
Abstract
After a stroke many neurons in the ischemic brain tissue die by a process called apoptosis, a form of cell death that may be preventable. The specific molecular cascades that mediate ischemic neuronal death are not well understood. The authors recently identified prostate apoptosis response-4 (Par-4) as a protein that participates in the death of cultured hippocampal neurons induced by trophic factor withdrawal and exposure to glutamate. Here, the authors show that Par-4 levels increase in vulnerable populations of hippocampal and striatal neurons in rats after transient forebrain ischemia; Par-4 levels increased within 6 hours of reperfusion and remained elevated in neurons undergoing apoptosis 3 days later. After transient focal ischemia in mice, Par-4 levels were increased 6 to12 hours after reperfusion in the infarcted cortex and the striatum, and activation of caspase-8 occurred with a similar time course. Par-4 immunoreactivity was localized predominantly in cortical neurons at the border of the infarct area. A Par-4 antisense oligonucleotide protected cultured hippocampal neurons against apoptosis induced by chemical hypoxia and significantly reduced focal ischemic damage in mice. The current data suggest that early up-regulation of Par-4 plays a pivotal role in ischemic neuronal death in animal models of stroke and cardiac arrest.
The profound neurologic disabilities that can result from a focal ischemic stroke or cardiac arrest are the consequences of dysfunction and death of neurons in the ischemic brain tissue (Dirnagl et al., 1999; Lipton, 1999, Mattson et al., 2000a). In focal cerebral ischemia, most of the cells in the ischemic core undergo necrosis, whereas in the border zone of the ischemic area (the penumbra), many neurons exhibit features of apoptosis, including caspase activation, DNA-laddering, and pyknosis (Linnik et al., 1993, 1995; Li et al., 1997, 1998; Velier et al., 1999). In transient global ischemia, specific populations of neurons, such as CA1 neurons in the hippocampus, undergo delayed cell death (Chen et al., 1998; Petito et al., 1997; Zhu et al., 1998). Therapeutic interventions aimed at blocking apoptosis by inhibition of caspases or preservation of mitochondrial function recently were reported to be effective in rescuing brain tissue from ischemic damage (Endres et al., 1997; Fink et al., 1998; Keller et al., 1998; Matsumoto et al., 1999; Schulz et al., 1999; Yu et al., 2000). In addition, some neurons may undergo a mixed form of cell death in which pathways typical of programed cell death occur early and are followed by secondary necrosis because of cellular adenosine triphosphate depletion (Dirnagl et al., 1999; Nicotera et al., 1999).
Prostate apoptosis response-4 (Par-4) is a 38-kDa protein initially identified as the product of a gene upregulated in prostate tumor cells undergoing apoptosis (Sells et al., 1994, 1997). Par-4 contains a leucine zipper in its C-terminus that is essential for the function of Par-4 in apoptosis (Sells et al., 1997; Guo et al., 1998a) and may mediate interactions with proteins known to modulate apoptosis, including protein kinase Cζ, Bcl-2, and components of the NF-κB signaling pathway (Diaz-Meco et al., 1996; Frutos et al., 1999; Camandola and Mattson, 2000). Par-4 is expressed at low levels in many different cell types including neurons (Boghaert et al., 1997; Guo et al., 1998a). Recent findings suggest roles for Par-4 in neuronal apoptosis induced in several experimental models, including trophic factor withdrawal and exposure to oxidative and metabolic insults (Mattson et al., 1999). Moreover, studies of postmortem tissues from patients and animal models of neurodegenerative disorders, including Alzheimer's, Parkinson's, and Huntington's diseases, amyotrophic lateral sclerosis, and HIV encephalitis, have documented increased levels of Par-4 in vulnerable neurons (Guo et al., 1998a; Duan et al., 1999a; Kruman et al., 1999; Pedersen et al., 2000). Par-4, which can be induced at the translational level, acts at an early stage of the apoptotic cascade before mitochondrial dysfunction and caspase-3 activation (Chan et al., 1999; Duan et al., 1999b). It is not known whether Par-4 participates in ischemic brain injury. The authors now report that Par-4 levels increase in vulnerable neurons before their death in two different rodent models of ischemic brain injury and further show that ischemic neuronal death is reduced by treatment with a Par-4 antisense oligonucleotide, suggesting an essential role for Par-4 in the cell death process.
MATERIALS AND METHODS
Transient forebrain ischemia in rats
Forebrain ischemia was performed in male Wistar rats (250 to 300 g) as reported previously (Zhu et al., 1998). Briefly, ischemia was induced under halothane/N2O/O2 (1.5%/28.5%/70%) anesthesia by occlusion of both common carotid arteries combined with depression of the mean arterial blood pressure to 40 mm Hg by trimethaphancamsylate (5 mg/kg, intravenously) and central venous exsanguination. After 10 minutes, the artery clips were removed and blood was rapidly reinjected to restore blood pressure. Arterial pH, PaCO2, PaO2, blood pressure, and plasma glucose were monitored throughout the surgical procedure. After surgery, animals were kept at an environmental temperature of 30°C for 2 hours. For immunocytochemistry, rats were perfused with a phosphate-buffered 4% paraformaldehyde solution in deep chloral hydrate anesthesia, and brains were removed and embedded in paraffin. For detection of DNA laddering, DNA was extracted from individual hippocampus and striatum at 1, 2, 3, and 4 days after global ischemia using QIAmp Tissue Kit (Qiagen, Hilden, Germany) as described previously (Zhu et al., 1998). After labeling the extracted DNA using a Dig-oligonucleotide 3′-end labeling kit (Boehringer Mannheim, Germany), electrophoresis was performed at 100 V for 3 hours in a 2% agarose gel, and the DNA was blotted on a nylon membrane. DNA fragments were detected on a chemiluminescence-sensitive film (Amersham, Freiburg, Germany) after incubation of the membrane with an anti-digoxigenin-alkaline phosphatase system and exposure to 1% disodium 3-(4-methoxyspirol1,2-dioxetane-3,2′-(5′-chloro)tricyclo [3.3.1.1(3,7)]decan-4-yl)phenyl phosphate) (Boehringer Mannheim).
Focal cerebral ischemia in mice
Reversible focal cerebral ischemia was performed in C57BL/6 mice by occluding the middle cerebral artery (MCA) for 1 hour with a 5–0 nylon thread that was introduced through the arteria carotis interna as described previously (Bruce-Keller et al., 1996). During surgery, animals were anesthetized with a mixture of xylazine (5 mg/kg) and chloral hydrate (350 mg/kg). Body temperature was regulated throughout surgery and for 2 hours after reperfusion using a heating lamp. Twenty-four hours after reperfusion, the mice were killed by a halothane overdose. Brains were removed, cut into 4 coronal slices (2-mm-thick), and stained with triphenyl-tetrazolium chloride (TTC; Sigma, St. Louis, MO, U.S.A.) at 37°C. The infarcted area remained white whereas surviving brain tissue showed a deep red color after 30 minutes. Brain damage was predominantly detected in the striatum and cortex, and to some extent in the hippocampus of the ipsilateral hemisphere. Infarct volume was calculated after measuring the infarct areas on the coronal brain sections as described previously (Culmsee et al., 1999). Par-4 antisense (AS, 5′-ATAGCCGCCGGTCGCCAT-GTT-3′) and missense (MS, 5′-CCGTGTCTGATCTTCGT-GCGT-3′) oligonucleotides (2 μg in 1 μL) were injected into the right lateral ventricle 5 hours before the onset of ischemia.
