
Abstract
The retinal vasculature supplies cells of the inner and middle layers of the retina with oxygen and nutrients. Photic stimulation dilates retinal arterioles producing blood flow increases, a response termed functional hyperemia. Despite recent advances, the neurovascular coupling mechanisms mediating the functional hyperemia response in the retina remain unclear. In this review, the retinal functional hyperemia response is described, and the cellular mechanisms that may mediate the response are assessed. These neurovascular coupling mechanisms include neuronal stimulation of glial cells, leading to the release of vasoactive arachidonic acid metabolites onto blood vessels, release of potassium from glial cells onto vessels, and production and release of nitric oxide (NO), lactate, and adenosine from neurons and glia. The modulation of neurovascular coupling by oxygen and NO are described, and changes in functional hyperemia that occur with aging and in diabetic retinopathy, glaucoma, and other pathologies, are reviewed. Finally, outstanding questions concerning retinal blood flow in health and disease are discussed.
INTRODUCTION
Oxygen and nutrients are supplied to the retina by a dual circulatory system (Figure 1). The high metabolic needs of the retinal photoreceptors (photoreceptors have the highest metabolic rate of any cell in the body 1 ) are satisfied by the choroidal vasculature. The more modest metabolic demands of the neurons and glial cells of the inner portions of the retina are met by the retinal vasculature. In lower vertebrates that have thinner retinas, including amphibians, reptiles, and some mammals, the retinal vasculature is absent and the choroidal circulation supplies the entire retina. 2
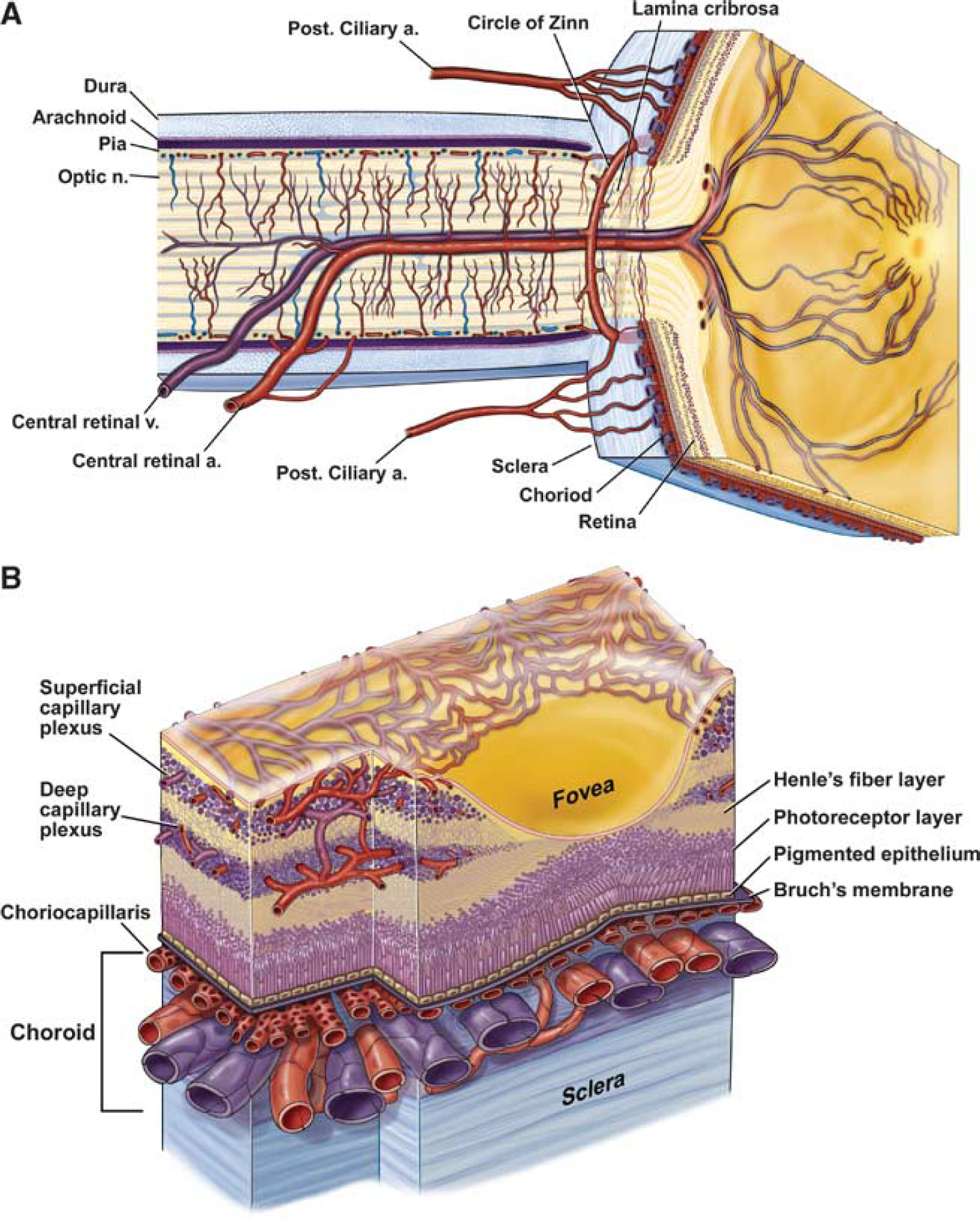
Anatomy of the ocular circulation. (
The choroidal circulation is supplied by the long and short ciliary arteries and the anterior ciliary arteries, which feed the large arteries in the outer portion of the choroid. 3 These arteries branch into smaller vessels, which, in turn, feed the highly anastomosed choriocapillaris network lying at the inner border of the choroid, adjacent to the retinal pigment epithelium and retinal photoreceptors. The total blood flow supplying the choroid is much greater than that supplying the retina (606 vs. 25 mg/min/whole tissue 4 ), to meet the high metabolic demands of the photoreceptors. 5
The retinal vasculature is supplied by the central retinal artery, which enters the retina with the optic nerve at the optic disc. The artery branches into radial arterioles and smaller vessels on the vitreal surface of the retina. 3 Pre-capillary arterioles and capillaries ramify from these surface vessels and form anastomotic networks in the ganglion cell layer, just beneath the retinal surface, and deeper in the inner nuclear layer, supplying horizontal cells, bipolar cells, amacrine cells, and Muller glial cells. Blood is returned through radial venules on the retinal surface that empty into the central retinal vein in the optic nerve.
The retinal and choroidal vasculatures differ in several respects. Retinal vessels lack autonomic innervation,6,7 whereas the choroidal circulation is innervated by both sympathetic and parasympathetic nerves.8-10 Also, autoregulation is present in the retinal circulation11,12 but is less well developed in choroidal vessels.13,14 Responses to light also differ. As will be described below, functional hyperemia is well developed in the retinal vasculature while the choroidal blood supply does not respond well to flickering light stimuli (but see Lovasik
When the retina is stimulated by a flickering light, blood flow in the retinal vasculature increases significantly. 18 This increase in blood flow, the functional hyperemia response, supplies oxygen and glucose to the active neurons in the inner and middle retinal layers. This hemodynamic response is present in the cerebral circulation as well and was described long ago in the cerebral cortex by Mosso 19 and Roy and Sherrington. 20
It is believed that the functional hyperemia response is critical for proper retinal function. 18 With increased neuronal activity in the retina, there is a need for enhanced oxygen and glucose supply and removal of metabolites. The increase in retinal blood flow serves this need. The loss of functional hyperemia that occurs in certain diseases may compromise retinal health and may contribute to the development of pathology.
