
Abstract
Apoptosis is the term given to programmed cell death, which has been widely connected to a number of intracranial pathologies including stroke, Alzheimer's disease, and more recently subarachnoid hemorrhage (SAH). Subarachnoid hemorrhage is a disease, without any form of effective treatment, that affects mainly the young and middle aged and as a result is responsible for severe disability in otherwise healthy and productive individuals. Despite intense research efforts in the field, we currently possess a very limited understanding of the underlying mechanisms that result in injury after SAH. However, a number of studies have recently indicated that apoptosis may be a major player in the pathogenesis of secondary brain injury after SAH. As a result, the apoptotic cascades present a number of potential therapeutic opportunities that may ameliorate secondary brain injury after SAH. Experimental data suggest that these cascades occur very early after the initial insult and may be related directly to physiologic sequela commonly associated with SAH. It is imperative, therefore, to obtain a thorough understanding of the early events that occur after SAH, which will enable future therapies to be developed.
Introduction
Spontaneous subarachnoid hemorrhage (SAH) is an important cause of premature death and disability worldwide. Even though SAH accounts for only 5% of all strokes, it has been estimated in autopsy studies that up to 6% of the population may harbor an intracranial aneurysm (McCormick and Nofzinger, 1965) and that each year approximately 10/ 100,000 people suffer from an aneurysmal SAH (Schievink et al, 2004). Subarachnoid hemorrhage, therefore, is a devastating disease carrying with it a mortality of 12% before receiving medical attention (Schievink, 1997). With a further 40% dying within a month of admission to hospital and a 30% morbidity in the survivors, the severity of this disease cannot be underestimated (Sekhar et al, 1988; Grosset et al, 1992; Sobey and Faraci, 1998; Kaptain et al, 2000).
Despite major advances in surgical techniques, radiology, and anesthesiology, the mortality and morbidity rates after spontaneous SAH have not changed in recent years (Schievink et al, 2004). In the past, research has concentrated primarily on vasospasm and its sequela, in an attempt to combat the high morbidity and mortality associated with SAH. To date, this has not resulted in a definitive treatment modality to prevent or ameliorate brain injury after SAH. Furthermore, it should be pointed out that early brain injury (EBI) is the primary cause of mortality in SAH patients (Broderick et al, 1994; Bederson et al, 1995). Therefore, it seems that EBI should be considered as a primary target for future research.
In this review, we intend to concentrate on EBI and the immediate pathophysiological events that occur after an SAH. We also intend to discuss the role of apoptosis in relation to SAH and the effects that these mechanisms have on the brain in the first 72 h after an SAH. We will also discuss the central role that apoptosis plays in relation to secondary brain injury in SAH. As a whole, apoptosis has been well described in the past and there are many fine reviews available (Vaux and Strasser, 1996; Cohen, 1997; Hetts, 1998; Thornberry and Lazebnik, 1998; Earnshaw et al, 1999; Reed, 2000; Strasser et al, 2000; Zamzami and Kroemer, 2001; Love, 2003). For this review, however, emphasis will be put on the apoptotic cascades as they relate to SAH. For an overall view of the schemata, see Figure 1.
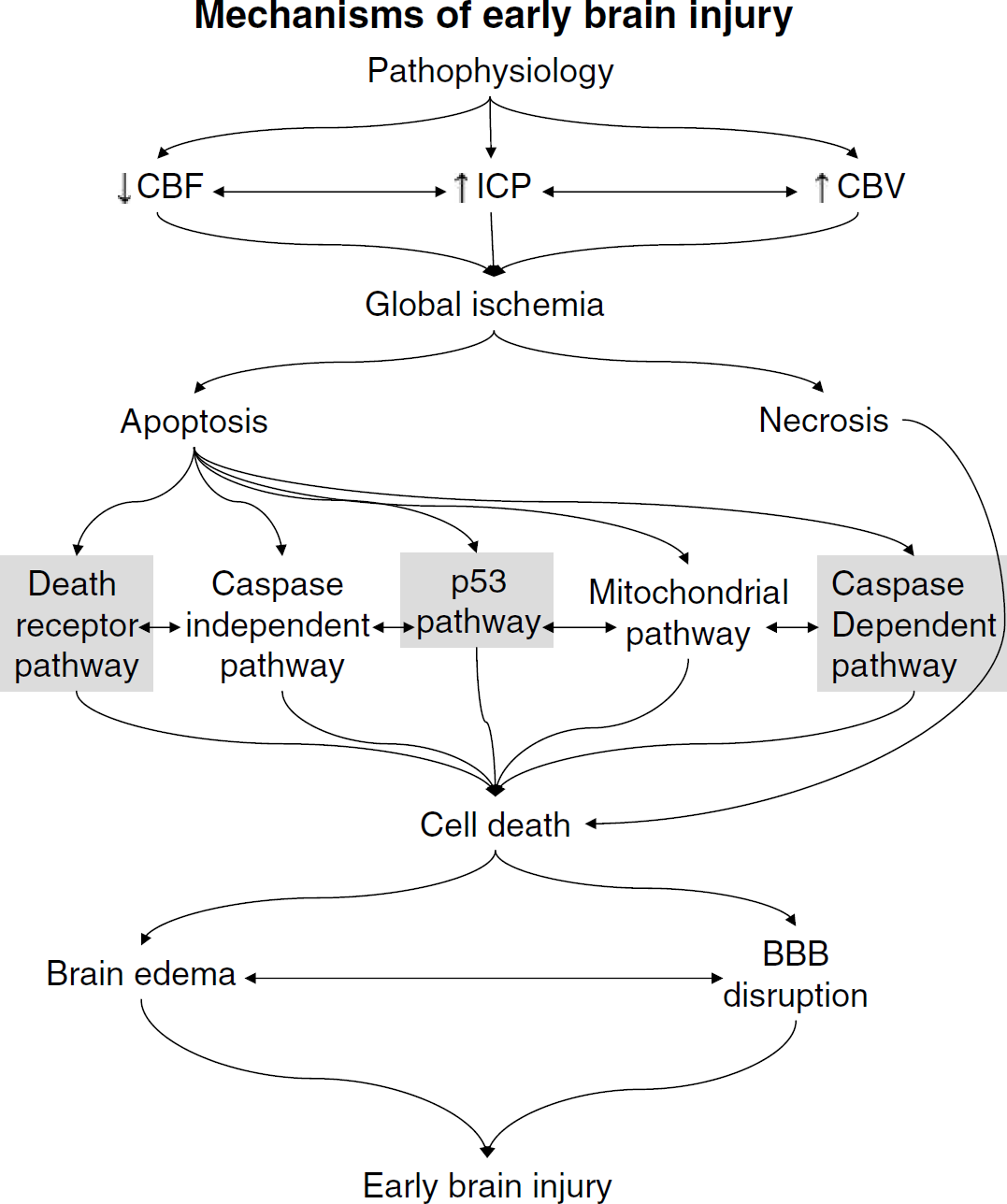
The overall scheme from subarachnoid hemorrhage to EBI. The apoptotic cascades highlighted are those that are known to be involved in SAH-induced apoptosis (Gules et al, 2003; Park et al, 2004; Zhou et al, 2005).