Primary rat hippocampal cultures
Hippocampi were removed from the brains of embryonic (day 18) Sprague-Dawley rats, and cells were dissociated by mild trypsination and trituration as described previously (Mattson et al., 1993). Hippocampal cells then were seeded onto polyethylenimine-coated, 35-mm culture dishes (for cell survival analysis) or glass coverslips (for Hoechst 33342 staining). Experimental treatments were performed on 7- to 8-day-old cultures. Par-4 antisense and missense oligonucleotide were added to the cultures at a final concentration of 25 μmol/L.
Neuron survival was quantified by methods described previously (Mattson et al., 1989, 1995). Briefly, viable neurons in pre-marked fields (10× objective) were counted before experimental treatment and 24 and 48 hours after treatment. Neurons that died in the intervals between examination points were usually absent, and the viability of the remaining neurons was assessed by morphologic criteria; neurons with intact neurites of uniform diameter and soma with a smooth round appearance were considered viable, whereas neurons with fragmented neurites and vacuolated cell bodies were considered nonviable. To quantify apoptosis, cells were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS), and membranes were permeabilized with 0.2% Triton X-100 and stained with the fluorescent dye Hoechst 33342 as described previously (Kruman et al., 1997). Hoechst-stained cells were visualized under epifluorescence illumination (340 nm excitation and 510 nm barrier filter) using a 60× oil immersion objective. Neurons with condensed and fragmented DNA were considered apoptotic. Two hundred cells per culture in at least four separate cultures per treatment condition were counted, and the percentage of apoptotic neurons was determined. Analyses were performed without knowledge of the treatment history of the cultures.
Immunhistochemistry and Western blot analyses
Fixed rat or mice brains were postfixed for 24 hours in 4% paraformaldehyde in PBS and then washed in 70% ethanol, paraffin-embedded, and 6-μm-thick sections were cut on a microtome. Sections then were treated for 30 minutes with 0.3% H2O2 and blocked for 1 hour at room temperature with 5% horse serum in PBS containing 0.4% Triton X-100. Sections were incubated with a monoclonal anti-Par-4 antibody (Santa Cruz, Santa Cruz, CA, U.S.A.) overnight at 4°C, washed twice with PBS, and then incubated for 1 hour in the presence of biotinylated secondary antibody at room temperature. After a 30-minute treatment with ABC reagent (Vector Laboratories, Burlingame, CA, U.S.A.), the sections were incubated for 5 minutes in a solution containing 0.1% H2O2, 