This review will focus on functional hyperemia in the retinal vasculature in health and disease. Special emphasis will be placed on the signaling mechanisms responsible for generating the functional hyperemia response. The reader is referred to recent reviews3,18,2,22 for information about other aspects of ocular circulation.
FUNCTIONAL HYPEREMIA IN THE RETINA
Functional hyperemia in the retina is typically studied by stimulating the eye with a flickering light, which maximally activates amacrine cells and ganglion cells in the inner retina. 23 A flickering stimulus dilates the primary arterioles on the retinal surface (Figure 2), leading to an increase in blood flow in arterioles, capillaries, and venules of the retina. Flickering light also increases blood flow in the capillaries of the optic disc (Figure 2). Increases in blood flow in retinal vessels supply active neurons in the inner and middle retinal layers with increased oxygen and glucose while blood flow increases in the capillaries of the optic disc supply the active axons of the retinal ganglion cells.
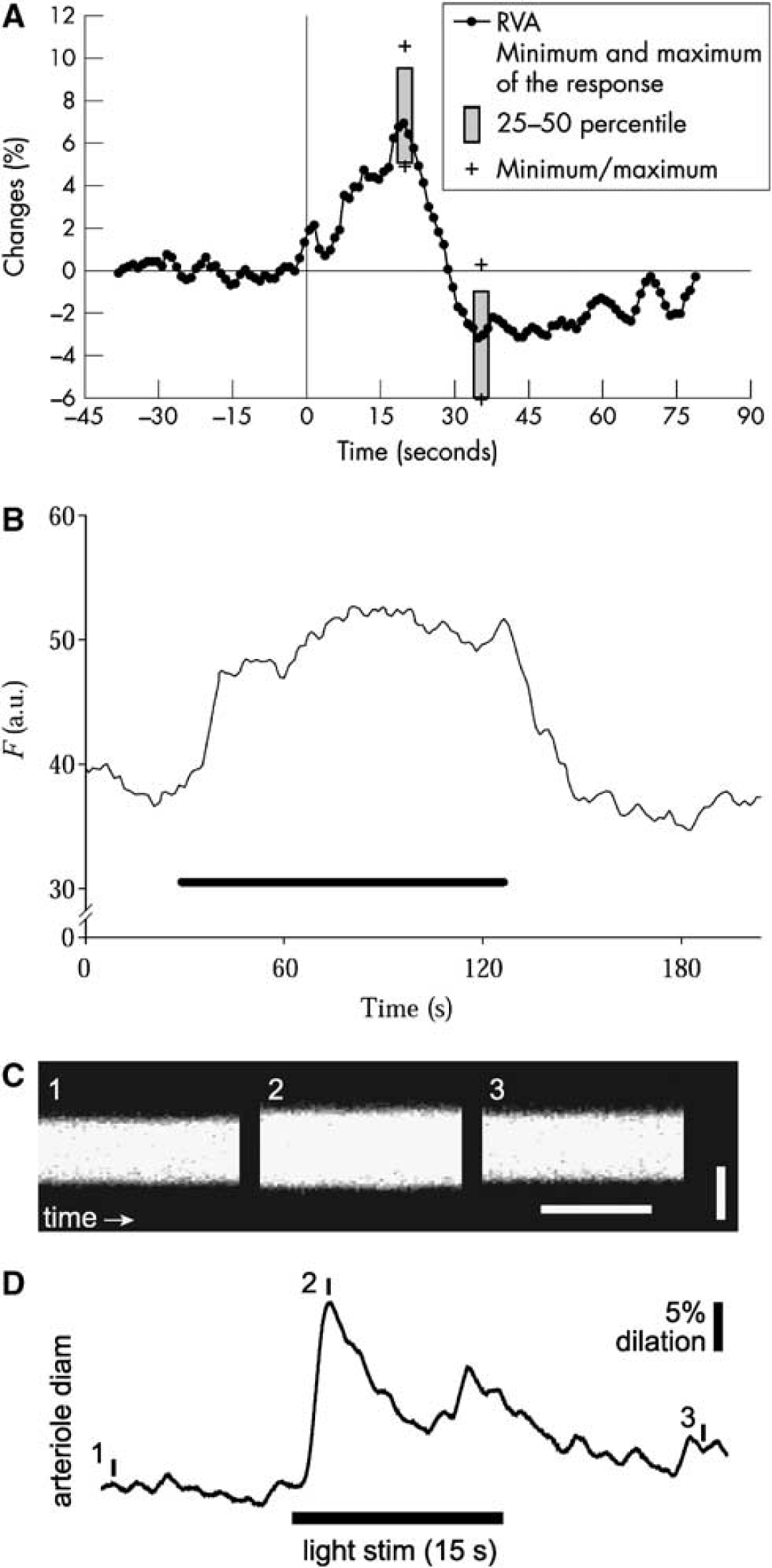
The functional hyperemia response in the retina. (
Arteriole dilation can be monitored with a modified fundus camera, such as the retinal vessel analyzer. 24 In humans, flickering light evokes sustained dilations ranging from 3% to 5%.24-26 The response has a latency of < 1 s. Primary venules in humans dilate to nearly the same extent as arterioles,24,25 although it is unclear whether this is an active or passive response. Similar light-evoked arteriole dilations occur in cats27,28 and rodents. 29 In rats and mice, light-evoked venule dilations are much smaller than those of arterioles (TE Kornfield, AI Srienc, and EA Newman, unpublished observations).
Arteriole dilation leads to increases in retinal blood velocity, which can be measured using techniques based on the Doppler effect. These measurement techniques include bi-directional laser Doppler velocimetry, which yields an absolute measure of red blood cell velocity, laser Doppler flowmetry, which measures relative red blood cell velocity, blood volume, and flux in the optic nerve head and choroid, and laser speckle flowmetry, which yields two-dimensional images of relative blood velocity on the retina surface. 18 Retinal blood flow can also be measured using magnetic resonance imaging, which has sufficient spatial resolution to distinguish retinal arterioles, venules, and choroidal vessels. 30 In humans, a flickering light results in blood flux increases of 30-38% in the optic nerve head31,32 and flux increases of 59% in retinal arterioles. 33 In the cat optic nerve head, flux increases of 27-59% are observed.27,34
As originally noted by Roy and Sherrington, 20 the localized nature of blood flow control is a hallmark of the functional hyperemia response. In the cortex, activation of a small parenchymal region results in an increase in blood flow that is largely restricted to that region. The localized nature of the response demonstrates that blood flow can be controlled locally in the cerebral vasculature. The few studies that have been conducted in the retina demonstrate that the retinal vasculature is also able to control blood flow locally in response to focal photic stimulation. When one hemi-field of the cat retina is stimulated with a drifting grating, blood flow increases, monitored with blood oxygen level-dependent functional magnetic resonance imaging, are restricted to the stimulated half of the retina. 35 Similarly, when the rat retina is stimulated focally with a flickering spot, blood velocity increases, monitored with laser speckle flowmetry, are greatest near the stimulated region. 36
Active control of blood flow in the retinal circulation is believed to occur principally in the arterioles on the surface of the retina.
