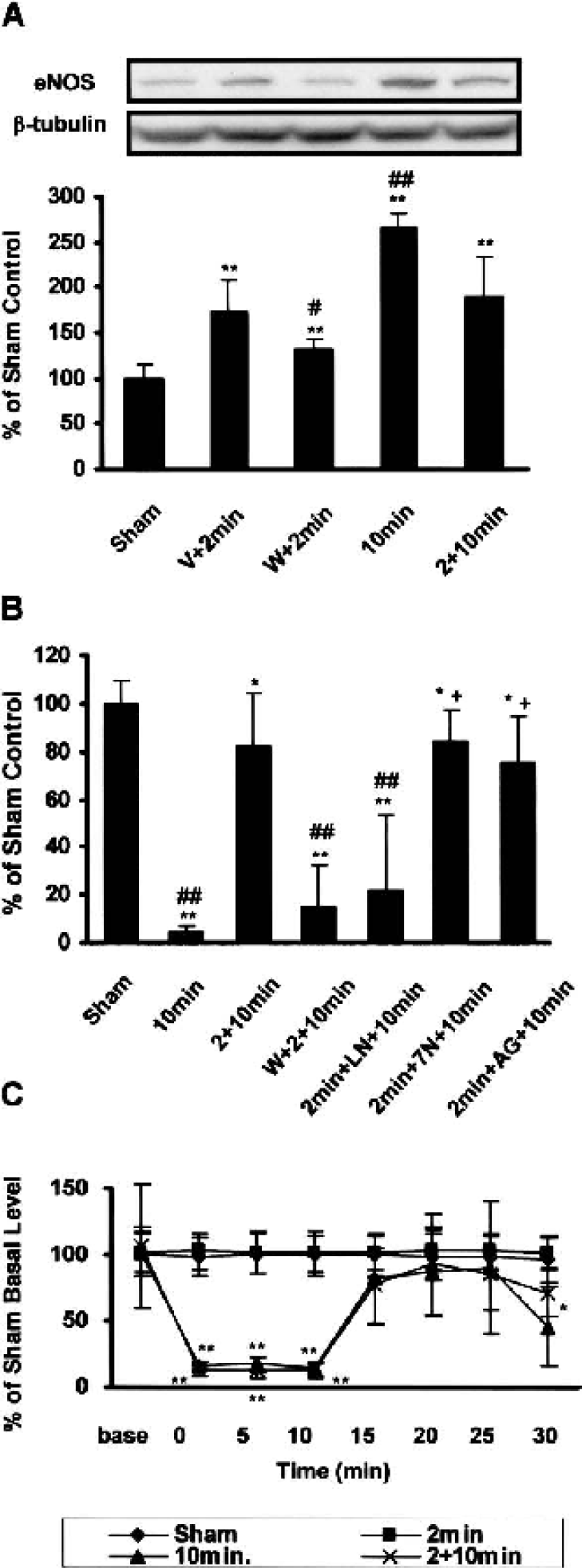
Up-regulation of eNOS by preconditioning ischemia and inhibition of preconditioning-induced ischemic tolerance by L-NAME. A, Representative images of eNOS immunoblots at 3 days after preconditioning, or at 48 hours after 10 minutes of ischemia with or without 2-minutes preconditioning. The densitometric analyses of the eNOS immunoblots were summarized (mean ± SD, n = 6). ∗∗ P < 0.01 versus sham-operated, ##P < 0.01, #P < 0.05 versus vehicle-treated preconditioned animals. When indicated, gerbils were intracerebroventricularly injected 2 μl vehicle or 100 μmol/L wortmannin 30 minutes before 2-minutes ischemia. B, Quantitative analyses of surviving neurons in the CA1 subfield at 7 days after 10 minutes of ischemia (mean ± SD, n = 8). When indicated, wortmannin was injected 30 minutes prior to 2-minute ischemia, whereas L-NAME (LN) or 7-nitroindazole (7N) was injected 30 minutes prior to 10-minute ischemia. Aminoguanidine (AG) was intraperitoneally administered immediately after, 24, and 48 hours after the preconditioning ischemia. ∗∗ P < 0.01, ∗ P < 0.05 versus sham-operated, ##P < 0.01 versus preconditioned animals with lethal ischemia, +P < 0.05 versus L-NAME-treated ischemic animals. C, Changes in CBF during ischemia. The CBF was monitored in sham-operated animals, in preconditioned animals or in 10-minutes ischemic animals with without preconditioning and expressed as percentages of the basal level of sham-operated animals. (mean ± SD, n = 6). ∗∗ P < 0.01, ∗ P < 0.05 versus the basal level of sham-operated animals.
