
Abstract
Vasodilatory responses to progressive reductions in intravascular pressure or to calcitonin gene-related peptide (CGRP) or cromakalim were determined in rodent middle cerebral arteries (MCAs) before and after treatment with peroxynitrite (ONOO−). Middle cerebral artery diameters in isolated, pressurized MCAs were measured as intravascular pressure was reduced from 100 to 20 mm Hg in 20-mm Hg increments before and after inactive ONOO−, pH-adjusted ONOO−, or 10, 20, or 40 μmol/L ONOO− was added to the bath. In other MCAs, responses to CGRP (1 × 10−9 −5 × 10−8) or cromakalim (3 × 10−8 −8 × 10−7) were measured before and after the addition of 25 μmol/L ONOO−. Inactive ONOO− (n = 6, P = 0.40), pH-adjusted ONOO− (n = 6, P = 0.29), and 10 μmol/L ONOO− (n = 6, P = 0.88) did not reduce vasodilatory responses to reduced intravascular pressure. Middle cerebral arteries treated with 20 (n = 6, P < 0.0001) and 40 (n = 6, P > 0.0001) μmol/L ONOO− constricted significantly when intravascular pressure was reduced. Vasodilatory responses to CGRP or cromakalim were reduced by ONOO− (P > 0.02, n = 6 and P > 0.01, n = 7, respectively). ONOO− had no effect on vasoconstriction in response to serotonin or vasodilation in response to KCl. These studies demonstrate that ONOO− reduces multiple cerebral vasodilatory responses.
Peroxynitrite is formed when nitric oxide (NO) reacts with the superoxide anion radical (·O2−) (Beckman et al., 1990). Superoxide is ubiquitous in the body and brain and as much as 5% total oxygen consumption may produce ·O2− (Beckman and Koppenol, 1996; Imlay and Fridovich, 1991). Superoxide is scavenged by superoxide dismutase (SOD) and, normally, the higher concentrations of SOD and ·O2− favor the reaction between them. However, if NO concentrations increase, such as after cerebral ischemia (Zhang et al., 1995), the faster rate of the reaction between NO and ·O2− will drive the formation of ONOO−.
The peroxynitrite anion (ONOO−) is a powerful oxidizing agent (Crow and Beckman, 1995; Beckman and Koppenol, 1996) that, when protonated, decays to form a highly reactive hydroxyl-type radical. Peroxynitrite in its anionic form is relatively stable, a factor that contributes to its toxicity by allowing it to diffuse farther from its site of formation to attack more distant cellular targets (Beckman, 1991). The hydroxyl radical, in contrast, can diffuse only a fraction (approximately 3 Å) of the typical diameter of a protein molecule (Beckman, 1994), thus making ONOO− the potentially more dangerous oxidant.
Peroxynitrite is capable of hydroxylation, nitration (+N), and nitrosation (or nitrosylation, +NO) reactions (Crow and Beckman, 1995). Phenols react very readily with ONOO−, and the most common phenol in biologic tissues is tyrosine, which forms long-lived nitrotyrosine combinations when exposed to ONOO− (Nakagawa et al., 1990; Crow and Beckman, 1995; Beckman et al., 1994a). Details of the chemistry of NO, ONOO−, and ·O2− are available in several excellent review articles (Ignarro et al., 1987; Beckman et al., 1994b; Crow and Beckman, 1995; Beckman and Koppenol, 1996; Koppenol, 1998; Pryor and Squadrito, 1995).
The effects of ONOO−, in vivo, are somewhat paradoxical. There is evidence (Vinten-Johansen, 2000) that low concentrations of ONOO− (1 to 2 μmol/L) may preserve coronary endothelial cells (Lefer et al., 1997; Nossuli et al., 1998). In addition, ONOO− may serve as a physiologic signaling molecule in the vascular endothelium (Go et al., 1999). In contrast, ONOO− contributes to cytokine-mediated myocardial damage (Kooy et al., 1997; Ferdinandy et al., 2000) and to mitochondrial damage and dysfunction in vascular endothelial and smooth muscle cells (Ballinger et al., 2000). Peroxynitrite reduces vasodilatory responses to prostacyclin and nitroglycerin in isolated, perfused human placental arteries (Kossenjans et al., 2000). In the brain, Nω-nitro-
In the cerebral circulation, ONOO− causes either vasodilation or vasoconstriction. Applied topically, ONOO− produced dose-dependent dilation (1 to 5 μmol/L) in both large and small pial arterioles in cats, in vivo (Wei et al., 1996). In isolated rodent middle cerebral arteries (MCAs), ONOO− produced dose-dependent constriction, except at high concentrations, that caused vasodilation and impaired vasoconstrictor responses to serotonin (Elliott et al., 1998).
The authors provided preliminary evidence that ONOO− reduces cerebral vasodilatory responses in vitro (DeWitt et al., 1999), but the effects of ONOO− on physiologic responses to changes in intravascular pressure or to cerebral vasodilators in isolated vessels have not been reported. To determine the effects of ONOO− on cerebral vasodilatory responses, arterial diameters were measured during progressive reductions in intravascular pressure or during the addition of calcitonin gene-related peptide (CGRP) or cromakalim, an activator of adenosine triphosphate-dependent potassium channels (K
MATERIALS AND METHODS
Adult, male Sprague-Dawley rats (300 to 400 g) were anesthetized with 4% isoflurane and decapitated. Brains were removed and MCAs were harvested and mounted in an arteriograph as previously described (Bryan et al., 1995). Briefly, a section (2 mm) of the artery was mounted in the arteriograph by inserting micropipettes into the lumen at either end and securing the vessel with nylon sutures (10-0). All side branches were tied off. The mounted arterial segments were bathed in a physiologic salt solution (PSS) of the following composition: 130 mmol/L NaCl, 4.7 mmol/L KCl, 1.17 mmol/L MgSO4·7H2O, 5 mmol/L glucose, 1.50 mmol/L CaCl2, 15 mmol/L NaHCO3. When gassed with a mixture of 74% N2, 21% O2, and 5% CO2, this solution has a pH of ≈7.4, a Pao2 of ≈150 mm Hg, and a Paco2 of ≈35 mm Hg. After mounting the arterial segment, the PSS was warmed from room temperature to 37°C and the arterial segments were allowed to equilibrate for 60 minutes, with intravascular pressure set at 50 mm Hg by raising reservoir bottles connected to the micropipettes. Leaks were detected by monitoring intravascular pressure with the stopcocks to the reservoirs closed. A decline in pressure indicated a leak, and vessels with leaks that could not be tied off were excluded from further study.