These vessels are surrounded by layers of smooth muscle cells 21 and display large dilations ranging from 3% to 8% 24,25,29 in response to photic stimulation. It is possible, however, that active control of blood flow also occurs in retinal capillaries, which are surrounded by contractile pericytes. Pericytes share many properties with vascular smooth muscle cells. Pericytes contain contractile proteins37,38 and have been shown to relax and contract in response to various signaling molecules.39,40 Pericytes cover a large fraction of the surface of capillaries in the retina, much larger than that in the brain,41,42 and active contraction of pericytes restricts blood flow through retinal capillaries after an ischemic insult. 43 However, it remains unclear whether pericytes actively control blood flow in the retina under normal physiologic conditions.
MECHANISMS OF NEUROVASCULAR COUPLING IN THE RETINA
Metabolic Feedback vs. Feedforward Mechanisms
Functional hyperemia is mediated, either directly or indirectly, by signaling from neurons to blood vessels. The mechanisms that mediate functional hyperemia, termed neurovascular coupling, have been of great interest for over a century. In their seminal work, Roy and Sherrington 20 proposed that metabolic products released from active neurons mediate functional hyperemia. According to this metabolic negative feedback mechanism, 44 neuronal activity leads to a drop in energy reserves in active neurons and to the generation of metabolic signals that dilate nearby blood vessels. The resulting increase in blood flow augments glucose and oxygen supplies and restores the energy reserves of the active neurons. Several metabolic signals could function as the neurovascular coupling signal. These include a drop in oxygen or glucose levels or increases in CO2 (leading to acidification), adenosine, or lactate levels. All of these changes could dilate arterioles. Recent work has demonstrated, however, that some of these signals do not mediate neurovascular coupling. Specifically, functional hyperemia can occur in the absence of a drop in oxygen45-47 or glucose 48 levels or acidification of the parenchyma,49,50 demonstrating that O2, glucose, and CO2 do not function as the neurovascular coupling signal. Still, neurovascular coupling could be mediated, at least in part, by adenosine or lactate metabolic signals. As discussed below, lactate may control blood flow indirectly by modulating prostaglandin E2 (PGE2) levels.
An alternate to the metabolic negative feedback mechanism of neurovascular coupling is a feedforward mechanism. 44 In a feedforward mechanism, active neurons release signaling molecules that directly or indirectly result in vasodilation. These signaling molecules are not directly related to cell metabolism, thus distinguishing this form of neurovascular coupling from metabolic negative feedback. Many signaling pathways may contribute to feedforward neurovascular coupling in the central nervous system. Active neurons release nitric oxide (NO) and PGE2, both of which relax vascular smooth muscle cells and result in vasodilation. Active neurons also release a number of transmitters that act on glial metabotropic receptors, evoking Ca2+ increases in glial cells. These Ca2+ increases lead to the release of vasodilatory agents from glial cells, including K+ and the arachidonic acid metabolites PGE2 and epoxyeicosatrienoic acids (EETs).
Both feedforward signals as well as metabolic negative feedback signals may contribute to functional hyperemia in the retina. Evidence supporting different neurovascular coupling signaling mechanisms is reviewed in the following sections.
Arachidonic Acid Metabolite-Mediated Neurovascular Coupling