Early Brain Injury
To date, the majority of research performed world-wide has focused on vasospasm and has resulted in a cornucopia of theories (Sako et al, 1993; Cook, 1995; Butler WE, 1996; Macdonald et al, 1997; Barbosa et al, 2001; Zubkov et al, 2001; Macomson et al, 2002; Sviri et al, 2003; Hansen-Schwartz et al, 2003; Borel et al, 2003; Sasaki et al, 2004; Macdonald et al, 2004; Zhou et al, 2005; Badjatia et al, 2005; Kessler et al, 2005). Although some are more widely accepted than others, the full mechanisms of action have yet to be elucidated. One area that has evolved as a result of this research is the area of EBI. The term EBI has only recently been coined and refers to the immediate injury to the brain as a whole, within the first 72 h of the ictus, secondary to a SAH (Kusaka et al, 2004). Therefore, EBI refers to the events that occur in the brain before the development of vasospasm, although it can be suggested that the etiology of vasospasm may be linked to that of EBI, because they certainly share many of the same characteristics. The timing of this injury is immediate, although the sequela can be seen in the long term. There is currently substantial interest in EBI and the mechanisms of injury are only just being clarified (Doczi et al, 1986; Glenn et al, 2002; Gules et al, 2003; Kusaka et al, 2004; Park et al, 2004; Claassen et al, 2004; Zhou et al, 2005). Therefore, EBI has great potential for the provision of treatment modalities, immediate and prophylactic, for patients with SAH, which may attenuate some of the devastating secondary injuries that SAH patients are prone to.
Pathophysiological Mechanisms of Early Brain Injury
The etiology of EBI lies with the initial bleed and the complex pathophysiological mechanisms that occur as a result, which predisposes the brain to secondary injury. Although these pathophysiological principles have only been shown in various animal models, they can be closely linked to clinical findings in humans; however, they have not been well studied to date (Claassen et al, 2004). Some evidence from patients with intracranial cerebral pressure (ICP) monitors already in situ at the time of a SAH has emerged, which follows the parameters found in experimental models (Nornes, 1978). At the moment of a SAH, blood is extravasated from a defect in an aneurysm, which under pressure leaks into the subarachnoid space (the bigger the defect, the bigger the blood volume) (Sobey, 2001). It has been shown both in clinical and experimental studies that there is an acute rise in the ICP, the extent of which reflects the severity of the bleed (Voldby and Enevoldsen, 1982) and a resultant decrease in the cerebral perfusion pressure (Fisher, 1975).
The precise mechanism behind this rise in ICP remains unknown, although the increase in volume secondary to the hemorrhage (Monroe-Kellie Hypothesis), vasoparalysis, and CSF drainage obstruction have been implicated (Grote and Hassler, 1988). In combination with the increase in ICP, cerebral blood flow (CBF) decreases (Bederson et al, 1995). The decrease in CBF and concomitant increase in ICP may also be a protective mechanism in an attempt to control blood loss from the aneurysm. Cerebral blood flow can decrease to almost zero in experimental models (Ostrowski et al, 2005). This is reflected by syncope in the clinical setting. Blood pressure also decreases, which again may be a protective mechanism to reduce blood loss, although this can be variable and clinically, by the time patients reach the hospital they tend to be hypertensive in an attempt to maintain cerebral perfusion pressure. Finally, the cerebral blood volume increases, perhaps as a result of vasodilatation.
Ultimately, these pathophysiological factors increase the ICP, which cannot be compensated for once it reaches critical levels. It is a combination of these factors that results in a global ischemic injury. This ischemic injury as a result of SAH can be seen in patients post mortem (Bederson et al, 1995) and is seen as a leading cause of morbidity in patients with SAH (Frykholm et al, 2004). Although, the severity of the SAH is different from patient to patient, they all to a greater or lesser degree experience these changes in intracranial pathophysiology. It has been well established that patients with a World Federation of Neurological Surgeons (WFNS) defined grade one SAH, who do not exhibit clinical or radiologic vasospasm and who do not suffer any obvious complications with regard to surgery or postoperative course, still show long-term psychosocial difficulties (Hutter et al, 1999; Kreiter et al, 2002; Claassen et al, 2004). It has also been estimated that among the survivors of SAH, up to 50% experience some level of cognitive dysfunction in the long term (Bonita and Thomson, 1985; Kreiter et al, 2002). These injuries are not minor and yet are not given the same emphasis as more immediate and obvious disabilities. Up to 50% of SAH survivors never return to their previous employment, which further indicates the often underdiagnosed injuries sustained at the time of the initial bleed (Hutter et al, 1999; Kreiter et al, 2002).
We believe that these subtle changes in behavior, memory, etc. are the result of EBI and represent long-term complications that cannot be explained by vasospasm alone. As a result of the global ischemic injury, secondary to raised ICP and decreased CBF as outlined above, apoptosis has been shown to be widespread in the brain after SAH. This has been shown in both animal models and in patients post mortem (Zubkov et al, 2000a, 2001; Park et al, 2004; Ostrowski et al, 2005; Zhou et al, 2005). Thus, it seems clear that an understanding of the apoptotic cascades in relation to SAH is vital to understand and treat SAH patients in the future.