0.05% diaminobenzidine tetrahydrochloride, 0.025% cobalt chloride, and 0.02% nickel ammonium sulfate; they then were washed extensively with water and air dried. Western blot analysis was performed as described previously (Guo et al., 1998a). Briefly, 100 μg of solubilized proteins was separated by electrophoresis in a polyacrylamide gel, transferred to a nitrocellulose sheet, and immunoreacted with monoclonal anti-Par-4 or anti-caspase-8 antibodies (0.1 μg/mL) overnight at 4°C. The nitrocellulose sheet then was processed using HRP-conjugated secondary anti-mouse antibody followed by a chemiluminescence detection method.
RESULTS
An increase of Par-4 levels precedes neuron cell death in the rat hippocampus and the striatum after transient forebrain ischemia
After 10 minutes of global cerebral ischemia, delayed neuron cell death was observed in the CA1 region of the hippocampus and in the dorsolateral striatum, areas known to be most sensitive to ischemic insults in the rat brain. In the hippocampus, essentially no CA1 neurons had died within 24 hours of ischemia, but 40% of the CA1 neurons were dead at 48 hours, and nearly all of the neurons were dead at 4 days (Fig. 1A). In the striatum, no significant neuronal loss occurred during the first 48 hours after ischemia. However, approximately 60% and 80% of the striatal neurons were dead at 3 and 4 days, respectively (Fig. 1B). In both the hippocampus and striatum, the amount of fragmented DNA was greatest at 3 and 4 days after ischemia and then was decreased at 7 days, presumably because the majority of dead cells had been cleared by that time point (Fig. 1).
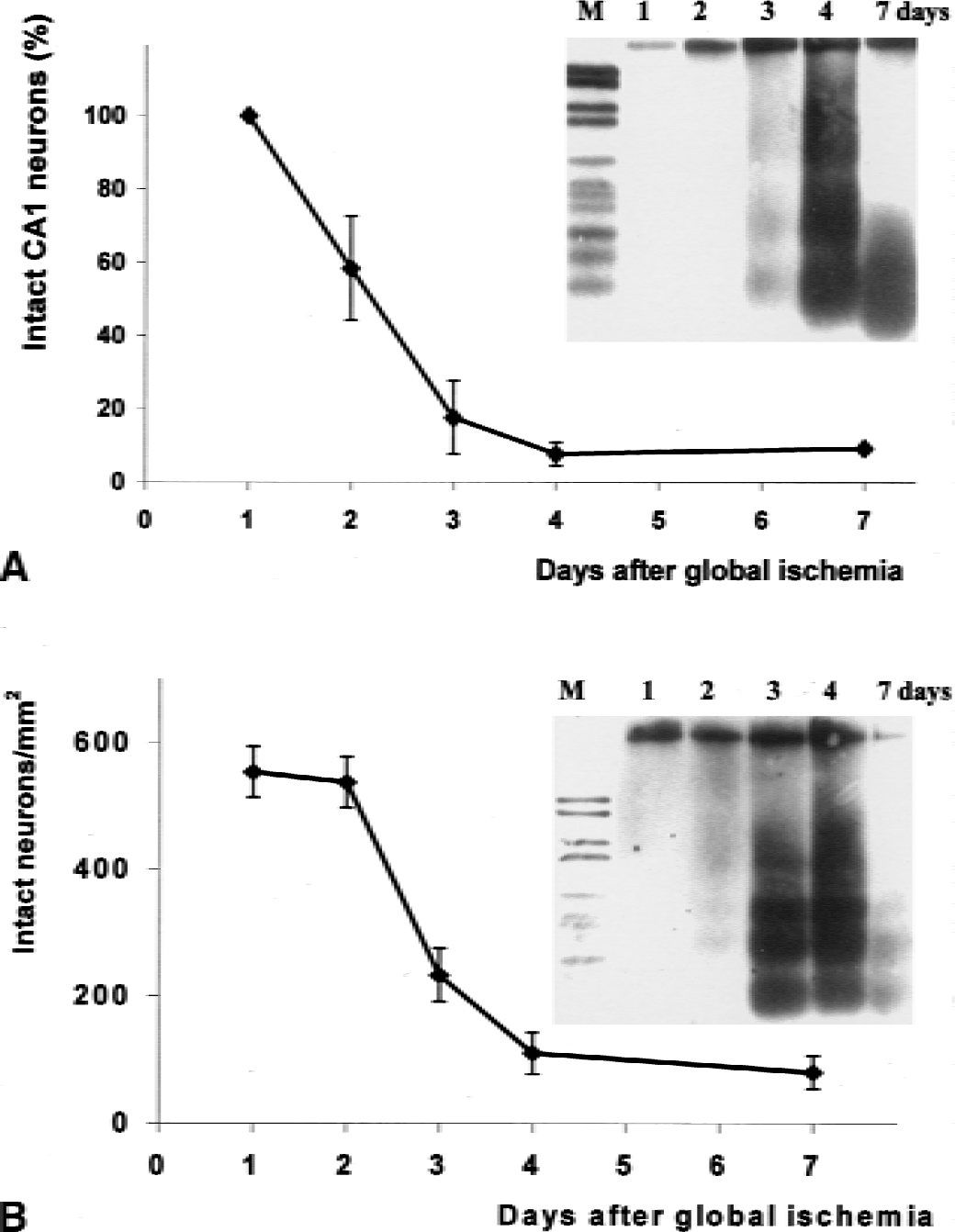
Time course of neuronal damage in CA1 subfield of the hippocampus
Immunohistochemistry performed in brain slices adjacent to those used for analysis of delayed cell death revealed a marked increase in Par-4 levels that was evident within 6 hours after ischemia in the hippocampus and striatum (Figs. 2 and 3). At 24 hours after ischemia, the increase in Par-4 immunoreactivity peaked in the ischemic brain tissue and then decreased thereafter and was restricted to dying cells at the 3 day time point (Figs. 2 and 3). Interestingly, at the peak of the increase of Par-4 levels, strong Par-4 immunoreactivity was present in neurites of CA1 neurons (Fig. 2). In the striatum, the sensitive neurons in the dorsolateral area exhibited increased Par-4 immunoreactivity 1 day after ischemia, and by day 3, Par-4 immunoreactivity was restricted to dying neurons (Fig. 3). The results obtained by immunostaining were confirmed by immunoblot analyses of protein extracts from hippocampus and striatum of animals killed at 