In recent years, persuasive evidence has emerged indicating that a feedforward neurovascular coupling mechanism mediated by signaling from neurons to glial cells to blood vessels is instrumental in generating functional hyperemia in the mammalian retina.
According to this model, neurovascular coupling occurs in several steps. The release of neurotransmitters from active neurons leads to Ca2+ increases in glial cells, which, in turn, results in the release of vasodilatory agents from the glial cells onto blood vessels.
As Ramon y Cajal 51 pointed out over a century ago, glial cells are ideally suited to regulate blood flow. Blood vessels are almost completely enveloped by the endfeet of glial cells. 52 Thus, glial cells, more than neurons, are well situated to communicate directly with vessels. In the retina, blood vessels are surrounded by the endfeet of both astrocytes, which lie near the vitreal surface of the retina, and Muller cells, which are the principal glial cells of the retina and extend from the vitreal surface to the photoreceptors.21,53
When neurons in the retina are activated by photic stimulation, intracellular Ca2+ increases are observed in glial cells (Figure 3). 54 The Ca2+ increases occur in Muller cells but not in astrocytes. This is not surprising as Muller cells directly contact neuronal synapses in the plexiform layers of the retina and thus will be stimulated by the release of transmitters from neurons. 54 Astrocytes, in contrast, are restricted to the vitreal surface of the retina and are far from neurotransmitter release sites. Light-evoked Ca2+ increases in Muller cells are mediated by the release of ATP from neurons and the activation of glial purinergic receptors. 54 The Ca2+ increases are blocked by purinergic antagonists but not by glutamatergic, GABAergic, or cholinergic antagonists. Light-evoked glial Ca2+ increases are also blocked by tetrodotoxin, 54 indicating that ATP release from ganglion cells and amacrine cells, the only retinal neurons which generate tetrodotoxin-sensitive action potentials, stimulate the Muller cells. Antidromic activation of ganglion cells also evokes Ca2+ increases in Muller cells, supporting the role of these neurons in the generation of glial Ca2+ increases. 54
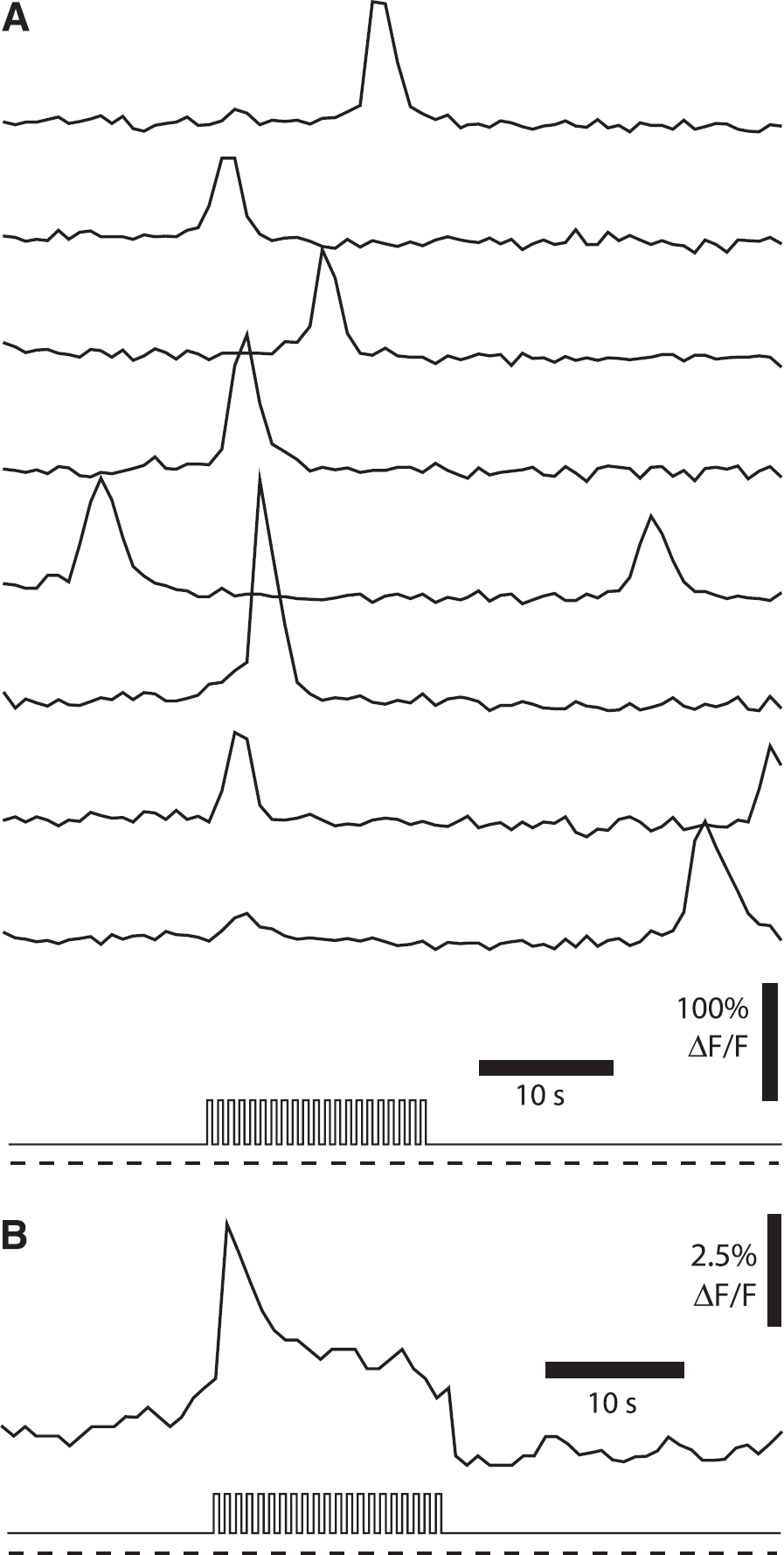
Cytoplasmic Ca2+ increases in Muller glial cells of the rat retina. (
Calcium increases in glial cells result in the release of vasoactive agents that dilate retinal arterioles. When retinal glial cells are directly stimulated by photolysis of caged Ca2+ or caged inositol 1,4,5 trisphosphate, resulting in increased glial Ca2+, neighboring arterioles dilate (Figure 4). 55 Signaling from glial cells to blood vessels is direct and does not involve neurons, as glial-evoked vasodilation is not reduced when transmitter release from neurons is blocked with tetanus toxin. 55 Glial-evoked vasodilation is mediated by the production of PGE2 and EETs. When the synthetic enzymes for these two arachidonic acid metabolites are inhibited, glial-evoked vasodilation is reduced by 88%.55,56
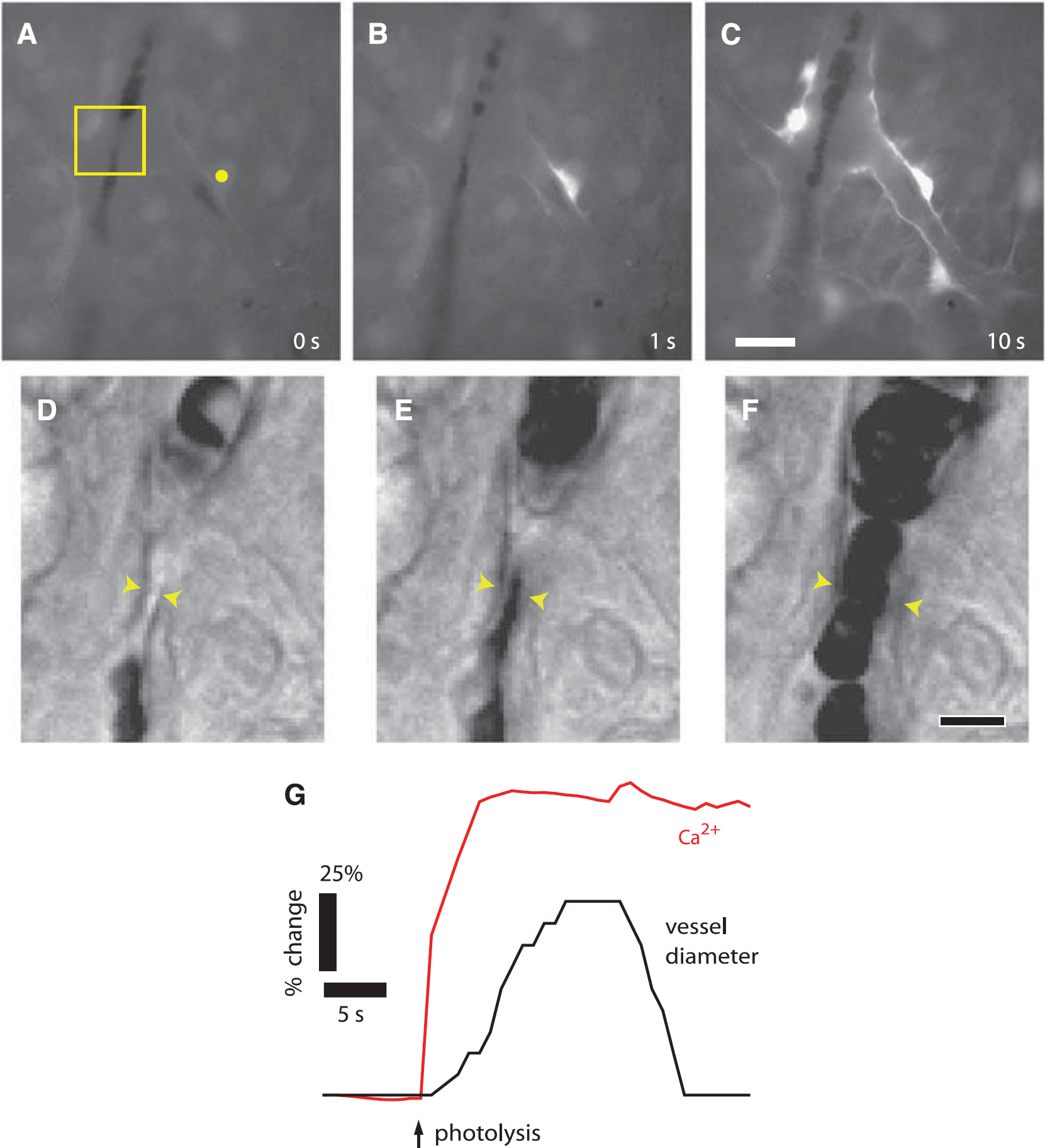
Calcium increases in glial cells evokes arteriole dilation in the rat retina. (
Calcium increases in glial cells can also result in the production of the vasoconstrictor 20-hydroxyeicosatetraenoic acid (20-HETE). 55 Under physiologic conditions, production of 20-HETE is outweighed by the production of the vasodilators PGE2 and EETs, and vasodilation results. However, as will be discussed below, under non-physiologic conditions, including hyperoxia and high NO levels, the vasodilatory mechanisms are suppressed, and 20-HETE-mediated vasoconstriction is observed.