After 1 hour of stabilization at an intravascular pressure of 50 mm Hg, each vessel was exercised to decrease mechanical hysteresis by changing the pressure between 20 and 40 mm Hg three times, allowing 5 minutes between pressure changes. The intravascular pressure then was increased to 100 mm Hg. Vessels were magnified with an inverted microscope equipped with a video camera and a monitor. Arterial inner diameter was measured using a video scaler calibrated with an optical micrometer. Vasodilatory responses were tested by decreasing intravascular pressure in 20-mm Hg increments with a 10-minute equilibration period at each pressure level before diameter measurements were made.
After the initial myogenic responses were evaluated, intravascular pressure was returned to 100 mm Hg and the MCAs were exposed to one of the ONOO− concentrations described below. Peroxynitrite was added to the PSS, and MCA diameters were measured 30 seconds, 1, 2, and 10 minutes afterwards. Peroxynitrite was added only once, and the concentrations listed below take into account the volume of the arteriograph bath. Fifteen minutes after the addition of ONOO−, vasodilatory responses again were tested by decreasing intravascular pressure in 20-mm Hg increments from 100 mm Hg. After the second measurement of vasodilatory responses to progressive reductions in intravascular pressure, serotonin (10−6 mol/L) and then KCl (15 mmol/L) were added. The Ca2+-containing PSS was then replaced with Ca2+-free PSS containing ethyleneglycoltetraacetic acid (2 mmol/L), and sequential responses to progressive reductions in intravascular pressure were assessed again.
Experimental design
For the studies of the effects of ONOO− on vasodilatory responses to reduced intravascular pressure, rats were randomly assigned to one of the following six groups (n = 6 rats per group):
Time control: two sequential measurements of MCA diameters during progressive reduction in intravascular pressure were performed to determine whether a first test of vasodilatory responses to reduced intravascular pressure impaired responses during a second test approximately 15 minutes later. Inactive ONOO−: MCAs were exposed to 20 μmol/L ONOO− that had been added to the PSS and allowed to remain at room temperature for 2 hours. This caused a decomposition of ONOO− that has been confirmed spectrophotometrically in previous studies (Elliott et al., 1998). pH-adjusted ONOO−: MCAs were exposed to 40 μmol/L ONOO− that had been added to the PSS, which immediately was pH adjusted to 7.46. 10 μmol/L ONOO−: MCAs were exposed to 10 μmol/L ONOO− added to the PSS. 20 μmol/L ONOO−: MCAs were exposed to 20 μmol/L ONOO− added to the PSS.
In studies of the effects of ONOO− on vasodilatory responses to CGRP or cromakalim, rats were assigned to one of the following two groups:
Calcitonin gene-related peptide (n = 6): MCA diameters were measured after the addition of 1 × 10−9, 5 × 10−9, 1 times10−8, or 5 × 10−8 mol/L CGRP. The dose responses to CGRP were measured again after 25 μmol/L ONOO− was added to the bath. In the absence of ONOO−, two successive dose-response curves for the concentrations of CGRP or cromakalim used in the current study were identical (data not shown). Cromakalim (n = 7): MCA diameters were measured after the addition of 3 × 10−8, 8 times10−8, 3 times10−7, or 8 × 10−7 mol/L cromakalim. Dose-responses to cromakalim were measured again after 25 μmol/L ONOO− was added to the bath.
To determine whether the immediate vasoconstrictor effects of the ONOO− solutions were because of increases in pH, in an additional group of MCAs from 6 rats, a small amount of 1 mol/L NaOH was added to the bath and MCA diameters were determined at 30 seconds, 1, 2, and 10 minutes later. To determine the pH and PO2 of the NaOH, inactive ONOO−, and 10, 20, and 40 μmol/L ONOO− solutions during the first 10 minutes of the experiment, NaOH or ONOO− was added to PSS in the arteriograph bath, after which samples were drawn 30 seconds, 2, 5, and 10 minutes later, and pH and PO2 were measured. This sequence was repeated three times (Table 1).
pH and PO2 of the PSS in the arteriograph bath after addition of 10 μmol/L, 20 μmol/L, 40 μmol/L, or pH-adjusted 40 μmol/L or inactive peroxynitrite (ONOO−), or 15 mmol/L NaOH
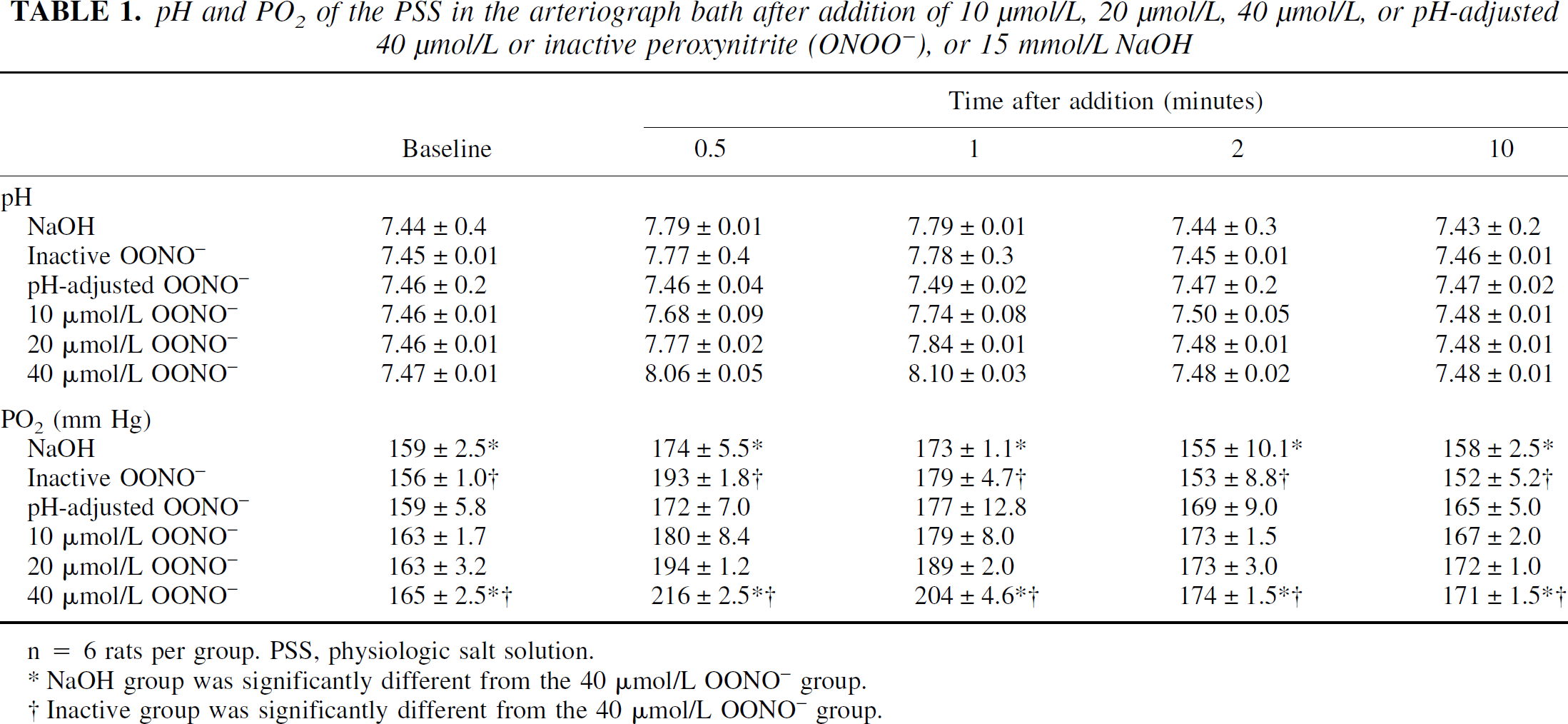
n = 6 rats per group. PSS, physiologic salt solution.