6 hours, 1 day, and 3 days after global ischemia (Fig. 4A). A large increase in Par-4 levels was measured at 6 hours after ischemia, in both the hippocampus and striatum, and levels remained elevated at the 1 day time point. At 3 days after ischemia, the increase in Par-4 levels was still detectable, but was diminished compared with day 1, possibly because of the deaths of many of the neurons with high Par-4 levels (Fig. 1). Thus, the increase in Par-4 levels preceded neuron cell death and DNA fragmentation induced by cerebral ischemia, suggesting a role for Par-4 at an early step in the apoptotic cascade.
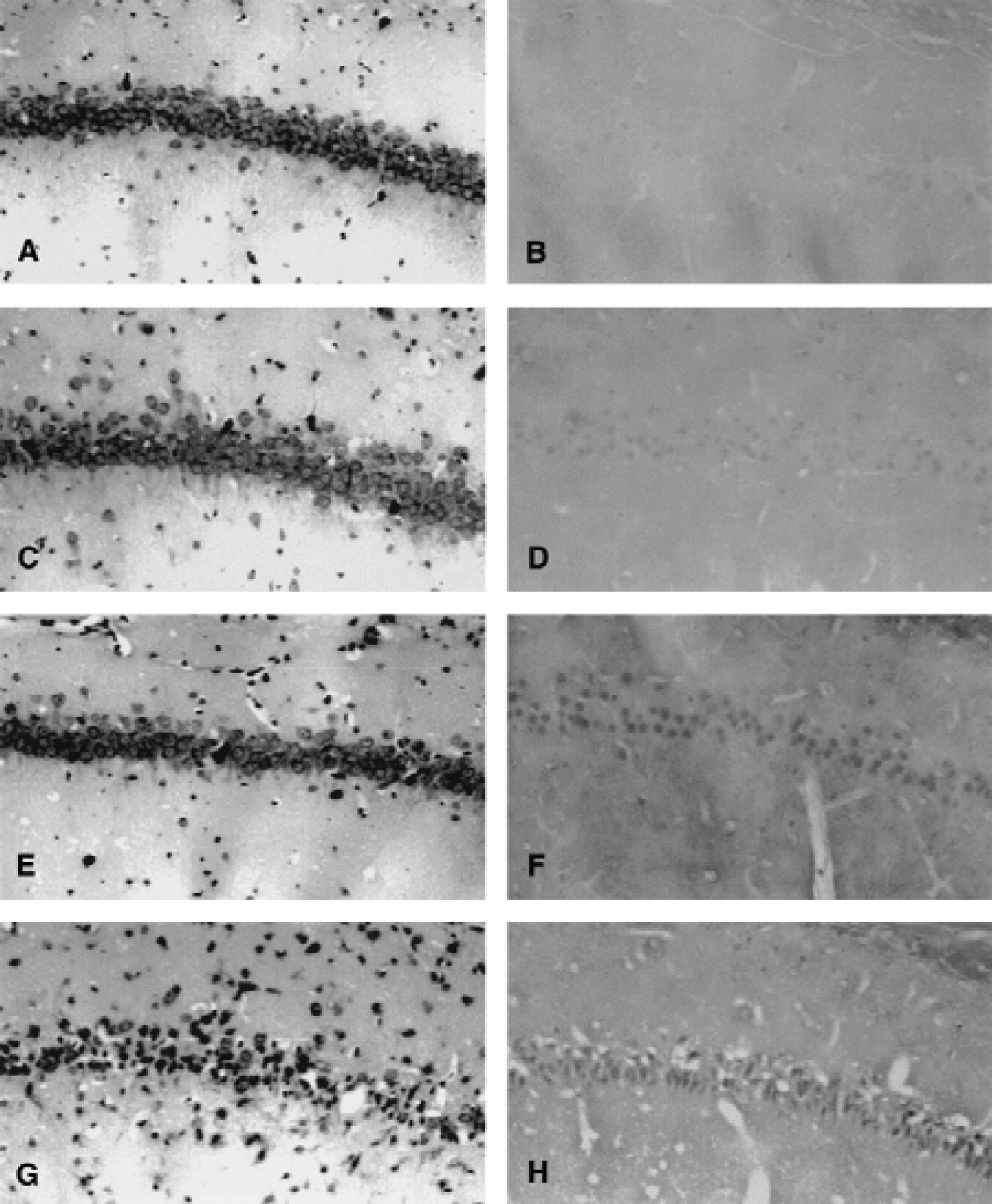
An increase in Par-4 levels precedes neuron cell death in the CA1 region of the rat hippocampus after transient forebrain ischemia. Nissl staining (left panels) and immunostaining for Par-4 (right panels) were performed in paraffin-embedded brain sections of nonischemic control rats
Par-4 levels increase in the striatal cells after global ischemia. Par-4 immunostaining was increased in the striatum of rats at 6 hours
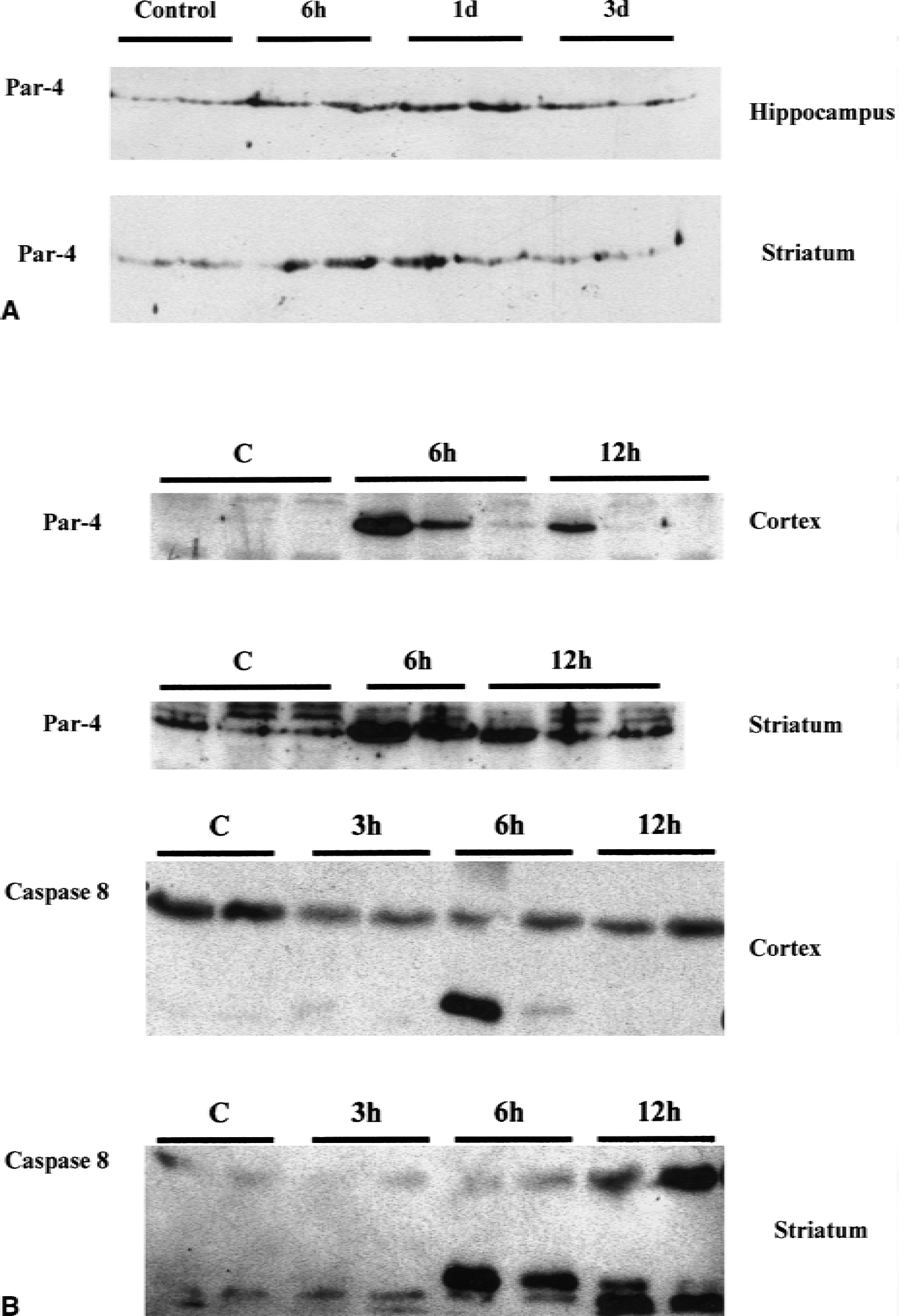
Immunoblot analysis of ischemia-induced Par-4 protein levels.
Par-4 levels increase before neuronal death after focal ischemia–reperfusion
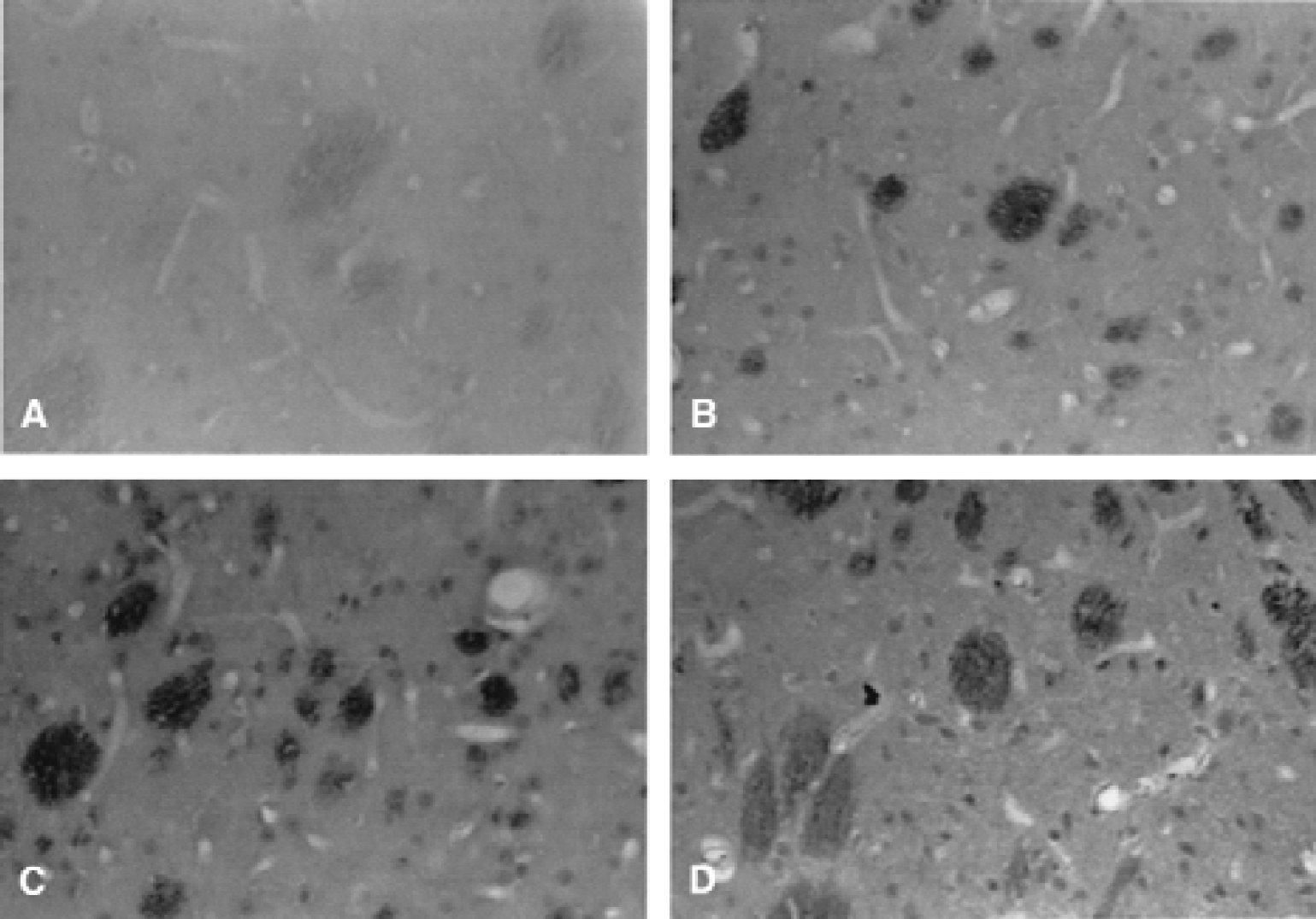
After transient middle cerebral artery occlusion (MCAO), tissue damage in the striatum and cortex was rapidly detectable at 6 to 12 hours after ischemia and an infarct was prominent after 24 hours. To investigate a possible role for Par-4 in the development of the tissue damage, Par-4 protein levels were analyzed at 6 and 12 hours after ischemia. Immunoblots revealed an increase in Par-4 levels in both cortex and striatum at 6 and 12 hours after ischemia, peaking at 6 hours after ischemia (Fig. 4B). The magnitude of the increase in Par-4 levels was variable in the cortex, but more consistent in the striatum, possibly because of interanimal variability in the severity of delayed reactions in the cortical tissue at the periphery of the infarct, whereas striatum is more consistently infarcted in this MCAO model. At a time point 2 hours after ischemia, no increase in Par-4 protein level was detected; whereas in some cases, increased Par-4 levels were sustained up to 24 hours after ischemia (data not shown). Consistent with the apoptotic cascade being underway within 6 hours of reperfusion, the authors found that caspase-8 was activated in bothcortex and striatum at 6 hours as indicated by a marked increase in a caspase-8 cleavage product detected onimmunoblots (Fig. 4B). Immunostaining revealed increased neuronal staining with Par-4 antibody inpyramidal neurons in the ischemic cortex (Fig. 5). In the striatum, Par-4 immunoreactivity was increased in the ischemic hemisphere at 6 and 12 hours after reperfusion, with additional staining of cells in the white matter.