The glial cell-mediated feedforward mechanism of neurovascular coupling, summarized in Figure 5, contributes significantly to functional hyperemia in the retina. When signaling from neurons to glial cells is interrupted by the purinergic antagonist suramin, light-evoked vasodilation is nearly abolished. 55 Suramin acts by preventing glial Ca2+ increases rather than by interfering with neuronal activity or intrinsic vascular responsiveness. Suramin neither reduces light-evoked neuronal activity nor does it block glial-evoked vasodilation when glial cells are stimulated by photolysis of caged compounds. 55
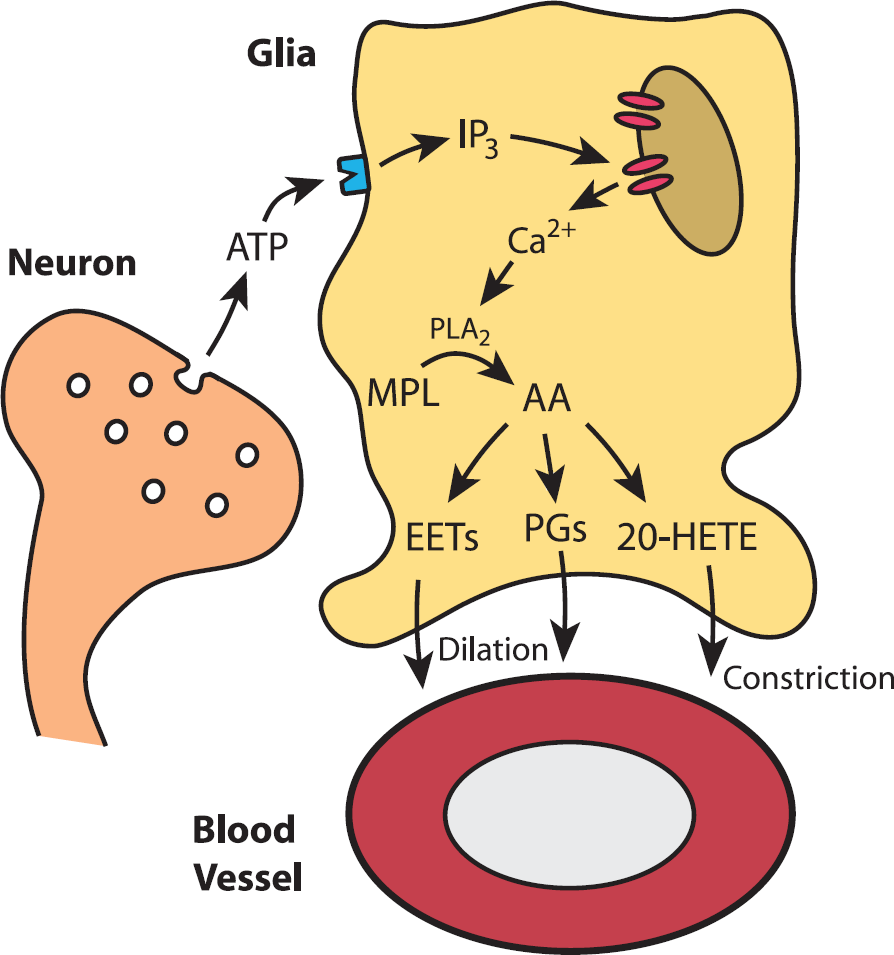
Summary of the arachidonic acid metabolite-mediated neurovascular coupling mechanism. ATP released from active neurons stimulates P2Y receptors on glial cells, resulting in the production of inositol 1,4,5 trisphosphate (IP3) and the release of Ca2+ from internal stores. Glial Ca2+ activates phospholipase A2 (PLA2) resulting in the production of arachidonic acid (AA) from membrane phospholipids (MPL). Increased AA levels lead to the production of its metabolites, including the vasodilators epoxyeicosatrienoic acids (EETs) and prostaglandin E2 (PGE2) and the vasoconstrictor 20-hydroxyeicosatetraenoic acid (20-HETE). Under physiologic conditions, the vasodilators have a stronger influence on vessels than the vasoconstrictor, leading to vessel dilation and increased blood flow. Drawing by Anusha Mishra, unpublished.
The role of prostaglandins in mediating neurovascular coupling is supported by additional observations. PGE2 dilates retinal arterioles in the
Potassium-Mediated Neurovascular Coupling
Neurovascular coupling in the retina may also be mediated by the release of K+ onto blood vessels. When extracellular K+ concentration ([K+]o) is raised from a resting level of 3-5 mmol/L up to 10-15 mmol/L, blood vessels dilate. Potassium-induced vasodilation is mediated by an increase in the conductance of inwardly rectifying K+ channels on vascular smooth muscle cells61,62 and by activation of the Na+ -K+ ATPase on the smooth muscle cells,63,64 both of which result in hyperpolarization and relaxation of the muscle cells. However, larger K+ increases, above ~ 15 mmol/L, depolarize vascular smooth muscle cells, resulting in vasoconstriction.
Depolarizing neurons in the retina release K+ into the extracellular space.
A number of years ago, it was proposed that neurovascular coupling is mediated by a feedforward glial cell K+ siphoning mechanism. 68 According to this model, 69 the diffusion of K+ from neurons to blood vessels is enhanced by a K+ current flow through Muller cells. The [K+]o increase due to neuronal activity generates an influx of K+ into Muller cells and results in Muller cell depolarization. Depolarization, in turn, induces a K+ efflux from cell endfeet, which have a very high density of K+ channels.70,71 Because Muller cell endfeet terminate on blood vessels, the K+ efflux occurs directly onto the vessels. Computer simulations of K+ dynamics 69 demonstrate that glial K+ siphoning results in a K+ increase at the vessel that is larger and more rapid than would occur solely by K+ diffusion through extracellular space. Potassium siphoned onto blood vessels could mediate neurovascular coupling.
More recently, the glial K+ siphoning hypothesis of neurovascular coupling was tested in the retina. 72 The results do not support the K+ siphoning hypothesis. Glial cells were depolarized by current injection through patch pipettes. This depolarization should evoke K+ efflux from the cell endfeet and result in vasodilation. Although vessels dilated in response to bath-applied increases in [K+]o, current injection did not induce vasodilation. In a second test of the K+ siphoning hypothesis, light-evoked vasodilation was assessed in mice where Kir4.1 K+ channels were genetically knocked out. Kir4.1 is the principal K+ channel in retinal glial cells, 73 and K+ siphoning fluxes should be nearly abolished in Kir4.1 knockout animals. However, light-evoked vasodilation was as large in the knockout animals as in wild-type controls. The experiments demonstrate that glial cell K+ siphoning does not contribute significantly to neurovascular coupling in the retina.
Although neurovascular coupling in the retina is not mediated by K+ siphoning, it could be mediated by a different K+ mechanism. Retinal Muller cells express BK Ca2+-activated K+ channels as well as Kir channels. 74 When neuronal activity evokes Ca2+ increases in Muller cells, BK K+ channels will open. 61 BK channels are also modulated by arachidonic acid metabolites, and Ca2+ -dependent increases in EETs will open the channels.75,76 BK channel opening will result in an efflux of K+ from glial cell endfeet onto blood vessels, which could lead to vessel dilation. This K+-glial BKchannel hypothesis of neurovascular coupling has received support in experiments in the cortex, 61 but the hypothesis has not been tested in the retina.
Nitric Oxide-Mediated Neurovascular Coupling
Nitric oxide is a potent vasodilator and is believed to have an important role in mediating neurovascular coupling in the cerebellum. 77 Activity-dependent Ca2+ increases within neurons activate neuronal nitric oxide synthase (NOS), leading to the production of NO. The membrane-permeant NO can then diffuse to blood vessels, where it opens K+ channels and relaxes vascular smooth muscle cells, 78 leading to vasodilation and increased blood flow.