Apoptosis in Early Brain Injury
Apoptosis was first described by Kerr et al (1972) and is essentially the term given to programmed cell death. It was first shown by Horvitz et al (1994) in the development of Caenorhabditis elegans. Since then, it has been shown to occur in many physiologic and pathologic states. To date, apoptosis has been extensively studied in stroke models and has been shown to be the dominant form of cell death in the penumbra (Li et al, 1997; Watanabe et al, 1999; Love, 2003). It has also been hypothesized that areas of tissue adjacent to dead or dying cells undergo apoptosis in an attempt to prevent overwhelming necrosis and inflammation. Although a core and penumbra cannot be identified per se in the SAH model, the brain as a whole is affected by global ischemia as discussed previously.
Following the global ischemia seen with SAH, apoptosis has been shown to occur in the hippocampus, blood-brain barrier (BBB), and vasculature with varying degrees of necrosis (Zubkov et al, 2001; Park et al, 2004; Ostrowski et al, 2005; Zhou et al, 2005). Apoptosis has been implicated in the development of vasospasm and smooth muscle cell proliferation in spastic arteries (Zubkov et al, 2000a). Apoptosis has even been demonstrated in aneurysms and has been implicated in aneurismal formation and rupture both in humans and in animal models (Kondo et al, 1998; Hara et al, 1998). However, when the injury is global, the degree of apoptosis can be more devastating than the injury itself. There are a number of apoptotic pathways that are believed to play a role in SAH: the death receptor pathway, caspase-dependent and -independent pathways, as well as the mitochondrial pathway (Figure 2).
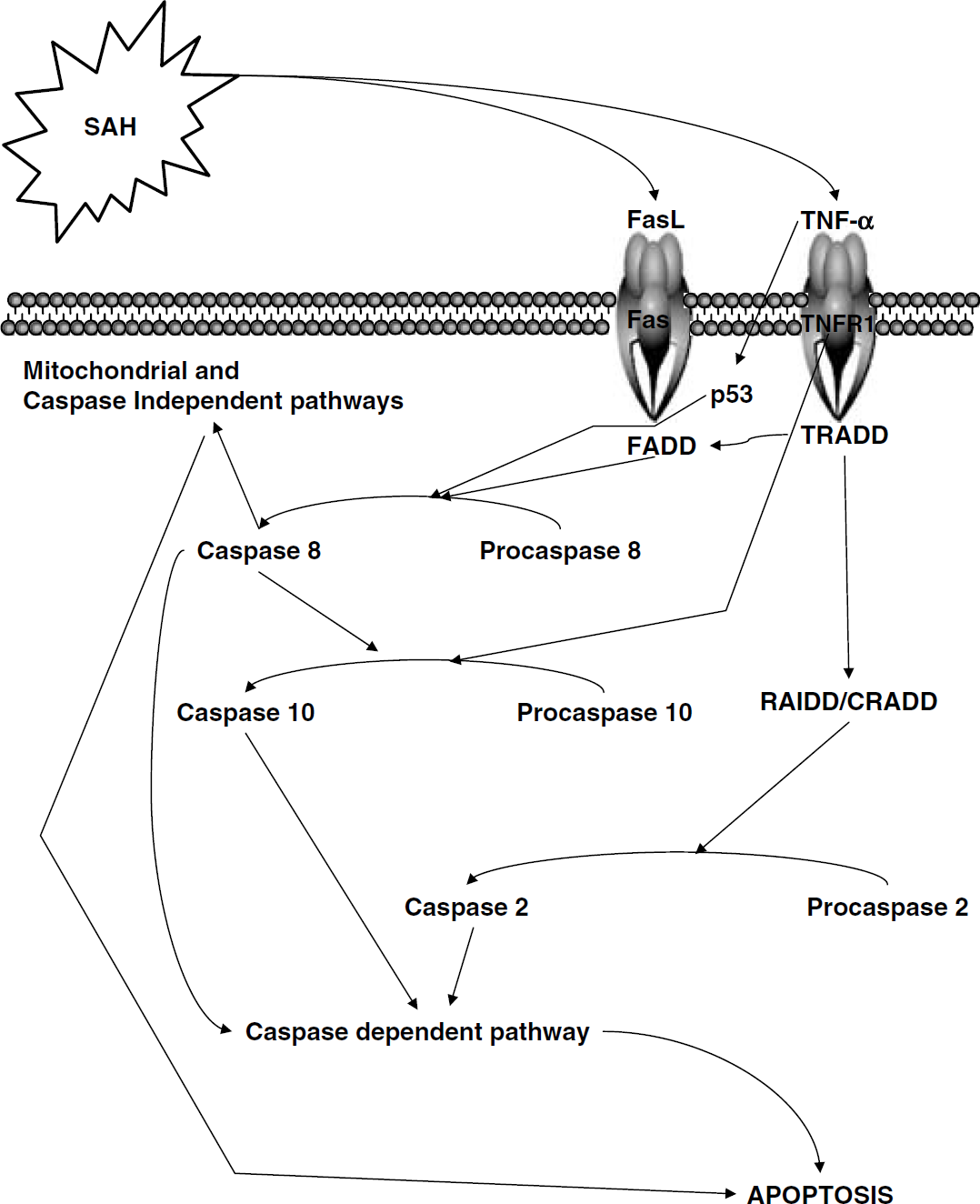
The overall apoptotic cascade in relation to SAH. Subarachnoid hemorrhage is considered to be an external stress event, which by a mechanism yet to be determined affects the death receptors. These in turn stabilize p53 and cleave procaspase 8, leading to apoptosis via the mitochondrial and caspase-independent pathways. Furthermore, the death ligands in conjunction with caspase 8 are also capable of triggering the caspase-dependent pathway leading to apoptosis.
There are many similarities with regard to apoptosis and the many disease processes in which it is involved. To date, apoptosis has been studied extensively in stroke and to a limited degree in SAH. Therefore, the question remains as to what are the differences, if any, between the apoptotic cascades and the pathology. It is known that the initiating event whether it be stroke, SAH, neurotoxins, etc. can influence which apoptotic cascade will become dominant in that disease process, as can the type of cell that is influenced by the event. For example, it has been shown that the caspase-dependent cascade may be particularly important in relation to ischemia, whereas the caspase-independent cascade relates more to neurotoxin-induced apoptosis (Dawson and Dawson, 2004). With regard to SAH, to date, the death receptor/p53 pathway has been described as being particularly important (Zhou et al, 2005). Of course, once initiated, all of the cascades come into play as the relationships between these proteins is extensive and intricately interwoven.