NaOH group was significantly different from the 40 μmol/L OONO− group.
Inactive group was significantly different from the 40 [μmol/L OONO− group.
Statistical analysis
Data were normalized to a percent of baseline diameters and analyzed using analysis of variance for a two-factor experiment with repeated measures on time after ONOO− exposure (0.5, 1, 2, and 10 minutes) or intravascular pressure (100, 80, 60, 40, and 20 mm Hg). The other factor was the 5 treatment groups-that is, pre- or postaddition of inactive ONOO−, pH-adjusted ONOO−, and 10 μmol/L, 20 μmol/L, and 40 μmol/L ONOO−. The studies involving CGRP or cromakalim were analyzed using the concentrations of CGRP or cromakalim and pre- and post-ONOO− treatment as factors for the repeated measures analysis of variance. Post hoc analyzes were performed using the Bonferroni-Dunn procedure for multiple comparisons. All data in the text, tables, and figures are expressed as means ± standard deviation.
RESULTS
Except in the pH-adjusted group, the pH of the PSS in the arteriograph bath increased transiently and then returned to baseline values within 5 minutes of the addition of NaOH, inactive ONOO−, or 10, 20, and 40 μmol/L ONOO− (Table 1). There were significant time effects on pH (P < 0.0001) but there were no significant differences in pH among the groups. The PO2 of the PSS in the arteriograph bath also increased after the addition of NaOH, inactive ONOO−, or 10, 20, and 40 μmol/L ONOO− (Table 1). PO2 increased significantly with time (P < 0.0001) after the addition of the test solutions, and the time versus group interactions were significant also (P < 0.0001). Post hoc testing revealed that PO2 was significantly greater in the 40 μmol/L ONOO− than in the NaOH or inactive ONOO− groups. The elevated PO2 observed after the addition of ONOO− containing solutions is likely because of the autooxidation of ONOO− to NO, NO2, and O2 (Joseph Beckman, personal communication).
In the time control group (n = 6), MCA diameters increased to 119% ± 13% of baseline (100 mm Hg) as intravascular pressure was reduced from 100 to 60 mm Hg and decreased slightly as intravascular pressure was reduced from 60 to 40 (117% ± 12% of baseline) and then to 20 mm Hg (110% ± 12% of baseline) during the first series of measurements (Fig. 1A). In the subsequent series, MCA diameters were 108% ± 19%, 112% ± 19%, 107% ± 12%, and 102% ± 19% of baseline (100 mm Hg) diameters at 80, 60, 40, and 20 mm Hg, respectively (Fig. 1A). In both the first and the second time control measurements, MCA diameters were greater than baseline (that is, the MCAs dilated) even at intravascular pressures of 20 mm Hg. There were no significant differences between the first and the second time control measurements (P = 0.11). Exposure to inactive ONOO−, which was maintained at room temperature for 2 hours after addition to the PSS before adding to the vessel bath, produced no significant (P = 0.405) change in the vasodilation produced by progressive reductions in intravascular pressure (Fig. 1B). Exposure to 40 μmol/L ONOO−, which was adjusted to a pH of 7.46 immediately after addition to the arteriograph bath, did not significantly (P = 0.29) reduce vasodilation in response to reduced intravascular pressure (Fig. 1C).