In the retina, NO levels have been measured
These results would appear to support NO as an important mediator of neurovascular coupling in the retina. However, additional studies indicate that the effects of NO are more complicated. In the
Lactate-Mediated Neurovascular Coupling
Lactate is a vasodilator that is a product of anaerobic glycolysis and is released by both glial cells and neurons. Lactate production increases substantially in the rabbit retina in response to flickering light.82,83 In the miniature pig, intravitreal injection of lactate near the retinal surface evokes arteriole dilation. 59 This effect is not due to acidification, as lactate injection at a neutral pH produces the same effect. A similar response is seen in isolated, pressurized retinal arterioles of the pig, which dilate in response to lactate. 84 In humans, increased serum levels of lactate, produced either by exercise or by intravenous injection, reduces flicker-evoked arteriole dilation. 26 This suggests that lactate contributes to neurovascular coupling, although the interpretation is complicated by the fact that lactate could have systemic effects and was administered on the luminal rather than on the abluminal side of blood vessels. Together these experiments demonstrate that lactate could contribute to neurovascular coupling as a metabolic negative-feedback mediator. However, definitive evidence for its role as a mediator of neurovascular coupling is lacking.
Adenosine-Mediated Neurovascular Coupling
Adenosine is another vasodilatory metabolic byproduct released by neurons and glial cells that could mediate functional hyperemia in the retina. Intravitreal but not intravenous administration of adenosine in the rabbit produces vasodilation and increased basal blood flow. 85 Adenosine also relaxes cultured pericytes isolated from the bovine retina. 86 In the cat, intravenous administration of adenosine increases baseline blood flow and enhances flicker-induced increases in blood flow. 34 This would argue against adenosine being a mediator of neurovascular coupling, as raising baseline levels of adenosine would be expected to decrease an adenosine-mediated vasodilation during flicker stimulation.
Summary of Neurovascular Coupling Mechanisms
As reviewed above, many vasoactive agents that are produced during photic stimulation dilate retinal arterioles and could mediate neurovascular coupling. It is likely that several of these agents act in concert to produce functional hyperemia in the retina. A similar phenomenon is seen in the brain, where a number of neurovascular coupling mechanisms are believed to operate concurrently to produce functional hyperemia. 45 However, recent studies suggest that one particular neurovascular coupling mechanism, the feedforward mechanism where glial cells release vasodilatory PGE2 and EETs, is a principal and perhaps dominant mechanism mediating functional hyperemia in the retina. Determining the relative importance of this and other neurovascular coupling mechanisms awaits further experimentation.
MODULATION OF FUNCTIONAL HYPEREMIA IN THE RETINA
Oxygen Modulation of Neurovascular Coupling
Oxygen as well as CO2 is a well-known modulator of blood flow. In humans,87,88 monkeys, 89 and miniature pigs, 22 hyperoxia constricts retinal arterioles and venules and reduces retinal blood flow. Similarly, hypoxia dilates arterioles and venules and increases blood flow in the retina.
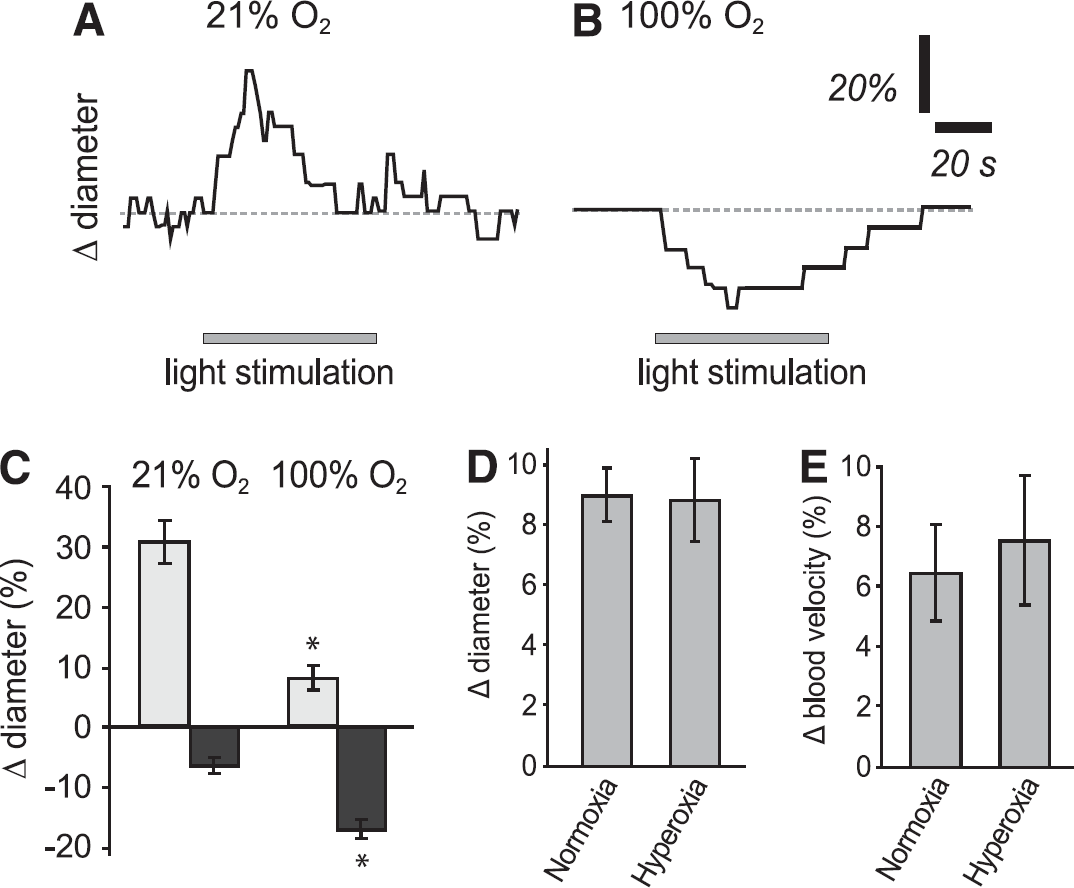
Oxygen depresses flicker-induced vasodilation in the
In addition to modifying basal blood flow, oxygen modulates neurovascular coupling in the retina. In the
A similar O2 inhibition of PGE2 signaling is seen in hippocampal slices. Under normoxic conditions, vasodilation evoked by neuronal activity is mediated by PGE2 release from astrocytes. 90 Under hyperoxic conditions, O2 inhibits PGE2 signaling by increasing the uptake of PGE2 into cells via the prostaglandin transporter, which exchanges PGE2 for lactate. When pO2 is raised, glycolytic production of lactate is reduced, lowering extracellular lactate levels. This enhances PGE2 transport into cells, lowering extracellular PGE2 levels and thus reducing PGE2-induced vasodilation.
Although O2 modulates neurovascular coupling in
Nitric Oxide Modulation of Neurovascular Coupling
NO, in addition to being a potent vasodilator, also exerts a strong effect on blood flow by modulating neurovascular coupling in the retina. The modulatory effects of NO have been characterized in the whole-mount rat retina. 55 When NO levels are < 100 nmol/L, flicker-evoked vasodilations, but not vasoconstrictions, are observed. As NO levels are raised, however, vasodilations become smaller and vasoconstrictions more common (Figure 7). At 10 mmol/L NO, large flicker-evoked vasoconstrictions, mediated by 20-HETE, occur. The mechanism by which NO suppresses flicker-evoked vasodilation is not known. The modulatory effect of NO could be due, however, to its inhibition of P450 epoxygenase, which synthesizes the vasodilator EETs. With less EETs being made, vasodilations will be smaller and vasoconstrictions will be unmasked.