The Death Receptor Pathway in Subarachnoid Hemorrhage
Subarachnoid hemorrhage can be considered to be an external stress event (Matz et al, 2000), which by a mechanism yet to be understood can, possibly through changes in the environment or physical structure of cells, lead to cellular suicide (Kidd, 1998). It has been shown in ischemic and hemorrhagic models that the injury if severe enough can result in DNA fragmentation (Matz et al, 2000, 2001; Matsushita et al, 2000). In the case of SAH, the cell membrane death receptors, for example, Fas, TNFR1, and DR3-5, are believed to be responsible for the translation of the signal across the cell membrane by activating the tumor necrosis factor receptor (TNFR) family, which appears to be the primary target in relation to SAH (Zhou et al, 2004, 2005). The members of the TNFR family are extensive and include tumor necrosis factor-α (TNF-α), TNF-related apoptosis-inducing ligand, and Fas ligand (Zheng et al, 1995; Ju et al, 1995; Kidd, 1998). We have shown in previous experimental investigations that TNF-α is upregulated after SAH (Zhou et al, 2004). It is these cytokines that probably through either an endocrine or paracrine mechanism provide the link between the external stress event and the internal apoptotic cascades. A second theory has also been suggested, which is that of an immune response acting through hematogenous cells resulting in activation of the death receptors (Jiang and Wang, 2004). However, further work is required in this area to determine which if any of these is correct.
All of the TNFR family members contain an intracellular death domain, which permits the transduction of the signal to the intracellular environment. In relation to the transduction of the SAH signal, the two cytokines identified as being the most influential are TNF-α and Fas ligand. Tumor necrosis factor-α activates TNFR1, whereas Fas ligand activates Fas. Tumor necrosis factor-α has been shown to induce p53 in U937 cells and has been identified as a significant downstream mediator of cell apoptosis (Yeung and Lau, 1998). Although experimental evidence exists showing the upregulation of TNF-α and p53 stabilization in SAH (Zhou et al, 2005), there is a paucity of information with regard to events occurring after this. We know from ischemic models that the death receptors are also capable of independently recruiting and activating caspases (Earnshaw et al, 1999). Fas ligand binds to its adapter molecule FADD, which cleaves procaspase 8 to caspase 8 (Muzio et al, 1998). TNFR1 binds to TRADD, which in turn binds to FADD resulting in caspase 8 cleavage as well. TNFR1 is also independently capable of cleaving procaspase 10, resulting in the formation of caspase 10 (Vincenz and Dixit, 1997). Finally, ligated TNFR1 can also bind to RIP and TRADD, which recruits RAIDD and CRADD, which influences procaspase 2 (Bergeron et al, 1998). Caspase 8 in turn activates Bid, which translocates to the mitochondria inducing cytochrome c release (Li et al, 1998). Additional experimental studies examining the effects of pancaspase inhibitors have shown a favorable outcome with regard to SAH, suggesting that p53 may work through either the caspase-dependent or mitochondrial pathways in SAH-induced apoptosis (Park et al, 2004; Zhou et al, 2004). For an excellent review of apoptosis and the death receptors, see Earnshaw et al (1999).
p53—'Guardian of the Genome'
p53 is a nuclear transcription factor that has been exhaustively studied over the past number of decades, particularly with regard to its role in tumorigenesis. Referred to as the ‘guardian of the genome’ (Lane, 1992) and designated ‘molecule of the year', for the year 1998 (Mowat, 1998), it contains an amino-terminal transactivation domain, a carboxy-terminal oligomerization domain, and a central DNA binding domain (Sheikh and Fornace, 2000; for review, see Prives and Hall, 1999). p53-mediated apoptosis has been identified in a host of intracranial pathologies, including ischemia, Alzheimer's, Parkinson's, and SAH (Daily et al, 1999; Mattson, 2000; Leker et al, 2004). p53 genes are responsible for the expression of a host of proteins, which can be transcription dependent or independent, depending on the cell type. We will be dealing with those proteins specifically known to influence the apoptotic cascades with regard to SAH (Sheikh and Fornace, 2000; Gules et al, 2003; Love, 2003; Leker et al, 2004; Hammond and Giaccia, 2005; Zhou et al, 2005).
One of the largest target groups of p53 is the BCL-2 family, named after B-cell lymphoma, which contains a multitude of pro- and antiapoptotic genes (for an excellent review, see Antonsson and Martinou, 2000). Some of these include Bax, Bid, Bik, Bak, Bcl-Xs, and Bim (proapoptotic) and Bcl-2, Bcl-X1, and Mcl-1 (antiapoptotic). However, it has been shown that it is the overall ratio of pro- to antiapoptotic signals that finally determines whether or not the cell dies (Philchenkov, 2004). Therefore, one can hypothesize that it may be dependent on the strength of the signal or extent of the injury. That said it is not known how exactly the Bcl-2 family influences apoptosis. One of the most widely accepted theories suggests it is the ability of Bcl-2 to inhibit caspases by binding to Apaf-1 and preventing cytochrome c release, thus preventing apoptosis (Reed, 1997; Hetts, 1998).