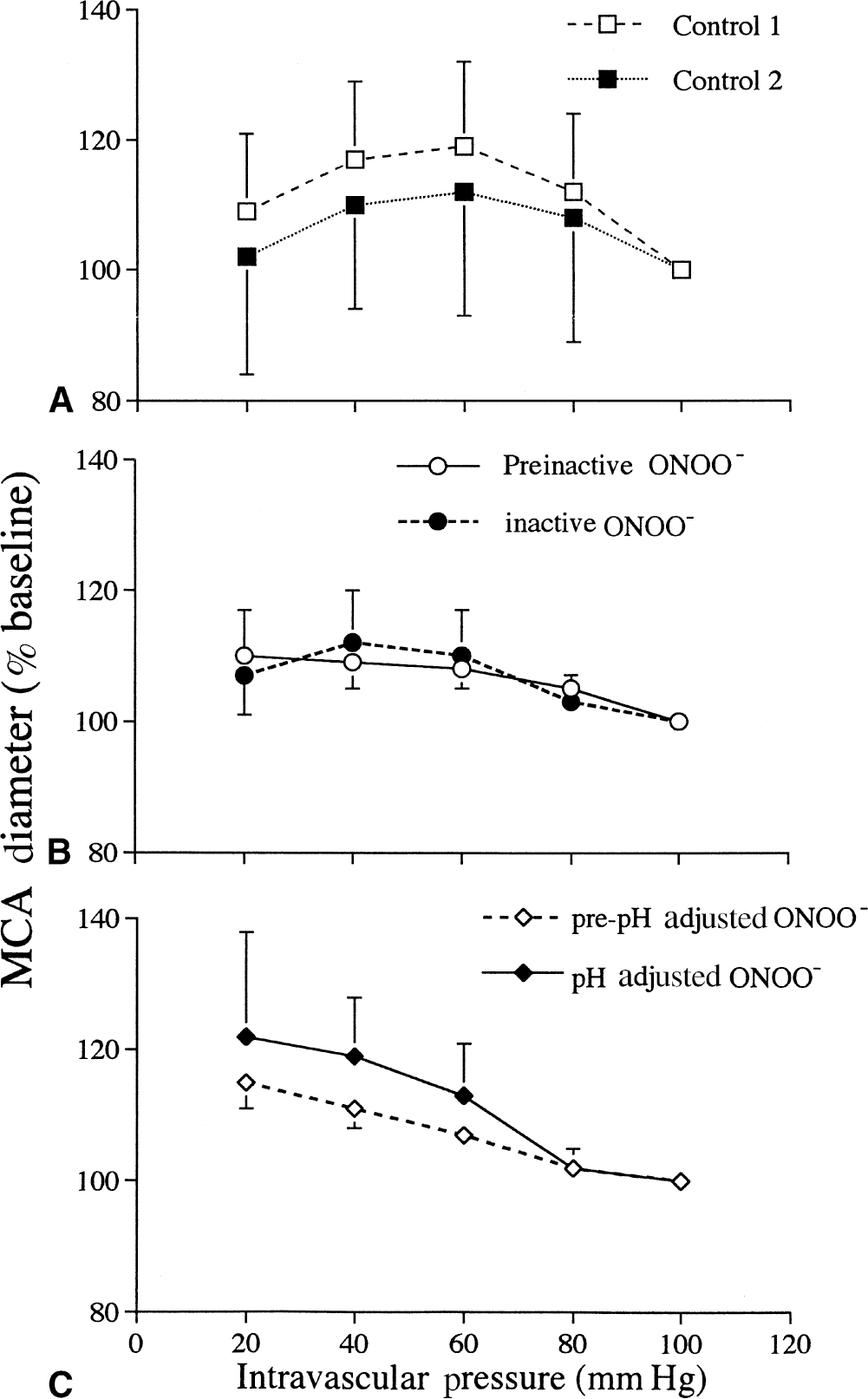
Middle cerebral artery (MCA) diameters (percent of diameter at 100 mm Hg) during 2 successive progressive reductions in intravascular pressure with no treatment
In all groups before ONOO− exposure, MCAs dilated as intravascular pressure was reduced from 100 to 40 mm Hg (Fig. 2A to 2C, pretreatment). There was a significant treatment group versus pressure interaction in the MCAs exposed to ONOO− (P < 0.0001). Exposure to 10 μmol/L ONOO− did not significantly (P = 0.617) reduce vasodilatory responses to progressive reductions in intravascular pressure when compared with inactive ONOO− (Fig. 2A). In contrast, vasodilatory responses were significantly reduced by exposure to 20 μmol/L (Fig. 2B, P < 0.0001) and 40 μmol/L (Fig. 2C, P < 0.0001) ONOO− when compared with inactive or 10 μmol/L ONOO−.
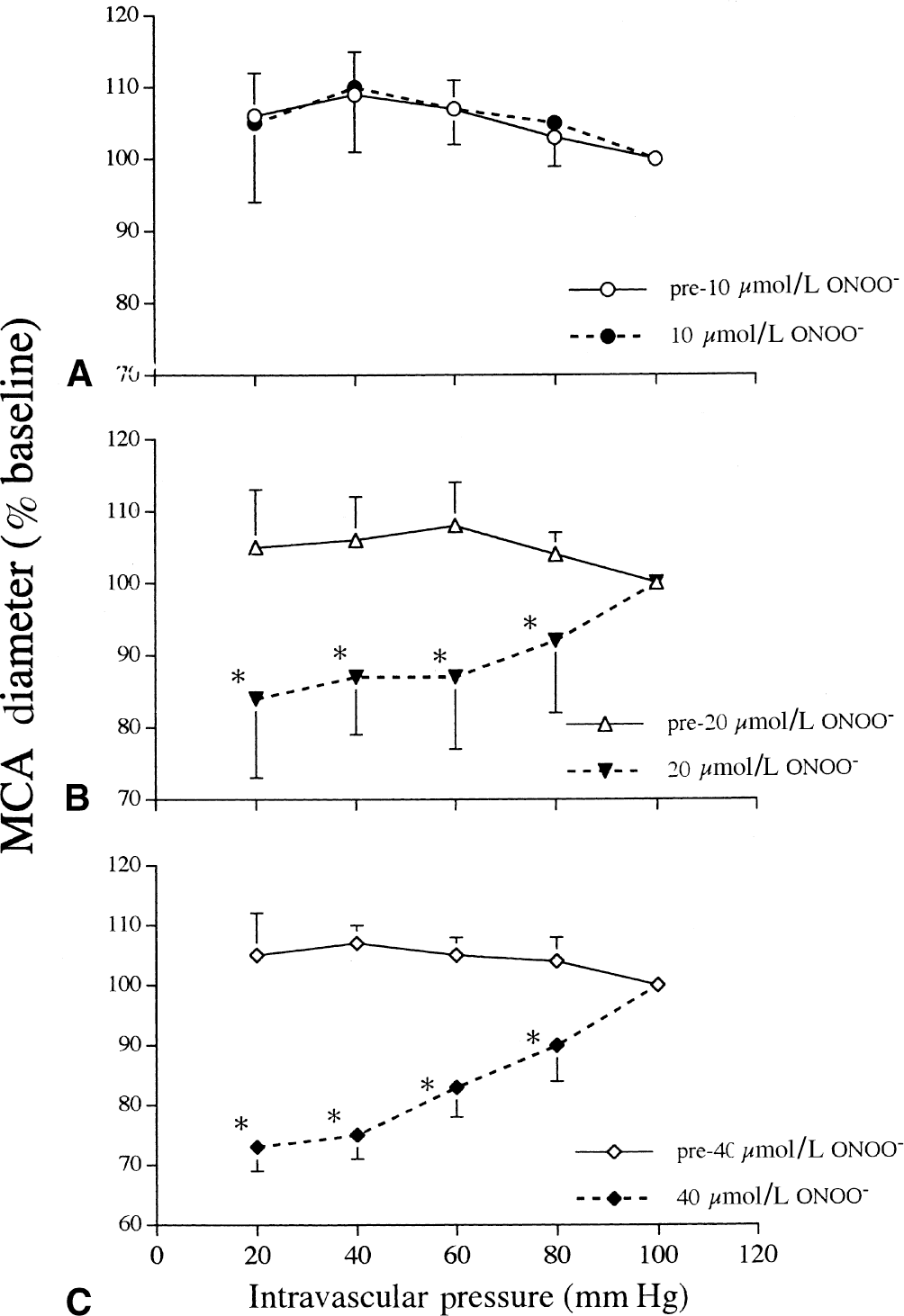
Middle cerebral artery (MCA) diameters (percent of diameter at 100 mm Hg) during progressive reductions in intravascular pressure after exposure to 10 μmol/L
Both CGRP and cromakalim produced significant (P < 0.0001), concentration-dependent increases in MCA diameters (Fig. 3A and 3B), whereas ONOO− significantly reduced the vasodilatory responses to CGRP (dose vs. group interaction P = 0.029; pre-ONOO− vs. post-ONOO− P = 0.019) and cromakalim (dose vs. group interaction P < 0.001; pre-ONOO− vs. post-ONOO− P = 0.007) (Fig. 3A and 3B).