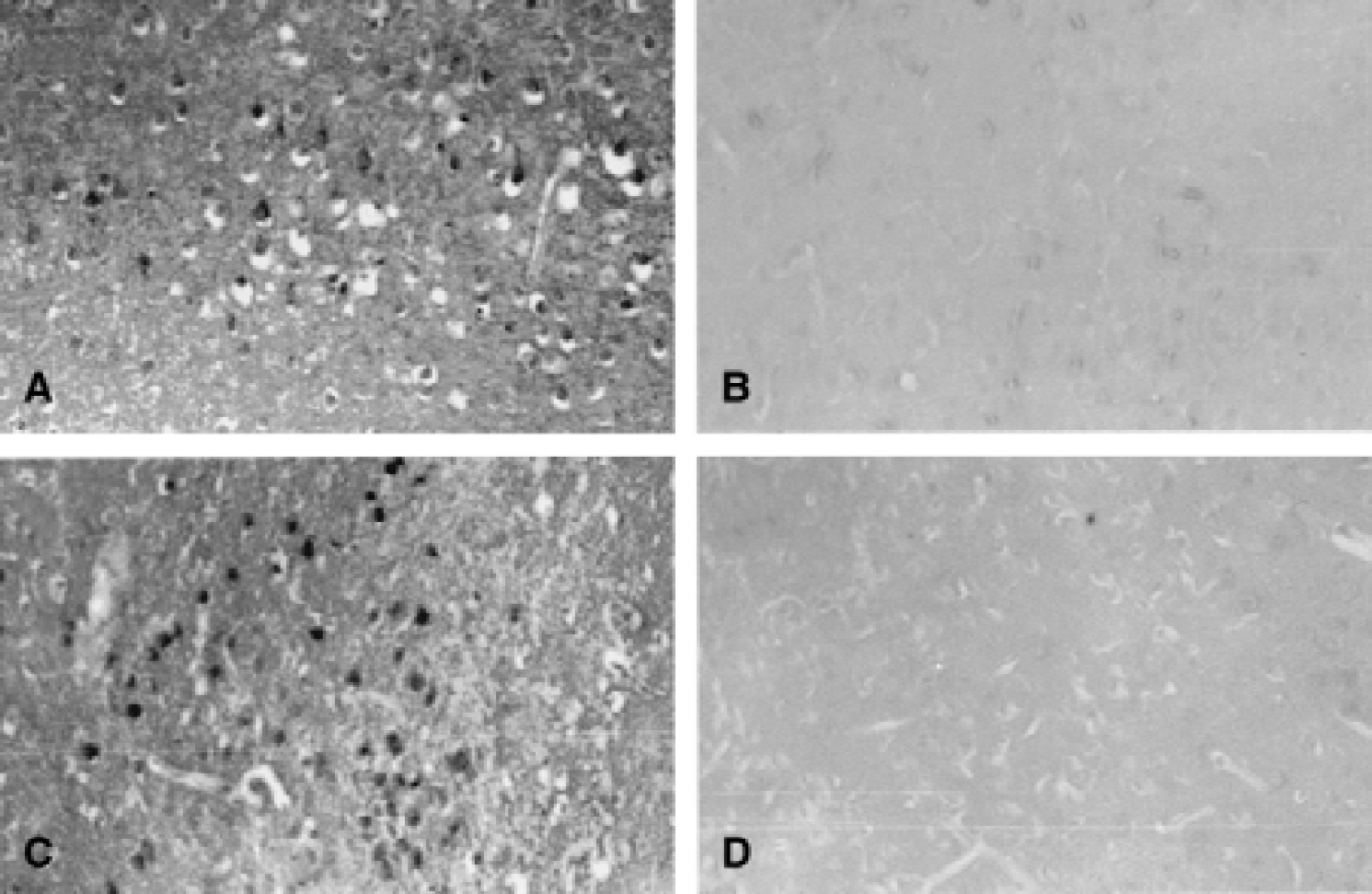
Par-4 levels increase in neurons after focal cerebral ischemia in mice. Par-4 immunohistochemistry was performed on mice brain slices obtained 6 and 12 hours after middle cerebral artery occlusion–reperfusion. Note increased neuronal Par-4 levels in the infarcted area 6
Par-4 antisense blocks apoptosis induced by chemical hypoxia in cultured hippocampal neurons and reduces ischemic brain damage after transient MCAO in mice
To determine whether the increased levels of Par-4 detected in neurons at early time points after transient forebrain ischemia and reversible MCAO contributed to neuronal death, the authors used a Par-4 antisense oligonucleotide that they had previously shown to be effective in suppressing Par-4 production and preventing neuronal death induced by oxidative insults when administered to cultured neurons or through intraventricular infusion in vivo (Guo et al., 1998a; Pedersen et al., 2000; and unpublished data). A cell culture model of ischemic neuronal injury involves exposing embryonic hippocampal neurons to sodium cyanide (NaCN), which induces chemical hypoxia and neuronal apoptosis (Mattson et al., 2000b). The mode of cell death induced by NaCN is concentration-dependent, with low concentrations inducing apoptosis and high concentrations inducing necrosis. In line with earlier findings in PC12 cells (Mills et al., 1996), the authors found apoptosis in cultured hippocampal neurons exposed to 0.1 mmol/L NaCN, whereas 1 mmol/L NaCN resulted in necrosis (Fig. 6). Immunoblot analysis revealed an increase in Par-4 levels 6 and 12 hours after exposure of hippocampal neurons to 0.1 mmol/L NaCN (Fig. 6C). Interestingly, only neuronal death induced by 0.1 mmol/L NaCN was attenuated in cultures treated with the Par-4 antisense oligonucleotide, whereas death induced by 1 mmol/L NaCN was not attenuated significantly (Fig. 6A). The Par-4 antisense oligonucleotide did not affect cell survival under basal culture conditions. These results were confirmed by counting apoptotic nuclei in Hoechst-stained cultures after exposure to 0.1 mmol/L NaCN (Fig. 6B). A control oligonucleotide with the same base composition as the Par-4 antisense oligonucleotide, but with a scrambled sequence, did not protect neurons against NaCN-induced apoptosis. The effectiveness of the Par-4 antisense was established by immunoblot analysis of protein extracts obtained from hippocampal neurons exposed to Par-4 for 6 hours (Fig. 6D).