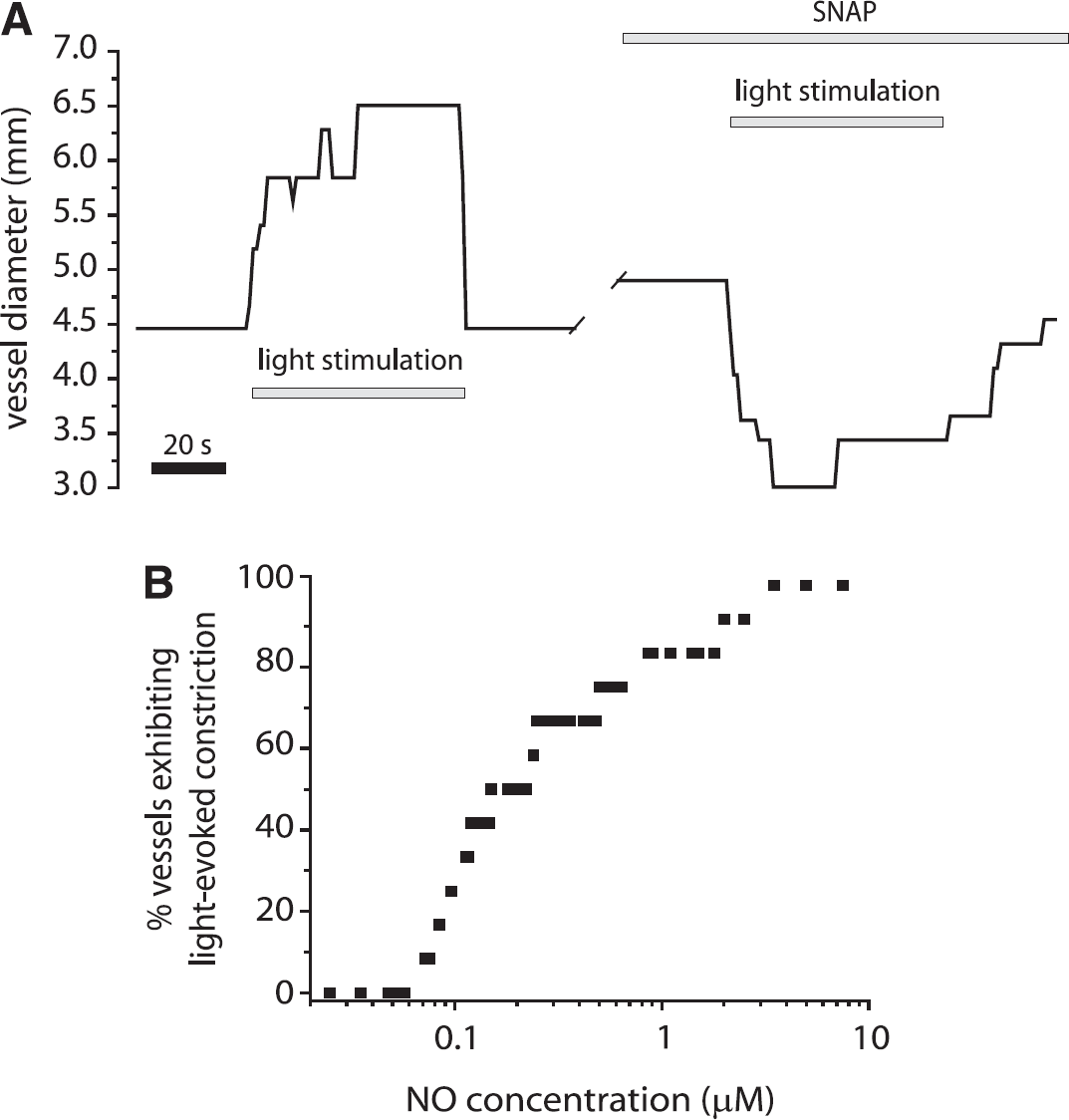
Nitric oxide (NO) depresses light-induced vasodilation and unmasks vasoconstriction in the retina. (
As discussed in the following section, NO modulation of neurovascular coupling also occurs
FUNCTIONAL HYPEREMIA IN THE PATHOLOGIC RETINA
Aging
Systematic changes in blood flow are observed as the retina ages. In humans, basal blood volume, velocity, and blood flow all decrease as subjects age.93,94 A reduction in flicker-evoked arteriole dilation is also observed in older subjects, although the effect was not significant. 95 Multiple factors could account for reductions in basal blood flow and light-evoked increases in blood flow in the aging retina. Age-dependent changes in the responsiveness of vascular smooth muscle cells could have a principal role in the reduction of basal and flicker-evoked blood flow.
Another possible mechanism may involve changes in Ca2+ signaling in retinal glial cells, which, as discussed above, mediate the release of vasodilators and vasoconstrictors onto blood vessels. Glial cells in the retina96,97 as well as the brain 98 generate spontaneous intercellular Ca2+ waves that propagate through networks of glial cells. These glial Ca2+ waves can dilate or constrict retinal arterioles as they propagate past vessels.55,96 In the rat, the frequency at which these glial Ca2+ waves are generated increases substantially with age. 96 This increase in glial Ca2+ signaling may influence retinal blood flow and contribute to the changes in basal and light-evoked blood flow that are observed as the retina ages.
Diabetic Retinopathy
Changes in both basal blood flow and flicker-evoked increases in blood flow occur during the course of diabetic retinopathy. Both increases and decreases in basal blood flow have been reported (reviewed in Pemp and Schmetterer 99 ). An increase in arteriole cross-sectional area and a decrease in blood velocity, leading to a net reduction in blood flow, are seen in patients in early stages of diabetic retinopathy. 100 However, another study showed increased blood flow in the earliest stages of the disease. 101 A trend towards increased blood flow is observed with longer duration of diabetes. 102 A decrease in basal blood flow is observed in the rat streptozotocin model of diabetic retinopathy just 2 weeks after induction of diabetes 103 while a decrease in both retinal and choroidal blood flow is seen in the Ins2Akita mouse model of diabetes at 7 months. 104 These inconsistencies in retinal blood flow changes could be due to several factors, including the stage of diabetic retinopathy at which the observations are made, glycemic control, the techniques used to monitor blood flow, and demographic parameters.