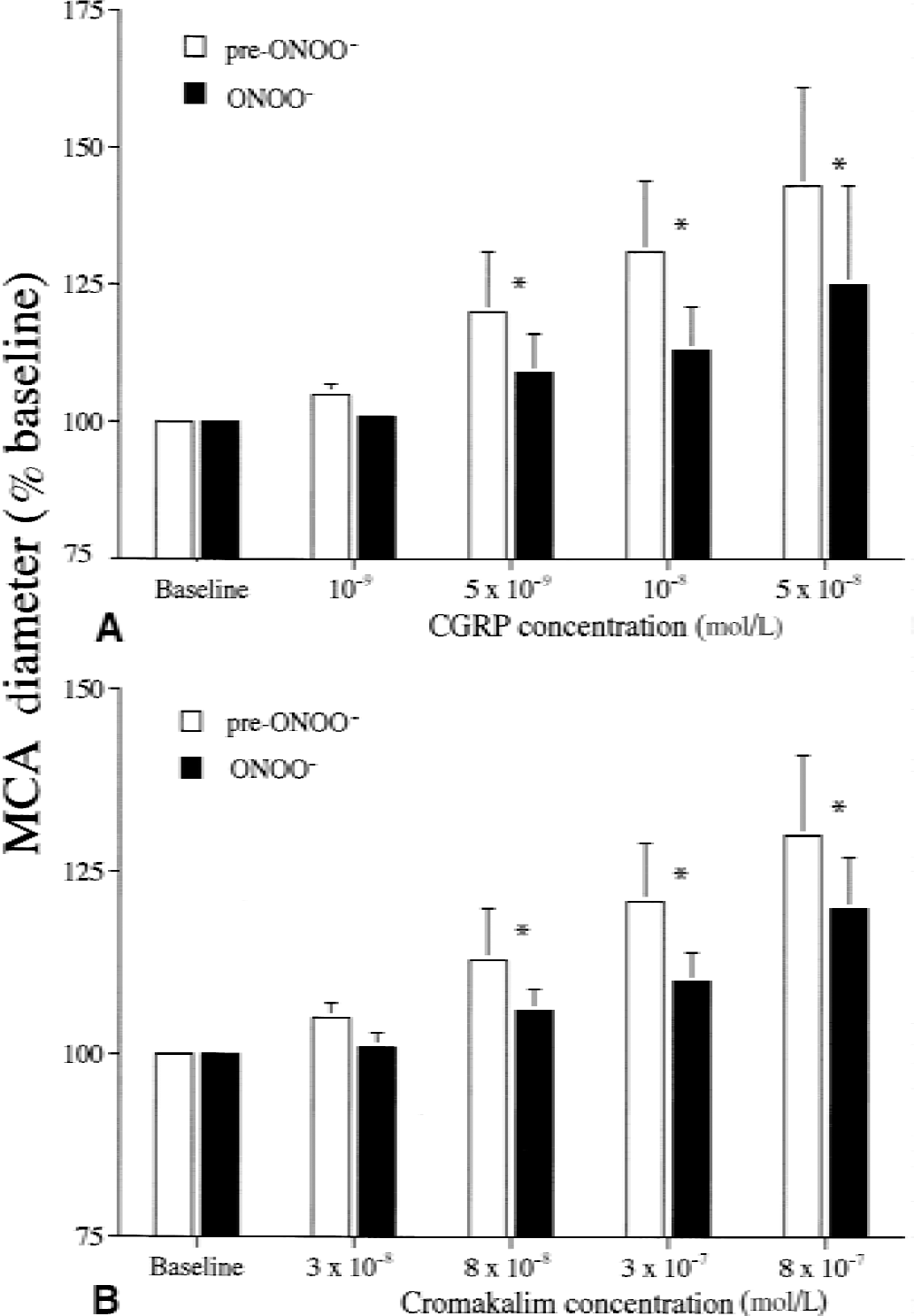
Middle cerebral artery (MCA) diameter (percent of baseline diameter) increases because of calcitonin gene-related peptide (CGRP, n = 6 rats,
After the second measurements of MCA diameters during progressive reductions in intravascular pressure, pressure was returned to 100 mm Hg and baseline diameters were measured again, and MCAs were exposed to serotonin and then KCl. Serotonin (1 × 10−6 mol/L) caused significant (P < 0.0001) reductions in the diameters (63 ± 19%, 61 ± 11%, 63 ± 4%, 66 ± 16% of pre-serotonin baseline) of the MCAs exposed to inactive ONOO− and 10 μmol/L, 20 μmol/L, and 40 μmol/L ONOO−, respectively. Potassium chloride (15 mmol/L) caused significant (P < 0.0001) vasodilation (196% ± 70%, 242% ± 45%, 188% ± 38%, 205% ± 87% of pre-KCl baseline) in preconstricted (with serotonin) MCAs exposed to inactive ONOO− and to 10 μmol/L, 20 μmol/L, and 40 μmol/L ONOO−, respectively.
Replacement of the bathing solution with Ca2+-free PSS caused marked vasodilation in all four groups (Table 2). Reductions in intravascular pressure caused progressive reductions in MCA diameters in all four groups. There were significant (P < 0.0001) pressure effects and a significant pressure versus treatment group interaction (P = 0.0144). The group exposed to 10 μmol/L ONOO− was significantly different (P = 0.001) from the group exposed to inactive ONOO−. The other groups were not significantly different from one another.