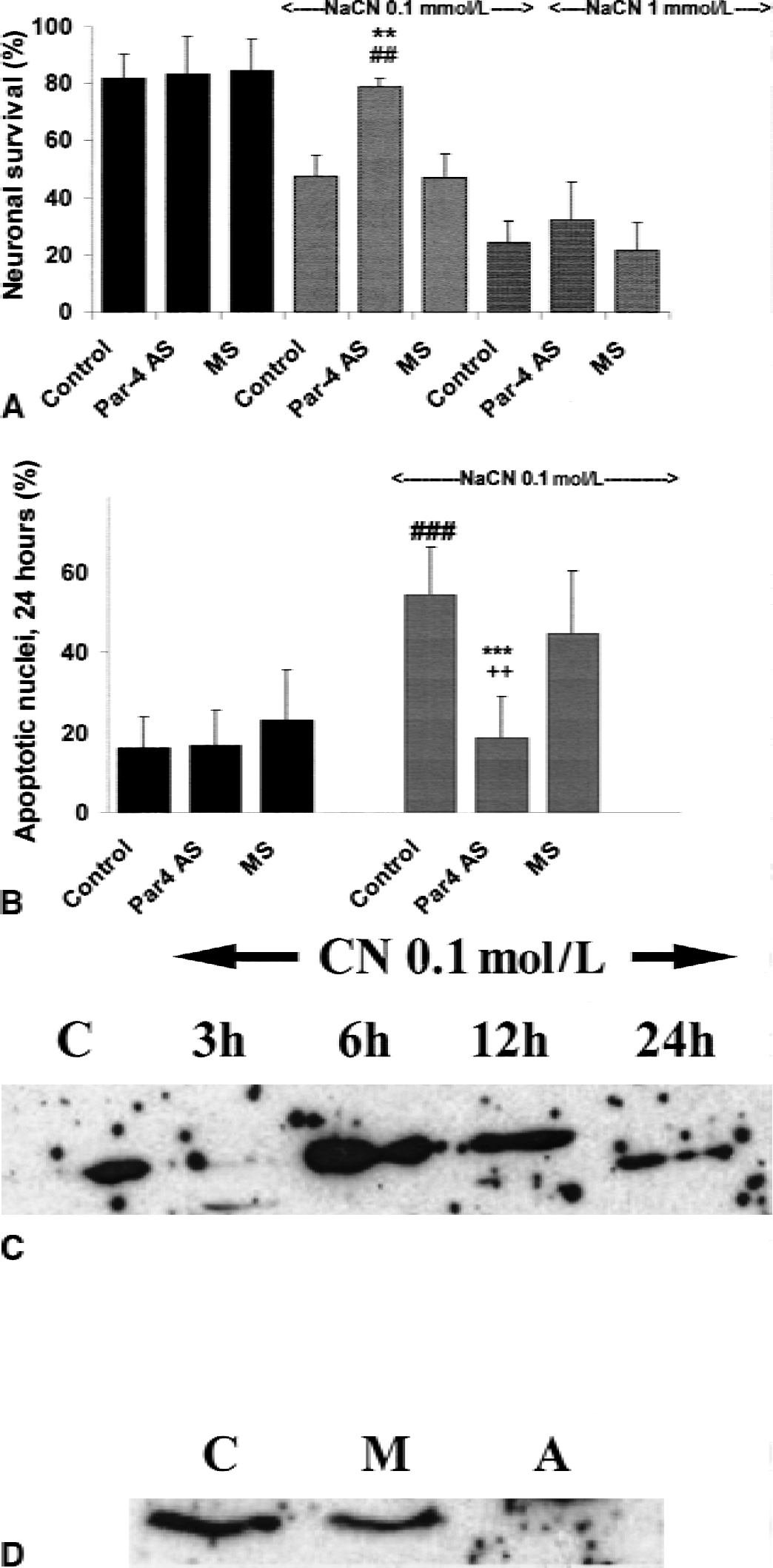
Par-4 mediates apoptosis, but not necrosis, induced by chemical hypoxia in cultured hippocampal neurons. Par-4 antisense (AS) or missense (MS) oligonucleotides (25 μmol/L) were added to cultures of rat embryonic hippocampal cells 4 hours before induction of chemical hypoxia. The Par-4 antisense oligonucleotide blocked neuronal apoptosis induced by 0.1 mmol/L NaCN, whereas necrosis induced by 1 mmol/L NaCN was not affected
The authors next tested whether the Par-4 antisense oligonucleotide could also affect ischemic brain damage in the MCAO model of focal cerebral ischemia. Par-4 antisense oligonucleotide or the control oligonucleotide were injected into the right ventricle of mice 4 hours before MCAO, and mice were killed 24 hours after reperfusion and infarct volume was quantified. There was a significant decrease (30%) in infarct volume in mice given the Par-4 antisense oligonucleotide compared with control groups of mice given either saline or control oligonucleotide (Fig. 7). The effect of Par-4 antisense oligonucleotide was most prominent at the level of the striatum (L2 in Fig. 7A). Immunoblot analysis of protein extracts obtained separately from the striatum and cortex 6 hours after transient focal ischemia revealed an attenuation of Par-4 up-regulation in animals pretreated with antisense oligonucleotides as compared with missense-treated groups (Fig. 7C). The antisense treatment completely blocked induction of Par-4 in the cerebral cortex and dramatically decreased its induction in the striatum. Par-4 up-regulation in the ischemic tissue at 6 hours after reperfusion was not affected by the missense oligonucleotide (Fig. 7C).