p53 is transported from the nucleus to the cytosol of the cell where it remains at very low concentrations, because it is unstable and quickly degraded, largely owing to murine double minute two (MDM2) or, in humans, HDM2 and CREB binding protein transcriptional cofactor/p300 (CBP/p300) (O'Brate and Giannakakou, 2003). The export of p53 from the nucleus is tightly regulated (Ryan et al, 2001) and occurs when MDM2 ubiquitinates p53 in the nucleus, which allows for its exportation from the cell (Stommel et al, 1999). Once in the cytosol, under normal conditions it is quickly degraded. However, the phosphorylation of p53 allows for its stabilization and accumulation within the nucleus, which can occur secondary to many stress events, including SAH (Giaccia and Kastan, 1998). N-terminal phosphorylation occurs at serines 15 and 20 (Shieh et al, 1997; Unger et al, 1999), which prevents MDM2 binding and subsequent export, thereby increasing nuclear concentration and increasing translational capacity (O'Brate and Giannakakou, 2003). Interestingly, the phosphorylation at these two sites typically causes cell cycle arrest and DNA repair, whereas severe DNA damage leads to phosphorylation of serine 46 in addition to serines 15 and 20, which causes apoptosis and cell death (Nakamura, 2004). Again, whether this phenomenon is a result of the strength of the signal or not remains unknown. It has been suggested that the accumulation of p53 in the nucleus leads to the translocation of p53 into the cytoplasm (Sansome et al, 2001; Chipuk et al, 2004; Erster et al, 2004), where it acts either directly or indirectly on the mitochondria resulting in cytochrome c release. The methods by which p53 translocates to the mitochondria is unknown; however, a Grp75-dependent mechanism has been suggested (Mihara et al, 2003). This laboratory has previously shown the presence of p53 in the cytoplasm in a canine double injection model of SAH (Zhou et al, 2005). With regard to SAH, we suspect that cytoplasmic p53 causes apoptosis by acting either directly on the mitochondria or through an intermediate such as Bax. The reason for this lies with the fact that SAH per se does not cause DNA damage, and therefore we speculate that apoptosis secondary to stress is orchestrated by cytosolic p53.
Recent work examining the role of p53 in hypoxic conditions has shown that p53 is stabilized in response to hypoxia (Hammond and Giaccia, 2005). It should be pointed out however that there is some dispute regarding this (Goda et al, 2003). It seems that MDM2 protein levels decrease in response to hypoxia, which of course leads to an accumulation of p53 in the nucleus (Koumenis et al, 2001). It is likely that the dispute lies with the degree of hypoxia experienced, with many authors agreeing that severe hypoxia is required to stabilize p53 (Schmid et al, 2004). This degree of hypoxia may be experienced transiently in SAH perhaps at the time of global ischemia, which may be enough to stabilize p53 (see above). However, the stabilization of p53 is further emphasized by the fact that the apoptotic signal is reinforced by hypoxia-inducible factor-1 (HIF-1), which is upregulated as a result of the global ischemic injury (Ostrowski et al, 2005).
Certainly it seems likely that HIF-1 is involved either directly, as a result of ischemia, or as a result of the SAH itself. Hypoxia-inducible factor-1 has been shown to rise acutely with respect to SAH, ischemia, and intracerebral hemorrhage (Bergeron et al, 2000; Ostrowski et al, 2005). Hypoxia-inducible factor-1 is known to upregulate the proapoptotic genes BNIP3 (Schmid et al, 2004) as well as vascular endothelial growth factor. Hypoxia-inducible factor-1 is also responsible for the pro-gression of many different cascades including that of apoptosis, possibly acting through p53 to upregulate the mitochondrial apoptotic pathway; therefore, it has been theorized that p53 stabilization in ischemic conditions is HIF-1 dependent (An et al, 1998). That said, there is currently considerable confusion in the literature concerning the link between p53 and HIF-1 (Wenger et al, 1998). There is mounting evidence that under certain hypoxic conditions HIF-1 is able to stabilize p53 (Hammond et al, 2002). The exact mechanism of this interaction is unknown; however, recently, the discovery of the Nucleophosmin gene (NPM), which has been found to interact with both HIF-1 and p53 (Li et al, 2004), may explain many of the confounding opinions currently in the literature regarding HIF-1 and p53 interaction and their role in apoptosis. Unfortunately, more work is required in this area to fully elucidate the relationship. This laboratory has shown that both p53 and HIF-1 are upregulated after SAH (Ostrowski et al, 2005; Zhou et al, 2005). To date, however, the interplay between the two, if any, has not been demonstrated.
Currently, it is difficult to foresee an animal model capable of testing many of these theories in relation to the strength of the signal or the severity of the injury. The monofilament puncture model does not allow the operator to consistently cause the same degree of injury and the injection model is not representative of a true SAH. Although attempts to score severity have been made, they have so far failed to be consistent. In the light of these facts, it is difficult to examine the link between severity and apoptosis, at least at the moment.
Apoptosis and the Mitochondria
As mentioned above, p53 activates the mitochondrial pathway through its actions on the Bcl-2 family. The Bcl-2 family are divided into pro- and anti-death members (Gross et al, 1999); examples of the pro-death proteins include Bax, Bid, Puma, Bim, Noxa, and Bak (Jiang and Wang, 2004; for review, see Adams and Cory, 1998). Of these, it is difficult to know which are important for SAH. It is known that Puma, Noxa, Bid, and Bax (Reed, 2000; van Loo et al, 2002) are upregulated by p53 after DNA damage.
It is also known that in neuronal cell death it is the upregulation of Bax that initiates the apoptotic cascade (Cregan et al, 1999). In fact, it has been shown that Bax is required for p53-induced caspase 3 activation in neuronal cell death (Cregan et al, 1999). Similar findings were observed in ischemic murine models subjected to focal ischemia (Benchoua et al, 2001). As a whole, the Bcl-2 family can either stimulate or inhibit cytochrome c release from the mitochondria depending on the dominant signal, that is, pro- or antiapoptotic dominance (Philchenkov, 2004). It is important to note that apoptosis is not an all or none mechanism (Vaux and Strasser, 1996). In fact, in situations of sublethal injury, an apoptotic cell can recover and necrotic cells have been shown to possess the ability to switch to apoptosis in certain conditions (Vaux and Strasser, 1996). In addition, p53 cleaves procaspase to form caspase 8, which in turn cleaves Bid to form truncated Bid. Truncated Bid then permits the release of cytochrome c from the mitochondria, which is further regulated by Bcl-2 and Bcl-xL (van Loo et al, 2002).