Discussion
Expression of eNOS in endothelium is up-regulated following shear stress, hypoxia, cell proliferation, or by stimulation with bacterial lipopolysaccharide, cytokines, growth factors, estrogens, oxidized low-density lipoproteins or LPC (Förstermann et al., 1998). Cerebral ischemia/reperfusion is associated with intraluminal shear stress, local hypoxia, and induction of hydrogen peroxide, growth factors and inflammatory cytokines by cerebral tissues, blood vessels, and blood cells (Förstermann et al., 1998; Govers and Rabelink, 2001). Limbourg et al. (2002) showed unchanged eNOS expression after transient focal ischemia, whereas three previous studies suggest that cerebral ischemia/reperfusion up-regulates eNOS expression (Zhang et al., 1993; Beasley et al., 1998; Veltkamp et al., 2002), which is in agreement with our results. Differences in animal species, in model of ischemia, and in the method of measuring eNOS expression may account for the discrepancies. Furthermore, the molecular signaling and functions of eNOS up-regulation following brain ischemia remain unclear. Among several signaling pathways underlying eNOS up-regulation by various pharmacological stimuli, we were particularly interested in PI-3K/Akt pathway. Because Akt phosphorylates eNOS at Ser1177, thereby activating the enzyme (Fulton et al., 1999; Gallis et al., 1999) and LPC up-regulates eNOS expression by enhancing transcriptional activity via PI-3Kγ-mediated pathway (Cieslik et al., 2001). However, several studies suggest that Akt activity is not required for gene expression of eNOS in cultured endothelial cells (Kureishi et al., 2000; Urbich et al., 2002; Ming et al., 2002). Unexpectedly, phospho-Ser473 Akt was not correlated with phospho-Ser1177 eNOS, but with the eNOS up-regulation in the present study. Erk1/2, which is possibly lying downstream to PI-3Kγ (Cieslik et al., 2001), remained dephosphorylated after reperfusion and unaffected by wortmannin injection (data not shown). It is well known that the expression of eNOS is regulated by a range of transcriptional and posttranscriptional levels (Fleming and Busse, 2003). The murine eNOS promoter possesses binding sites for numerous transcription factors (Teichert et al., 1998), including Sp-1 system, which can be activated via PI-3K pathway (Cieslik et al., 2001). Experiments of DNA mobility shift with oligonucleotides containing the sequence of the eNOS promoter using nuclear extracts from postischemic hippocampal tissues showed slight increase in binding activity for activator protein-1 (AP-1), but not for Sp-1. However, the increase in AP-1 binding activity was not significant, and was not affected by wortmannin injection (our personal observation). By contrast, the wortmannin injection, which completely inhibited Akt phosphorylation as previously described (Yano et al., 2001), significantly inhibited the up-regulation of eNOS mRNA and its protein, implying that PI-3K pathways are required for induction of eNOS at least partly in the transcriptional level. Consistent with our results, Gobeil et al. (2002) reported that an increased Akt phosphorylation is well-correlated with PGE2-induced eNOS mRNA induction. Inconsistent with the previous report, in which eNOS was expressed in the hippocampal pyramidal neurons (Dinerman et al., 1994), eNOS immunoreactivity was mainly detected in the endothelium in the present study. In addition, eNOS up-regulation following brain ischemia was also evident in the endothelium of the CA1 subfield lacking living neurons. Thus, eNOS up-regulation following ischemia is independent of presence of neurons in the CA1 subfield.
PI-3K pathways contribute to induction of ischemic tolerance by preconditioning as reported previously, but the downstream molecules remain to be determined (Yano et al., 2001). Interestingly, pretreatment with 30 mmol/L L-NAME, which was reported to exhibit neuroprotective effects (Kohno et al., 1995), abolished the acquired ischemic tolerance in the present study. NO production is reported to be required for cerebral ischemic tolerance (Nandagopal et al., 2001; Kirino, 2002; Dirnagl et al., 2003). Acquisition of tolerance in newborn rat also required NO production by eNOS (Gidday et al., 1999). Likewise, tolerance against ischemic neuronal injury induced by volatile anesthetics depends on iNOS in neuronal cells as a mediator (Kapinya et al., 2002). As for cardiac ischemic tolerance, NO plays dual roles in the acquisition of delayed ischemic tolerance. For example, eNOS-derived NO initially functions as a trigger and iNOS-derived NO subsequently functions as a mediator of the adaptive response (Bolli et al., 1998; Bolli, 2001). NO produced by eNOS is also an effector of delayed coronary endothelial tolerance (Laude et al., 2003). It is noteworthy that iNOS induction is a mediator of ische-mic tolerance not only in the heart but also in both kidney and intestine (Bolli, 2001; Park et al., 2003). However, neither change in nNOS nor iNOS expression was apparently detected in the CA1 subfield following transient forebrain ischemia in our model (data not shown). Furthermore, neither 7-NI nor AG had any effect on the acquired tolerance. Thus, up-regulation of eNOS by preconditioning may account for the ischemic tolerance in the brain. However, physiological relevance of the eNOS expression in the ischemic tolerance remains unclear. The expression of eNOS did not affect the CBF changes during ischemia/reperfusion, in consistent with previous reports (Alkayed et al., 2002; Dirnagl et al., 2003). To define physiological relevance and molecular mechanisms underlying neuroprotection by eNOS up-regulation, further extensive studies are required using eNOS-knockout animals.
In summary, a sustained increase in eNOS protein levels in the blood vessels of gerbil hippocampus is observed at 6 to at least 168 hours after transient forebrain ischemia. The PI-3K pathway, which might be mediated through Akt or other downstream molecules, is underlying the postischemic eNOS up-regulation. The up-regulation of eNOS may partly account for acquisition of ischemic tolerance and neuroprotection by preconditioning.