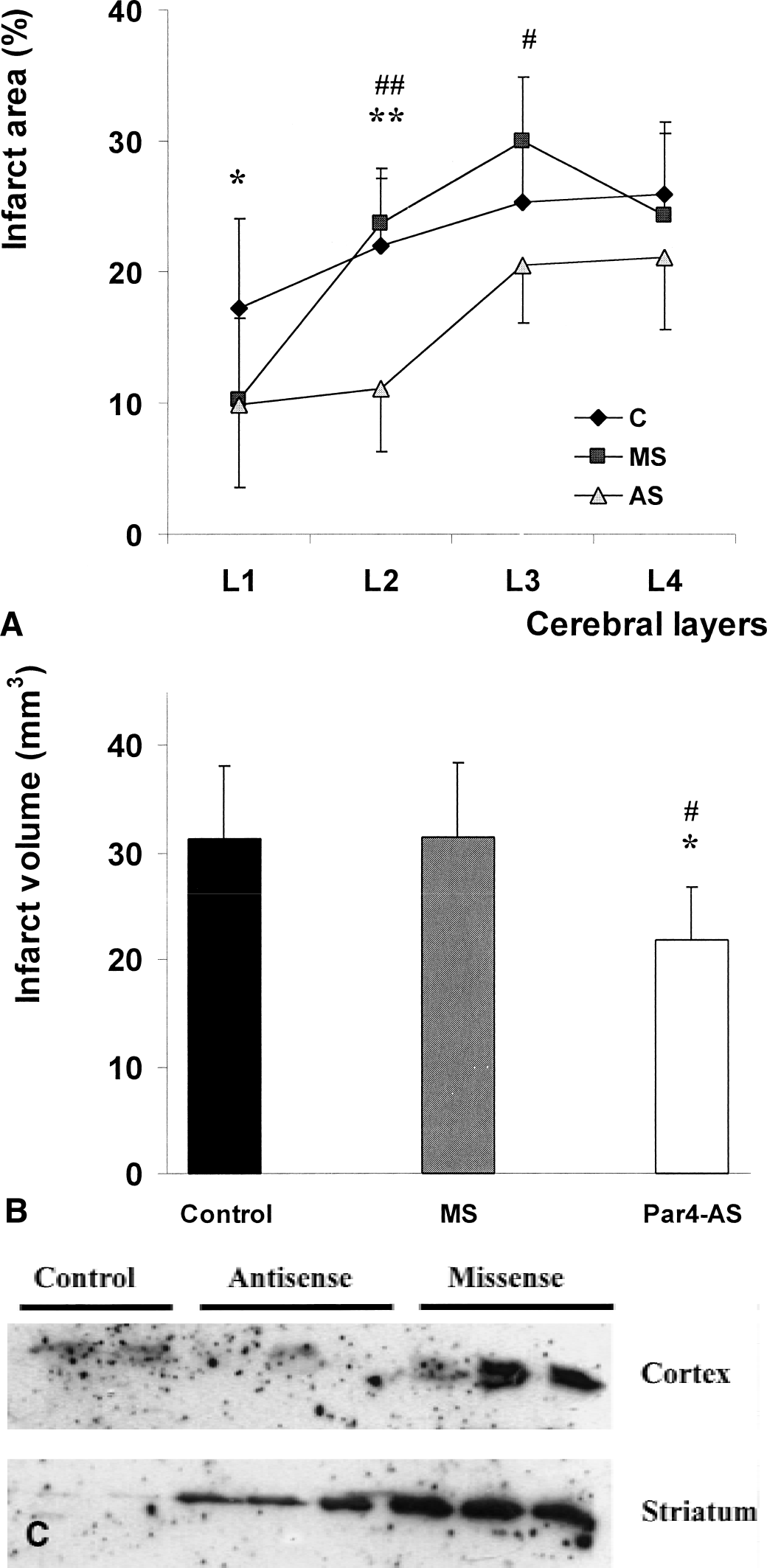
Par-4 antisense oligonucleotide suppresses Par-4 regulation and attenuates focal ischemic brain injury in mice.
DISCUSSION
The current findings obtained in rodent models of stroke and chemical hypoxia in cultured neurons revealed a substantial role for Par-4 in mediating neuron cell death induced by ischemia. Neuronal apoptosis has been described as a major feature of ischemic brain damage in both the transient global forebrain ischemia model (Zhu et al., 1998, 1999) and in MCAO models (Linnik et al., 1995; Li et al., 1998; Yu et al., 2000). The authors' immunohistochemical and immunoblot analyses establish an association between increased levels of Par-4 and subsequent neuronal death in CA1 hippocampal neurons after transient global forebrain ischemia and in cortical and striatal neurons after MCAO-reperfusion. The sustained increase in Par-4 levels in CA1 neurons undergoing apoptosis 3 days after transient global ischemia suggests that the maintenance of elevated Par-4 levels contributes to the death of those neurons. Increases in levels of Par-4 also preceded and accompanied neuronal death in cerebral cortex and striatum after MCAO-reperfusion. The ability of a Par-4 antisense oligonucleotide to protect hippocampal neurons against chemical hypoxia-induced apoptosis in culture and to protect cortical and striatal neurons against focal ischemic injury in vivo strongly suggests that Par-4 production is a critical link in the chain of events leading to ischemic neuronal death. The protective effect of Par-4 antisense in the model of reversible focal ischemia in the mouse was significant, but not dramatic, which may reflect the fact that necrosis is a major cause of neuron death after reperfusion in this model (Li et al., 1997). Recent studies have provided evidence that Par-4 also participates in neuronal death in experimental models of Huntington's, Parkinson's, and Alzheimer's diseases (Guo et al., 1998a; Duan et al., 1999a, 2000). The current findings add cerebral ischemia to the list of neurodegenerative conditions that involve Par-4. When taken together with data suggesting that Par-4 also plays a role in physiologic neuronal death induced by trophic factor withdrawal (Chan et al., 1999), it appears that Par-4 is a conserved component of biochemical death cascades similar to more well-established effectors of apoptosis, such as proapoptotic Bcl-2 family members and caspases (Bergeron and Yuan, 1998; Chan and Mattson, 1999; Marks and Berg, 1999).
Previous data obtained in studies of cultured rat hippocampal neurons suggest a role for Par-4 at an early, premitochondrial stage of neuronal apoptosis (Guo et al., 1998a; Chan et al., 1999; Duan et al., 1999b). The current findings are consistent with involvement of Par-4 early in the cell death process induced by cerebral ischemia in that Par-4 levels increased well before evidence of nuclear DNA cleavage and cell death. It has been shown that Par-4 can promote mitochondrial permeability transition and caspase-3 activation (Chan et al., 1999; Duan et al., 1999b), and the underlying mechanism is beginning to be understood (Fig. 8). Par-4 contains a leucine zipper domain towards its C-terminus that is essential for its proapoptotic activity (Sells et al., 1997; Guo et al., 1998a). Data suggest that Par-4 can bind to and inhibit PKCζ (Diaz-Meco et al., 1996), which may contribute to its death-promoting action, possibly by suppressing activation of the cell survival-promoting transcription factor NF-κB (Camandola and Mattson, 2000). Suppression of NF-κB activity may result in a rapid down-regulation of genes encoding the antiapoptotic proteins Bcl-2, calbindin, and inhibitor of apoptosis proteins (Mattson and Camandola, 2001). Bcl-2 protects mitochondrial membrane potential and integrity by blocking the proapoptotic effect of Bax (Murphy et al., 1996; Myers et al., 1995), calbindin prevents apoptosis by stabilizing cellular calcium homeostasis (Guo et al., 1998b; Wernyj et al., 1999), and inhibitor of apoptosis proteins may act by directly inhibiting caspases (Robertson et al., 2000).