Arterial inner diameters (μm) during progressive reductions in intravascular pressure in middle cerebral arteries in Ca2+-free PSS after exposure to 10 μmol/L, 20 μmol/L, 40 μmol/L, or inactive peroxynitrite (ONOO−)
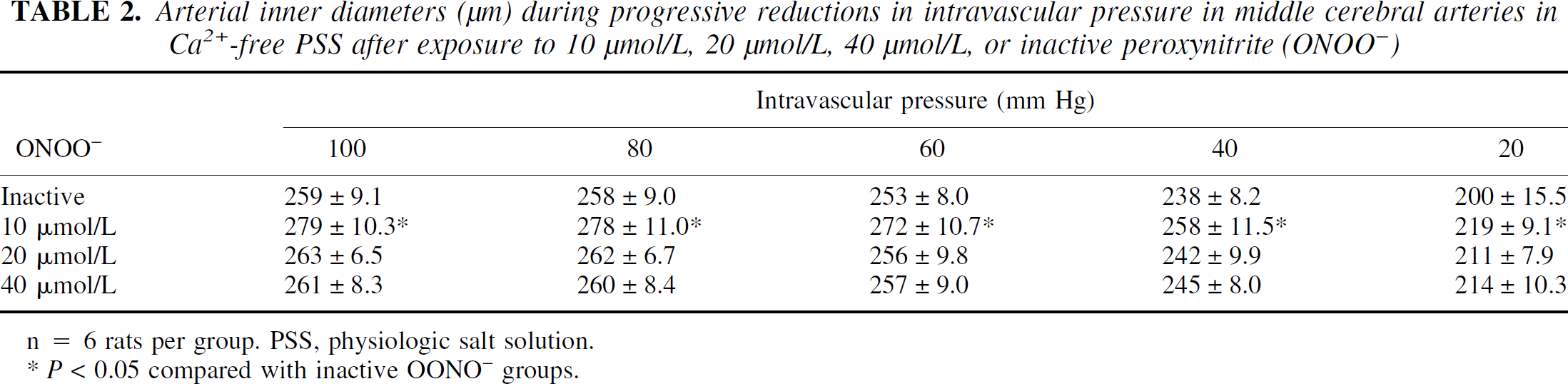
n = 6 rats per group. PSS, physiologic salt solution.
P < 0.05 compared with inactive OONO− groups.
When first added to the arteriograph bath, the ONOO− containing solutions caused transient reductions in MCA diameters followed by increases in MCA diameters in the 20 μmol/L, 40 μmol/L, and pH-corrected ONOO− groups (Table 3). There were significant time (P < 0.0001) and time vs. group (P < 0.0001) effects for time after ONOO− exposure and ONOO− concentration (Fig. 4). Middle cerebral arterial diameters in the 40 μmol/L ONOO− group (n = 6) were significantly different from MCA diameters in the 10 μmol/L (P = 0.010) and 20 μmol/L (P = 0.0003) ONOO− concentrations. Exposure to any of the ONOO− concentrations caused constriction followed by some level of vasodilation. Middle cerebral arteries exposed to 10 μmol/L ONOO− (n = 6) constricted (82% ± 12% of baseline) and then returned to pre-ONOO− diameters (97% ± 9% of baseline) within 10 minutes (Fig. 4). A vasoconstriction (77% ± 6% of baseline) was caused by 20 μmol/L ONOO− (n = 6) at 1 minute after the addition of ONOO−, followed by a slight (8% ± 7%) dilation (108% ± 17% of baseline) by 10 minutes after ONOO− addition. Similarly, 40 μmol/L ONOO− caused vasoconstriction (74% ± 10% of baseline) 1 minute after addition, followed by vasodilation to 127% ± 28% and 117% ± 15% of baseline at 2 and 10 minutes after addition, respectively. Exposure to inactive ONOO− (n = 6) and NaOH (n = 6) produced transient reductions in diameter (79% ± 8% and 64% ± 17% of baseline, respectively), followed by a return toward baseline diameters. Exposure to pH-adjusted 40 μmol/L ONOO− caused MCAs to dilate significantly (P = 0.0033 vs. 20 μmol/L ONOO−) after ONOO− addition (Fig. 4). No transient vasoconstriction was observed in the pH-adjusted ONOO− group.
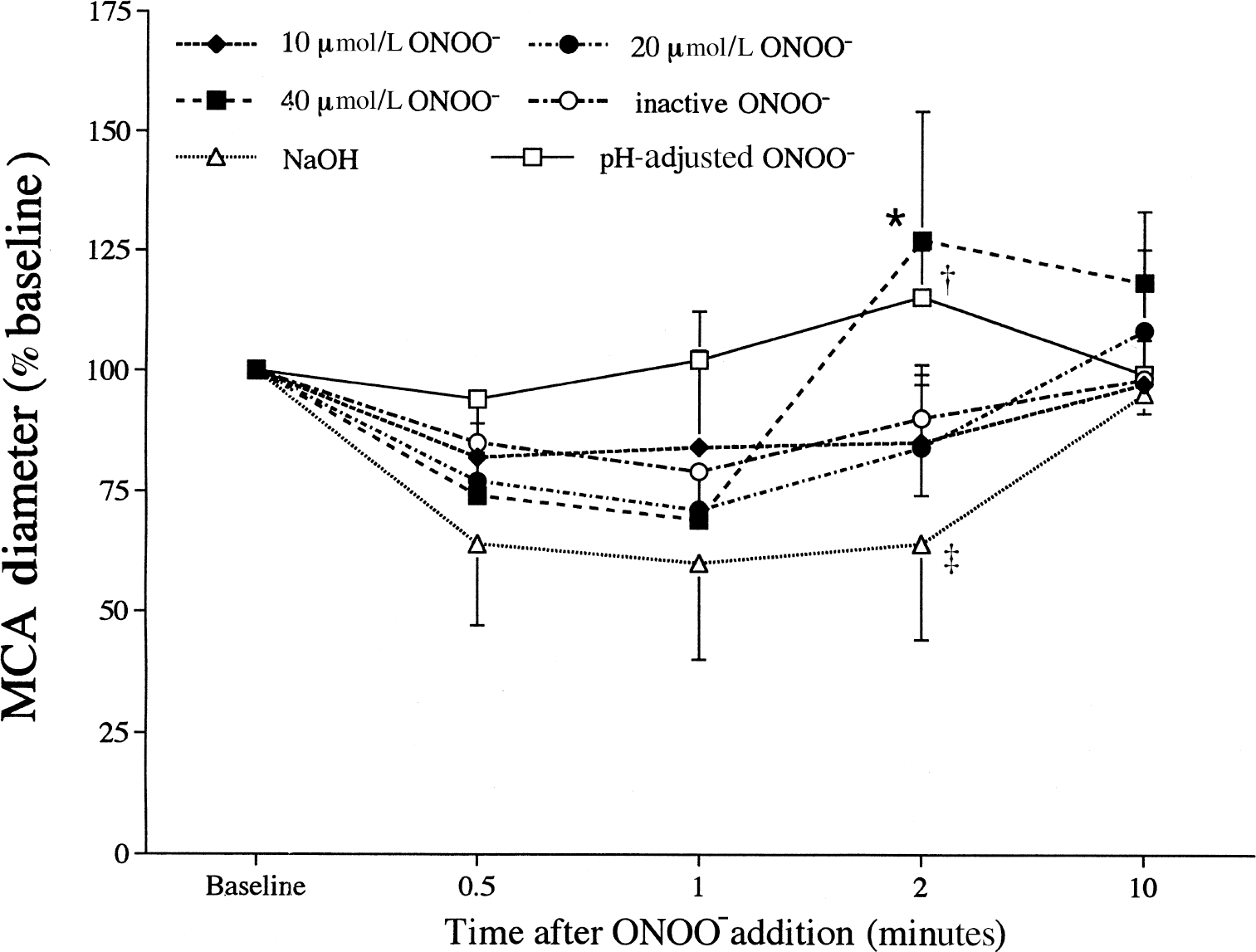
Middle cerebral artery (MCA) diameters (percent of pre-ONOO− baseline) 0.5, 1, 2, and 10 minutes after exposure to inactive peroxynitrite (inactive ONOO−), physiologic salt solution with sodium hydroxide to raise the pH to that of the inactive ONOO− (NaOH), 40 μmol/L ONOO− adjusted to pH 7.48 (pH-adjusted ONOO−), or 10 μmol/L, 20 μmol/L, or 40 μmol/L ONOO−. *Significantly (P < 0.05) different from NaOH group. †Significantly (P < 0.05) different from 20 μmol/L ONOO− group. ‡Significantly (P < 0.05) different from the pH-adjusted ONOO− group. n = 6 rats per group.