Changes in the functional hyperemia response in diabetic patients are less ambiguous. There is a pronounced decrease in flicker-evoked vasodilation in patients in the early stages of diabetic retinopathy105-107 (Figure 8A). The loss of vasodilation occurs before overt signs of clinical retinopathy are observed. A similar reduction in flicker-evoked vasodilation is seen in a rat model of type 1 diabetes, 92 where light-evoked vasodilations are reduced from 11% to 4% 29 (Figures 8B and 8C). As in patients, this decrease in functional hyperemia in the rat occurs in the early stages of retinopathy, before morphologic changes in the vasculature are seen. The loss of the functional hyperemia response observed in both diabetic patients and in an animal model of diabetes could result in retinal hypoxia and may contribute to the development of retinopathy. 29
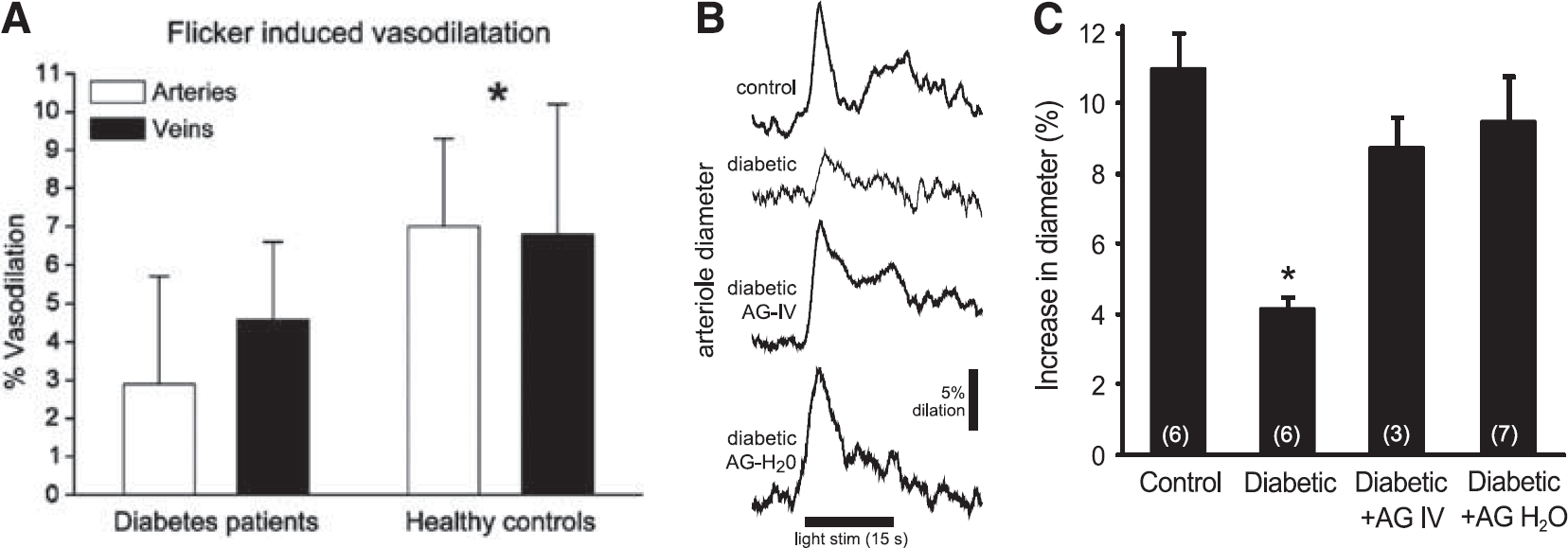
Flicker-induced vasodilation is depressed in diabetic retinopathy. (
Another change that occurs in the early stages of diabetic retinopathy in animal models of diabetes is an upregulation of inducible NOS (iNOS). iNOS increases are seen in both glial cells and neurons in the retina92,108 and result in increased NO levels.108,109 As described above, NO, in addition to acting on vessels to induce vasodilation, is a modulator of neurovascular coupling that suppresses flicker-evoked vasodilation. 55 The upregulation of iNOS and the resulting increase in NO could be responsible for the decrease in flicker-evoked vasodilation observed in diabetic retinopathy. If this is the case, then inhibition of iNOS should result in the restoration of the functional hyperemia response in the diabetic retina.
The effect of inhibiting iNOS has been tested in an animal model of type 1 diabetes. In the rat
Aminoguanidine, by reversing the loss of functional hyperemia, could be a useful therapeutic tool for treating diabetic retinopathy. Indeed, aminoguanidine has been shown to dramatically slow the progression of diabetic retinopathy in animal models of diabetes.108-112 A human trial of aminoguanidine also demonstrates a slowing of diabetic retinopathy progression, although the trial was terminated early due to side effects of aminoguanidine when administered at high doses. 113 In addition to inhibiting iNOS, aminoguanidine also blocks formation of advanced glycation endproducts and its beneficial effect on diabetic retinopathy could be due to this effect on advanced glycation endproducts. However, aminoguanidine slows the progression of diabetic retinopathy without affecting advanced glycation endproduct levels,111,112 suggesting that it acts by inhibiting iNOS. It remains to be determined whether the beneficial effect of iNOS inhibitors in slowing the progression of diabetic retinopathy is due to the restoration of the functional hyperemia response.
Other Pathologies
Changes in retinal blood flow have been observed in a number of other pathologies, although relatively little research has been done in this area. In patients with open-angle glaucoma, a reduction in basal blood flow in the optic nerve head and retina is observed.114-116 Similarly, both retinal and choroidal blood flow is reduced in the DBA/2 J mouse model of glaucoma. 117 Also, in the early stages of the disease, a reduction in flicker-evoked dilation of retinal venules but not arterioles is observed in patients. 118 These observations support the hypothesis that the pathologic changes associated with glaucoma may be due, at least in some instances, to retinal ischemia caused by the increase in intraocular pressure and the resulting decrease in perfusion pressure, leading to a decrease in retinal blood flow. 119 It should be noted, however, that glaucoma can develop in patients with normal intraocular pressure.119,120
In patients with age-related macular degeneration, reductions in choroidal blood flow, but not retinal flow, have been observed.121-124 These reports support the hypothesis that a primary cause of age-related macular degeneration may be a diminished supply of oxygen and nutrients and a decrease in the clearance of waste products from the pigment epithelium and the outer retina caused by reduced choroidal blood flow, possibly arising from stiffening of the sclera and Bruch's membrane due to lipid deposition. 125
Changes in functional hyperemia occur in hypertensive patients, 126 where a reduction in flicker-evoked arteriole dilation is seen. In Alzheimer's patients, a narrowing of retinal venules and a reduction in blood flow occurs. 127 A more detailed characterization of blood flow changes in retinal diseases awaits additional research.
FUTURE DIRECTIONS
Although much is known about blood flow and functional hyperemia in the retinal vasculature, many questions remain to be addressed.
Research during the past decades has implicated many different mechanisms that may mediate neurovascular coupling in the retina. These mechanisms include production of arachidonic acid metabolites by glial cells, release of K + from glial cells and neurons, release of NO, and production of neuronal and glial metabolites, including lactate and adenosine. The relative importance of each of these signaling mechanisms in generating the functional hyperemia response remains to be determined.
Photic stimulation of the retina results in large, rapid dilations of retinal arterioles. These dilations are presumably responsible for generating the increases in blood flow that occur in response to flickering light. However, pericytes, which are intrinsically contractile and surround capillaries, could contribute to generating the functional hyperemia response in the microvasculature. Future research will determine whether capillaries and their associated pericytes actively contribute to generating functional hyperemia.
Reductions in basal blood flow and in flicker-evoked increases in blood flow are observed in aging and in many retinal pathologies. These reductions could result in retinal hypoxia and possibly contribute to the development of retinal pathology. The retina is particularly susceptible to hypoxia caused by compromised blood flow, as it has an extremely high metabolism and O2 consumption. Arden128,129 has proposed that compromised retinal blood flow coupled with the increase in O2 consumption of photoreceptors in the dark may be a primary cause of diabetic retinopathy. Similarly, the decrease in basal blood flow observed in glaucoma and the resulting hypoxia has been suggested to be a primary cause of retinal damage seen in the disease. 115 It remains to be determined whether these changes in retinal blood flow are responsible for the development of diabetes, glaucoma, and other retinal pathologies. An additional challenge will be to determine whether therapies based on reversing reductions in basal blood flow and functional hyperemia are successful in treating these retinal diseases.
DISCLOSURE/CONFLICT OF INTEREST
The author declares no conflict of interest.
Footnotes
ACKNOWLEDGEMENTS
The author thanks Gerhard Garhöfer for his helpful comments on the manuscript.