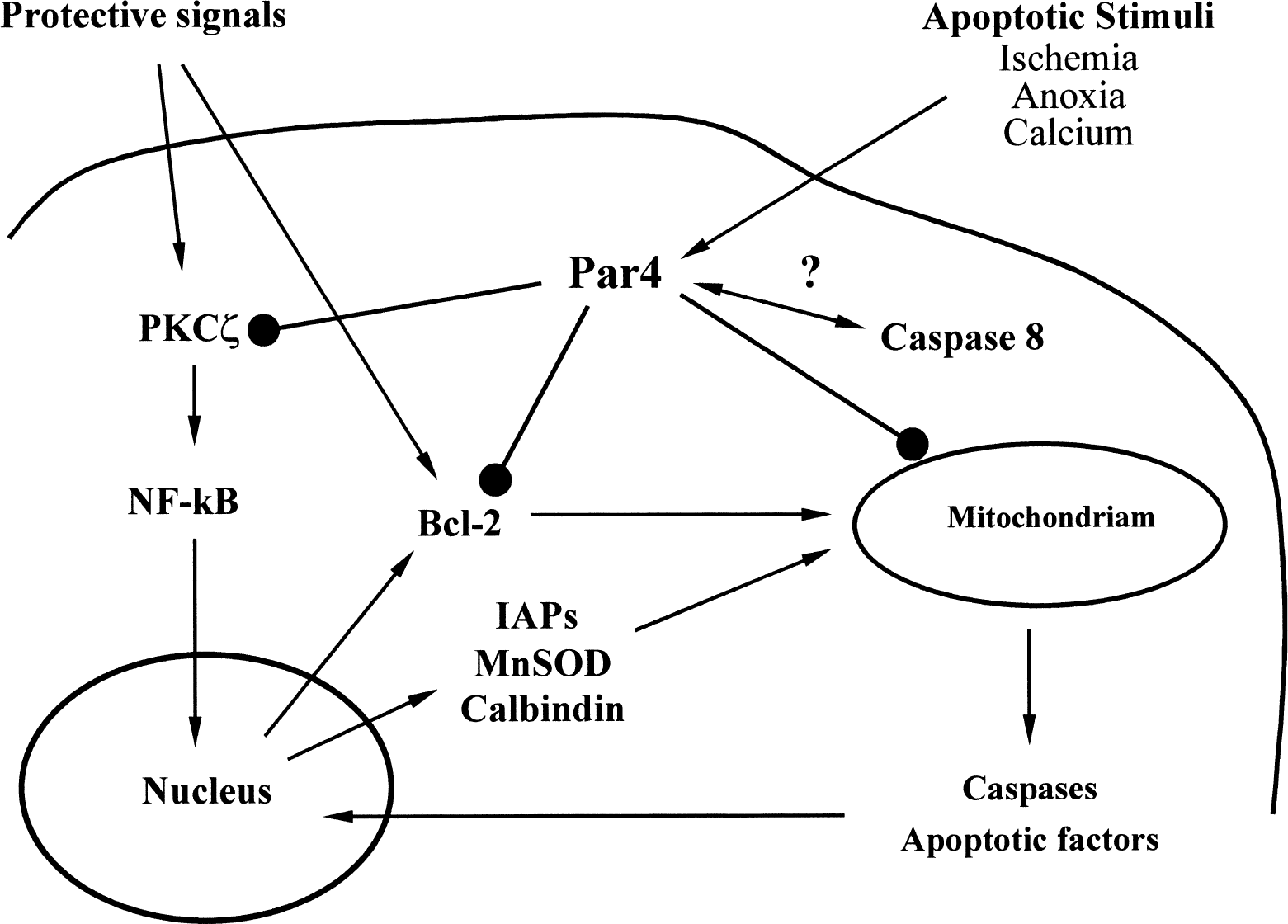
Possible mechanisms underlying the proapoptotic action of Par-4. Par-4 is upregulated by various apoptotic stimuli on translational level upstream the apoptotic cascade that leads to mitochondrial dysfunction, activation of caspases, and DNA damage. Par-4 may act by suppressing PKCζ and NF-κB activities resulting in down-regulation of Bcl-2, inhibitor of apoptosis proteins (IAPs), and calbindin. Par-4 may also interact with caspase-8 at a premitochondrial step in the cell death process.
However, suppression of NF-κB activity is unlikely to be the only mechanism through which Par-4 promotes apoptosis, because studies of synaptosomes have clearly shown that Par-4 can induce mitochondrial dysfunction by a posttranscriptional mechanism (Duan et al., 1999b). Recent studies have established that many of the components of the apoptotic machinery in neurons (including Par-4, Bcl-2 family proteins, and caspases) are present not only in cell bodies, but also in neurites and synaptic terminals (Mattson and Duan, 1999). Previous studies of cultured neurons and synaptosome preparations have shown that Par-4 can be produced locally in dendrites and postsynaptic terminals (Duan et al., 1999b). Immunohistochemical analysis in the current study indicated that Par-4 levels increase in dendrites of CA1 hippocampal neurons after transient global forebrain ischemia, consistent with a role for Par-4 in promoting dendritic degeneration. The authors also observed that caspase-8 cleavage occurred roughly in parallel with increased Par-4 production after focal ischemia. In addition to its leucine zipper domain, Par-4 also contains a domain in its C-terminus that is homologous to the death domain of TNF-receptors, Fas and some caspases, that function in cell death signaling pathways (Sells et al., 1997; Wallach et al., 1999). This suggests the possibility that Par-4 promotes apoptosis by interacting with initiator caspases such as caspase-8, which is an upstream caspase that has been shown to initiate apoptosis by cleaving effector caspases, such as caspase 3 (Peter and Krammer, 1998; Scaffidi et al., 1998; 1999; Liu et al., 2000). After cerebral ischemia, an increased activity of caspase-8 occurs and inhibitors of caspase-8 exhibit potent neuroprotective activities (Felderhoff-Mueser et al., 2000; Velier et al., 1999). Therefore, it will be of considerable interest to understand interactions between Par-4 and caspase-8 during neuronal apoptosis.
In conclusion, the current data suggest that up-regulation of the proapoptotic factor Par-4 participates in neuron cell death induced by cerebral ischemia. Therefore, Par-4 could become a target for therapeutic strategies to prevent neuronal apoptosis induced by acute brain insults.