Middle cerebral artery diameters (μm) after addition of 10 μmol/L, 20 μmol/L, 40 μmol/L, or inactive or pH-adjusted 40 μmol/L peroxynitrite (ONOO−), or 15 mmol/L NaOH
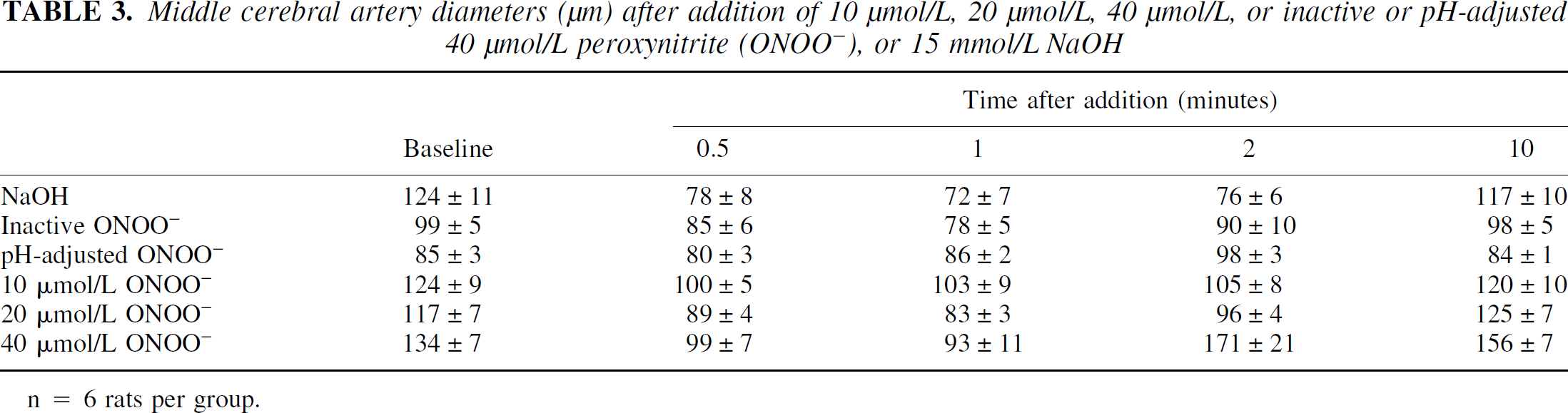
n = 6 rats per group.
DISCUSSION
The current results indicate that ONOO− impairs normal vasodilatory responses to reduced intravascular pressure, to CGRP, and to cromakalim in isolated rodent MCAs. The degree of impairment of the vasodilatory responses was related to the concentration of ONOO− added. Although 10 μmol/L ONOO− produced no significant reductions in vasodilation, 20 μmol/L and 40 μmol/L ONOO− caused significant (P < 0.0001) reductions in MCA diameters during progressive reductions in intravascular pressure (Fig. 2). Twenty μmol/L and 40 μmol/L ONOO− impaired vasodilation to reduced intravascular pressure without influencing the vasoconstrictory or vasodilatory responses to serotonin or KCl, respectively. The current study is the first report of the effects of ONOO− on cerebral vasodilatory responses to reduced intravascular pressure or to K+-channel activation in vitro.
The most novel observation of the current study is that ONOO− produces a concentration-dependent impairment of vasodilatory responses to reduced intravascular pressure in isolated, pressurized segments of the rodent MCA. The cerebral vasculature maintains a resting state of contraction (tone) and is capable of intrinsic, “myogenic” vasodilation or vasoconstriction in response to changes in intravascular pressure in vitro (McCarron et al., 1989; Meininger and Davis, 1992). During the initial (that is, pretreatment) reductions in intravascular pressure, MCAs dilated as intravascular pressure was reduced to 40 mm Hg and then constricted somewhat, but remained greater than baseline diameters, as intravascular pressure was reduced to 20 mm Hg (Fig. 2A to 2C). In the cerebral circulation, the degree of vasodilation is dependent on the pressure range and the size of the arteries or arterioles studied, both in vivo (Kontos et al., 1978) and in vitro (Golding et al., 1998). In the current study, the relaxation observed as intravascular pressure was reduced from 100 to 20 mm Hg was 5% to 15% greater than baseline (100 mm Hg). These values are very similar to those reported in previous studies of cerebral arteries in vitro (Osol and Halpern, 1985; Osol et al., 1989; Mathew et al., 1999) or in vivo (Kontos et al., 1978; Harper et al., 1984).