Cytochrome c is a transcription protein located in the mitochondrial intermembranous space. During apoptosis, this membrane becomes permeable to cytochrome c, possibly through pore formation or membrane destruction (Vander Heiden et al, 1997, 1999; for review, see Zamzami and Kroemer, 2001). As a result, cytochrome c is released into the cytosol where it binds to Apaf-1 (Yakovlev et al, 2004). The cytochrome c/Apaf-1 complex referred to as the apoptosome then recruits and cleaves procaspase 9, which activates the downstream caspase cascade (Nijhawan et al, 2000) (see Figure 3). The critical step in this process is that of cytochrome c release, which is mediated by the Bcl-2 family, which in turn is controlled by p53 (Yakovlev et al, 2004). Caspase is a prerequisite for the cleavage of procaspase 3 to caspase 3, which is known to be involved in p53-mediated apoptosis (Cregan et al, 1999). Interestingly, the intrinsic pathway (mitochondrial pathway) is energy dependent and probably only occurs in areas where ATP is available, for example the penumbra (Benchoua et al, 2001). In areas where energy is not available, that is, ATP-depleted areas, the extrinsic pathway, that is, caspase 8, which is capable of self-cleavage, with direct activation of caspase 3, occurs. Therefore, in SAH brains, either of these cascades can occur depending on the severity of the insult and the area of the brain being examined. For example, the hippocampal cells are far more prone to injury than other areas because of their sensitivity to ischemia and ATP requirements (Park et al, 2004). Similarly, it has also been shown that cells undergoing either necrosis or apoptosis are capable of switching depending on the energy reserves available (Leist et al, 1997; Eguchi et al, 1997). It has been shown that the translocation of cytochrome c from the mitochondria can be arrested through the inhibition of p53. Similarly, caspase 8 was also shown to decrease in experimental models of SAH-induced apoptosis after the prevention of p53 stabilization in the cytosol, suggesting that the caspase-dependent pathway and mitochondrial release of cytochrome c are important in SAH (unpublished data). Further work in this area is required to elucidate the importance of these pathways in SAH.
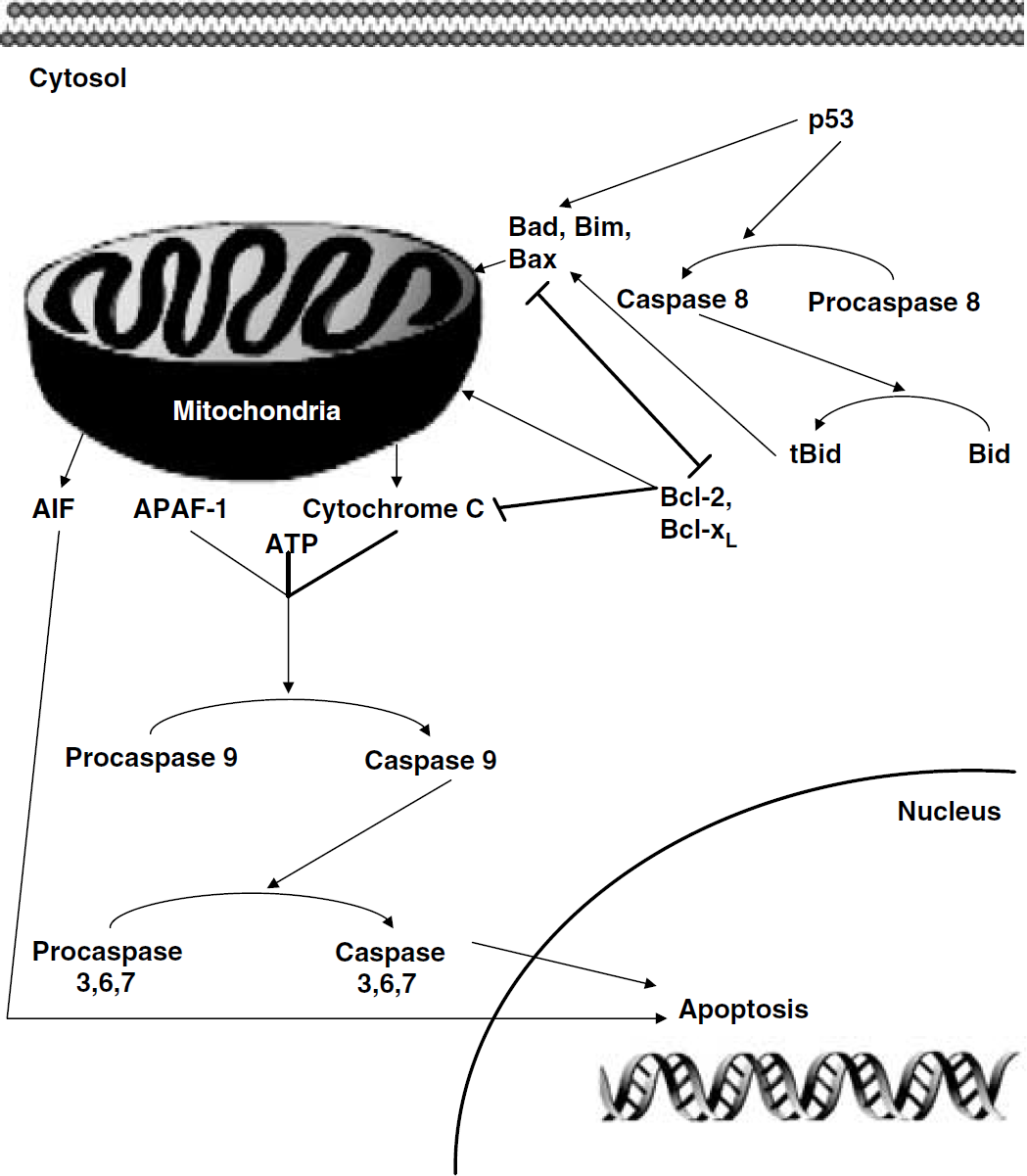
The mitochondrial and caspase-independent pathways. p53 cleaves procaspase 8 to form caspase 8, which cleaves Bid to form truncated Bid. Bax is required for p53 stimulation of apoptosis and truncated Bid is instrumental in the release of cytochrome c, which is under the control of the Bcl-2 family of proteins. Depending on the overall concentration of pro- versus antiapoptotic members of the Bcl-2 family, cytochrome c is released. Cytochrome c along with Apaf-1 and ATP cleaves the apoptosome, resulting in the activation of the caspase cascade. In addition, the caspase-independent cascade is represented here, as AIF can directly induce apoptosis. Other proteins in the independent cascade such as SMAC/DIABLO and endonuclease G are not represented.