There were no significant differences in vasodilation between two successive measurements of MCA diameters during progressive reductions in intravascular pressure (Fig. 1A). Middle cerebral arteries exposed to inactive (Fig. 1B), pH-adjusted (Fig. 1C), or 10 μmol/L ONOO− (Fig. 2A) dilated normally during progressive reductions in intravascular pressure. In contrast, exposure to 20 and 40 μmol/L ONOO− led to vasoconstriction during progressive reductions in intravascular pressure (Fig. 2B and 2C). Vasoconstriction during reductions in intravascular pressure was not because of direct ONOO−induced vasoconstriction because MCA diameters in the group exposed to 20 μmol/L ONOO− were not different from pre-treatment diameters and because 40 (iμmol/L ONOO− produced a vasodilation (Fig. 4) within 2 minutes of the addition of ONOO− to the arteriograph bath. These observations indicate that ONOO− produces a concentration-dependent abolition of vasodilatory responses to decreases in intravascular pressure in MCAs. The interpretation that impaired vasodilatory responses are caused by the effects of ONOO− is supported by the authors' observations that the pH adjustment of the 40 μmol/L ONOO− solution, which would rapidly inactivate ONOO− (Beckman et al., 1990; Beckman and Koppenol, 1996), prevented the impairment of vasodilatory responses (Fig. 1C).
The mechanisms through which ONOO− reduces “myogenic” vasodilatory responses to reduced intravascular pressure are unknown, partially because the mechanisms of myogenic responses to changes in intravascular pressure are not known (Meininger and Davis, 1992; Schubert and Mulvany, 1999). In the rodent cerebral arteries, the K
The effects of ONOO− on vasodilatory responses to CGRP and cromakalim are also unknown. In the cerebral circulation, CGRP-mediated vasodilation involves the activation of K
Inactive and 10, 20, and 40 μmol/L ONOO− and NaOH produced transient vasoconstriction (Fig. 4), perhaps because of the effects of the alkaline pH of the test solutions. The pH of the PSS in the arteriograph bath increased significantly (P < 0.0001) after the addition of ONOO− or NaOH but then returned to baseline levels within 10 minutes (Table 1). Inactive ONOO− and PSS treated with NaOH to raise the pH to that of the ONOO− solutions produced vasoconstriction 0.5, 1, and 2 minutes after addition to the same degree as did ONOO−, suggesting that the transient vasoconstriction caused by ONOO− was because of the alkalinity of the solutions rather than to a direct effect of ONOO−. Alkalinization of cerebrospinal fluid causes vasoconstriction in feline pial arterioles in vivo (Kontos et al., 1977) and reductions in PCO2 in the bathing medium cause membrane depolarization and vasoconstriction in vitro (Harder and Madden, 1985). This hypothesis is supported by the authors' observation that the dose of 40 μmol/L ONOO− that was pH adjusted to 7.48 immediately after addition to the arteriograph bath produced vasodilation 5 and 10 minutes after addition with no initial vasoconstriction. Elliott et al. (1998) observed reductions in MCA diameter after the addition of 10 μmol/L ONOO− but attributed the vasoconstriction to the ONOO− because NaOH did not change MCA diameter in their study. However, Elliott et al. (1998) did not state the pH of the PSS in the arterial baths in their isolated MCA experiments so it is difficult to compare the effects of pH versus ONOO− in the two studies. In contrast, Wei et al. (1996) reported that low concentrations of pH-corrected ONOO− (1 to 5 μmol/L) dilated feline pial arterioles in vivo. The lowest concentration of ONOO− used in the current study (10 μmol/L) caused transient reductions in MCA diameter, but these reductions likely were caused by the effects of the high pH of the ONOO− solution.
High concentrations of ONOO− (40 to 150 μmol/L) cause cerebral arteriolar vasodilation. A 43% dilation of MCAs was produced by 50 μmol/L ONOO−, similar to the 27% dilation that the authors observed after the addition of 40 μmol/L ONOO− (Fig. 3). The pH-adjusted 40 μmol/L ONOO− used in the current study also vasodilated MCAs, but to a lesser extent than did 40 μmol/L ONOO− that was not pH adjusted. Wei et al. (1996) reported vasodilation of small and large pial arterioles in cats in vivo after the superfusion of high concentrations of ONOO− (100 to 150 μmol/L).
An important question is whether ONOO− levels in vivo reach the concentrations that impaired myogenic responses, in vitro. Because ONOO− levels currently cannot be measured in vivo, the concentrations reached during ischemia and TBI are not known. Nitric oxide may reach 4 μmol/L in MCA occlusion in rats (Malinski et al., 1993), and because ·O2− radicals react with NO more quickly than they react with SOD, micromolar concentrations of ONOO− could be present in ischemia (Beckman et al., 1990, 1994a). The concentrations of ONOO− that reached the MCAs in the current study would have been less than the added concentrations. Adding 100 μmol/L ONOO− to an extracellular buffer solution results in an effective concentration of 15 μmol/L ONOO− in the solution (Elliott et al., 1998). Therefore, the addition of 20 μmol/L ONOO−, a concentration that significantly reduced myogenic responses to reduced intravascular pressure, would likely produce an effective concentration of 3 μmol/L at the MCA segments. Finally, the addition of a single bolus of ONOO−, which would have degraded within a few seconds (Beckman et al., 1994b), would result in exposure of the MCA segments to ONOO− for only a few seconds. In contrast, the elevation of NO and ·O2− after ischemic or traumatic brain injury lasts for many minutes (Fabian et al., 1995; Malinski et al., 1993; Peters et al., 1998; Cherian et al., 2000), potentially exposing cerebral arteries to ONOO− for longer periods than were used in the current study.
Conclusion
The authors demonstrated that ONOO− produces concentration-dependent reductions in vasodilatory responses to progressive reductions in intravascular pressure, CGRP, and cromakalim. These observations indicate that ONOO− reduces physiologic vasodilation to reduced intravascular pressure and receptor-mediated vasodilation, perhaps through actions on membrane K+ channels.
Footnotes
Acknowledgments
The authors are grateful to Joseph S. Beckman, Ph.D., Department of Anesthesiology, University of Alabama, Birmingham, for providing the peroxynitrite used for these studies. The authors thank Jordan Kicklighter for valuable editorial and secretarial support. The authors also are grateful to their colleagues in the Department of Anesthesiology at the University of Texas Medical Branch for their support of the Charles R. Allen Research Laboratories.