As a result of the ischemic insult induced by SAH, we have shown that apoptosis occurs in the endothelial cells of vessels. Experimentally, caspase 3 can be inhibited, which ameliorates the degree of apoptosis. Of interest however is that the prevention of apoptosis in the cerebral vasculature also attenuated the degree of vasospasm (Zhou et al, 2004).
Endothelial cell death may result in thrombus formation, induce cell proliferation as well as migration (McGirt et al, 2002; Borel et al, 2003; Zhou et al, 2004), in both penetrating and major cerebral vessels (Zubkov et al, 2000b, 2002b). Furthermore, apoptosis was identified in a patient who died from an SAH (Zubkov et al, 2000a). For a full review of the activation of the caspase cascades and mechanisms of action, see Thornberry and Lazebnik (1998).
Caspase-Independent Pathways
As discussed above, p53 seems central to the apoptotic cascades in SAH. Recently, a new role for p53 has been found in the caspase-independent cascade (Cregan et al, 2002). In many experimental models of stroke and SAH, inhibition of caspases has been shown to afford some protection; however, apoptosis still occurs (Zhan et al, 2001; Park et al, 2004; Zhou et al, 2004). Therefore, it seems clear that another caspase-independent cascade may be involved. Apoptosis-inducing factor (AIF) is a mitochondrial intramembranous flavoprotein that has been shown to be released from the mitochondria and translocate to the nucleus in response to various death signals (Daugas et al, 2000; Cregan et al, 2002). p53 has been shown to trigger the release of AIF in the absence of Apaf-1, resulting in a caspase-independent apoptotic cascade (Cregan et al, 2002).
Interestingly, in a similar way to cytochrome c, AIF appears to be under the control of the Bcl-2 family and in fact the release of both AIF and cytochrome c is inhibited if Bcl-2 members are blocked, suggesting that the Bcl-2 family may be solely responsible for the caspase-dependent and -independent cascades (Xiang et al, 1996; Cregan et al, 2002). The Bcl-2 family is also responsible for the inhibition of second mitochondria-derived activator of caspase/direct IAP (inhibitor of apoptosis protein) binding protein with low pI (Smac/Diablo) (Du et al, 2000; Verhagen et al, 2000), yet another mitochondrial protein similar to cytochrome c, which depresses procaspase-9 through the inhibition of inhibitor of apoptosis protein (Adrain et al, 2001). This makes the Bcl-2 family a powerful target for future therapeutic intervention. The role of Smac/Diablo in SAH has not to date been identified nor has that of endonuclease G, yet another mitochondrial protein.
Consequences of Apoptosis
One of the first complications related to both the pathophysiological aspects of SAH and the apoptotic cascades discussed above is the disruption of the BBB (Germano et al, 2000; Kusaka et al, 2004; Park et al, 2004). It is likely that the immediate pathophysiological upsets manifest themselves as early BBB disruption (Peterson and Cardoso, 1983), whereas late BBB disruption is caused by the apoptotic phenomena (Gules et al, 2003). The evidence for this is sparse, because there is very limited information available from human studies regarding the time course of BBB disruption (Germano et al, 2000). Even in animal models, the time course is dependent on the animal model used (Peterson and Cardoso, 1983; Sasaki et al, 1985; Davis et al, 1986; Doczi et al, 1986; Johshita et al, 1990; Germano et al, 1998). Results from experimental models have found BBB changes ranging in time from 1 h to 6 days. However, the overall pattern appears to be a biphasic response of the BBB to SAH in the short and long term (Doczi, 1985). Although one can suggest tentatively that a similar biphasic effect can be seen in humans, it is far from categoric.
Damage to the endothelial cells as well as leading to BBB disruption, may also lead to a decrease in the production of endothelial-dependent relaxing factors, and this has been speculated to aggravate vasospasm locally, if not generally (Kassell et al, 1985; Zhang et al, 1998). This is further aggravated by denuding the vessels of endothelial cells, which exposes the vessel to a host of vasoactive and toxic metabolites, which can also aggravate vasospasm (Miranda et al, 1996; Zhou et al, 2004). Clinically, it is probably a combination of these factors and others that results in BBB disruption and vasospasm. The destruction of the BBB and subsequent edema have been implicated as one of the major predicators of cognitive dysfunction in the long run after SAH (Kreiter et al, 2002).
Brain Edema
Brain edema is a major component of EBI as a direct consequence of the disruption to the BBB (Laszlo et al, 1995; Doczi, 2001) and not as a result of vasospasm (Claassen et al, 2002). Although brain edema secondary to SAH has been largely ignored in the literature, Classen and colleagues showed that 8% of patients had global cerebral edema detected by CT scan on admission and that an additional 12% developed appreciable edema over the first 6 days (Kassell et al, 1990a, b; Claassen et al, 2002). The destruction of the BBB after a SAH is not well understood although a number of different mechanisms have been proposed as outlined above.
In patients with SAH, classic vasogenic edema has been described, which is a direct result of BBB breakdown and which was also shown in experimental models (Doczi, 1985). However, recently, cytotoxic edema in combination with vasogenic edema has been described using MRI techniques (Sibon et al, 2004; Orakcioglu et al, 2005). The presence of cytotoxic edema indicates the global ischemic injury that occurs at the time of a SAH, as cytotoxic edema occurs largely because of the failure of energy-dependent Na+/K+ pumps. The role of brain edema in relation to EBI has come to the forefront of research using MRI. MRI has been described as a powerful tool for the noninvasive detection of EBI (Busch et al, 1998). The use of apparent diffusion gradients demonstrates cellular swelling after a propagating wave of ischemia, which could be seen spreading throughout the ipsilateral and contralateral hemispheres. Experimental models using these MRI techniques have shown a fall of the apparent diffusion gradient as early as 2 mins after the SAH, confirming a global ischemic insult and demonstrating global cytotoxic edema (Busch et al, 1998).
Therefore, as mentioned above, the first arm of the biphasic response results in immediate brain edema. Through the mechanisms previously described, there is a resultant rise in the ICP, which further reduces CBF, leading to further ischemia (Fukuhara et al, 1998). As a result, there is more damage to the BBB and the apoptotic cascades are initiated, which leads to further breakdown in the BBB, suggesting a biphasic response. It is the disruption of the endothelial cells because of cell death that allows for the acute rise in both cerebral edema and ventricular volumes (Laszlo et al, 1995). Therefore, brain edema contributes to the rise in ICP seen immediately after a SAH (Hayashi et al, 1977). It is also believed to result in acute vasoconstriction, which combined with the edema leads to a further reduction in CBF, resulting in global ischemic injury (Doczi, 2001). The mechanism by which this occurs has not to date been fully elucidated. Unchecked, this cycle will repeat itself, leading to further edema and eventually death.
This laboratory has shown brain edema in SAH models after SAH in the monofilament rat model. We have also shown a reduction in edema and preservation of the BBB with the use of caspase 3 inhibitors (z-VAD-FMK) (Park et al, 2004) and p53 inhibitors (Pifithrin) (Zhou et al, 2005). Furthermore, delayed global edema has been shown to be an independent predictor of death (Claassen et al, 2002).
The psychological problems faced by SAH survivors have been well described in the past (Vilkki et al, 1990, 2004; Jarvis, 2002; Powell et al, 2004; Jarvis and Talbot, 2004). Even patients with a relatively benign SAH and postoperative course describe long-term memory and concentration problems. The etiology behind this phenomenon remains to be clarified. Animal studies using SAH models have demonstrated profound hippocampal neuronal loss within a relatively short period of time. Histologic data confirm over 30% loss in some studies. There have been few if any long-term studies examining memory and learning capacity after SAH. It is believed that the loss of hippocampal neurons occurs secondary to the global ischemia, which occurs at the time of a SAH. This is probably exacerbated by BBB breakdown and brain edema. Although some degree of global ischemia occurs in all patients who have had a SAH, it seems incredulous that grade one patient's who complain of headache only with no syncope have experienced such a degree of ischemia as to lose hippocampal neurons. The hippocampal cell loss can be prevented using various apoptotic inhibitors to some degree, which appears to lessen the long-term neurologic sequelae in animal models at least (Park et al, 2004).
Necrosis and Subarachnoid Hemorrhage
Apoptotic and necrotic pathways often occur simultaneously and in fact it can be difficult to differentiate between the two. In general, apoptosis can be regarded as an energy-dependent process whereas necrosis is not. As a result, whether a cell becomes apoptotic or necrotic is dependent on the strength of the initial injury, which if severe will consume energy quickly, leading to necrosis. However, in SAH, if the initial bleed were severe enough to prevent blood flow to the brain as in a global stroke, it is unlikely that the brain tissue would survive. As a result, necrosis is not a major factor in SAH, unlike stroke. In addition, it is generally believed that necrosis is not a reversible entity, making it an undesirable target for therapeutic intervention.
However, necrosis has been implicated in two major areas, which are the vasculature and the circumventricular regions. Some studies have shown the presence of necrotic cells in the vasculature. Necrotic cells can be found in the major vessels, particularly in the smooth muscle layer, where they are believed to contribute to vasospasm (Hughes and Schianchi, 1978; Ogihara et al, 2001). Cell culture studies have suggested that oxyhemoglobin may play a leading role with regard to necrosis in the smooth muscle layer (Ogihara et al, 2001). However, what is not clear from the available data is whether necrosis in the vessel contributes to vasospasm or is in fact the result of vasospasm, as it has been well established that the energy metabolites within a vessel experiencing vasospasm are significantly diminished (Yoshimoto et al, 1993). Lastly, necrosis has been implicated in brain edema after SAH. It has been shown that necrosis occurs after SAH in circumventricular regions, which contribute to fluid and electrolyte balance (Akpinar et al, 2005). In particular, the subfornicial organ and organum vasculosum lamina terminalis were found to be susceptible. Additional work is needed in this area to fully explore the consequences of these findings.
Conclusions
The future of this topic lies in a complete elucidation of the apoptotic cascades in relation to SAH.
To date, the apoptotic pathways believed to play a major role in SAH-induced apoptosis are the death receptor and TNF-a, p53, and the caspase-dependent cascades. Additional work is required to examine the caspase-independent and mitochondrial cascades in relation to SAH. In addition, several different inhibitors have been used such as caspase 3 and p53 inhibitors (Zubkov et al, 2002a; Park et al, 2004; Zhou et al, 2004, 2005). However, to the best of our knowledge, multiple level inhibitors have not been used. An example of this might be to selectively block the limiting steps of the caspase-independent and -dependent cascades and the Bcl-2 family proteins. This would inhibit all known apoptotic routes. Such an approach would be controversial and fewer numbers of inhibitors would be used initially. It must be remembered that such a degree of inhibition of such an important pathway could be disastrous for the model, as was shown in gene knockout mice, where Apaf-1, caspase 9, and caspase 3 knockouts resulted in severe developmental defects incompatible with life (Kuida et al, 1996, 1998; Yoshida et al, 1998).
All of these pathways however have the same result, which is of course the death of the cell. Cell death after SAH has important implications not only for vasospasm but also for the long-term sequelae of SAH. What is interesting and unexplained is why perimesencephalic bleeds are not associated with similar sequelae. Although many theories exist to explain this phenomenon, it could be suggested that a perimesencephalic hemorrhage is not a stress event severe enough to initiate the apoptotic cascade. Current research activities in this area are focused on the inhibition of various mediators of these cascades to determine if cell survival can be increased and to look at the outcomes in these models. Such inhibitors have had some success and there is certainly potential to inhibit some if not all of these cascades, which may improve patient outcome through their effects on brain tissue as well as the vascular supply.
In summary, it appears that EBI is a combination of physiologic insults to the brain, resulting in global ischemia, BBB breakdown, edema, and cellular death signaling. These changes occur acutely and chronically although after 72 h vasospasm becomes the main protagonist. The consequences of EBI can be seen immediately and in the long term. There are currently few studies examining the role of EBI in the short- and long-term outcome of SAH models. There has recently been renewed interest in cell death mechanisms regarding SAH and as these are innately tied in with SAH, research is continuing in these areas. It is also difficult to extrapolate a lot of this information to humans and this has not been carried out to date. The future of this area of research is clearly to fully elucidate the appropriate pathways, to relate these to the human condition, and finally to design potential pharmaceutical treatment options with single or multiple inhibitors, which may be able to suppress many of the devastating secondary injuries associated with SAH.