
Abstract
Cortical spreading depression (CSD) is accompanied by hyperemia followed by long-lasting hypoperfusion and impaired cerebrovascular reactivity. The authors show that vasodilation to extraluminal acidosis (pH 7.0) and increased concentrations of extraluminal potassium (12, 20, 40 mmol/L) was significantly reduced in isolated rat middle cerebral arteries after CSD in vivo before the artery was isolated, compared with sham-operated controls. Application of 80-mmol/L potassium induced vasoconstriction after CSD. Therefore, the impairment of vascular reactivity after CSD in vivo occurs, at least in part, at the vascular level itself.
Keywords
Cerebrovascular reactivity is impaired after global and focal cerebral ischemia (Cipolla et al., 1997; Schmitz et al., 1997), subarachnoid hemorrhage (Alabadi et al., 1997), cortical injury (Golding et al., 2000), migraine (Olesen et al., 1981), and CSD (Wahl et al., 1987). CSD is a short-lasting depolarization wave followed by transient depression of electrical activity slowly propagating over the cortex (Leao, 1944). It is thought to play a role in the pathophysiology of migraine and cerebral ischemia (Back et al., 1994; Olesen et al., 1981; Strong et al., 1983). As shown in animal experiments, the electrophysiologic event of CSD is accompanied by hyperemia and local hyperoxygenation of blood and tissue (Back et al., 1994; Wolf et al., 1996). After hyperemia, a long-lasting hypoperfusion occurs, and cerebrovascular reactivity to dilating stimuli such as hypercapnia, potassium, and somatosensory stimulation is severely impaired for hours (Busch et al., 1999; Lauritzen, 1984; Wahl et al., 1987). The CSD theory of migraine postulates CSD as the electrophysiologic and pathophysiologic correlate of the migraine aura (Lauritzen, 1994). Focal hyperemia followed by spreading oligemia with impaired neurovascular coupling and CO2 reactivity as well as magnetic field correlates of CSD have been shown in patients with classic migraine attacks during aura (Bowyer et al., 2001; Cutrer et al., 1998; Hadjikhani et al., 2001; Lauritzen, 1994; Olesen et al., 1981).
The following study was designed to discriminate parenchymal from vascular mechanisms impairing vascular reactivity after CSD. The authors present, for the first time, strong evidence for an impairment of vascular smooth muscle function in vitro in isolated rat MCA after CSD in vivo.
MATERIALS AND METHODS
Male Wistar rats (240–280 g; n = 27 in the CSD group, n = 27 in the sham-operated group) were anesthetized with isoflurane (1% in N2O/O2). A small craniotomy was performed at the right parietal cortex (3 mm lateral, 1 mm rostral to bregma), and a single CSD was elicited by pinprick in animals in the CSD group. Sham-operated animals without CSD elicitation served as controls. The propagating wave of CSD is accompanied by transient strong hyperemia, which can be used to control for CSD in vivo. To control for the CSD wave to reach the tissue in the cortical area of the distal part of the main trunk of MCA, which was later used to study isolated vascular reactivity, the CSD-induced transient hyperemia was measured in the microcirculation in the vicinity of the main trunk of MCA by laser-Doppler flowmetry through the thinned bone. After CSD, the animals were decapitated, the brain rapidly removed, and the right MCA carefully dissected. The MCA was transferred to an organ chamber filled with cold (4°C) 3-(N-morpholino) propanesulfonic acid (MOPS)-buffered saline, cannulated with glass pipettes, perfused (MOPS-buffered saline containing 1% albumin), pressurized (75–80 mm Hg), and warmed to 37°C (for a detailed description of this method, see Lindauer et al., 2001). During equilibration (30 minutes), the vessels developed spontaneous tone.
In separate experiments, vessel reactivity to extraluminal acidosis (pH 7.0) (n = 7), and to extraluminal elevation of K+ to 20 mmol/L followed by 12 mmol/L (n = 10), to 40 mmol/L (n = 5), or to 80 mmol/L (n = 5) was tested after equilibration. Vascular reactivity in each CSD group was compared with the reactivity in the same number of sham-operated control experiments.
Data are presented as mean ± SD. Vascular reactivity is expressed in absolute values in micrometers or as percent change from resting diameter at baseline. Reactivity of isolated MCA after CSD is compared with sham-operated controls using unpaired Student's t-test. When normality test failed (comparison of reactivity to 12- or 80-mmol/L K+ in absolute values), nonparametric comparison is performed using Mann-Whitney rank sum test. A probability value of P>0.05 was considered statistically significant.
RESULTS
Because of the time involved with preparation and equilibration (30 minutes each), the reactivity of isolated MCA was tested approximately 1 hour after CSD in vivo. Resting diameter in CSD and sham-operated control groups (n = 27 each) decreased from 241 ± 32 μm and 245 ± 33 μm before equilibration to 135 ± 13 μm and 133 ± 12 μm, respectively, after 30 minutes of pressurizing. Vascular reactivity to changes of pH value of extraluminal buffer from 7.4 to 7.0 (acidosis) was reduced in MCAs from rats in the CSD group compared with sham-operated animals. In vessels from sham-operated animals, diameter increased by 53 ± 12 μm, whereas vessels from CSD animals dilated by 33 ± 17 μm (P = 0.02) (Fig. 1).
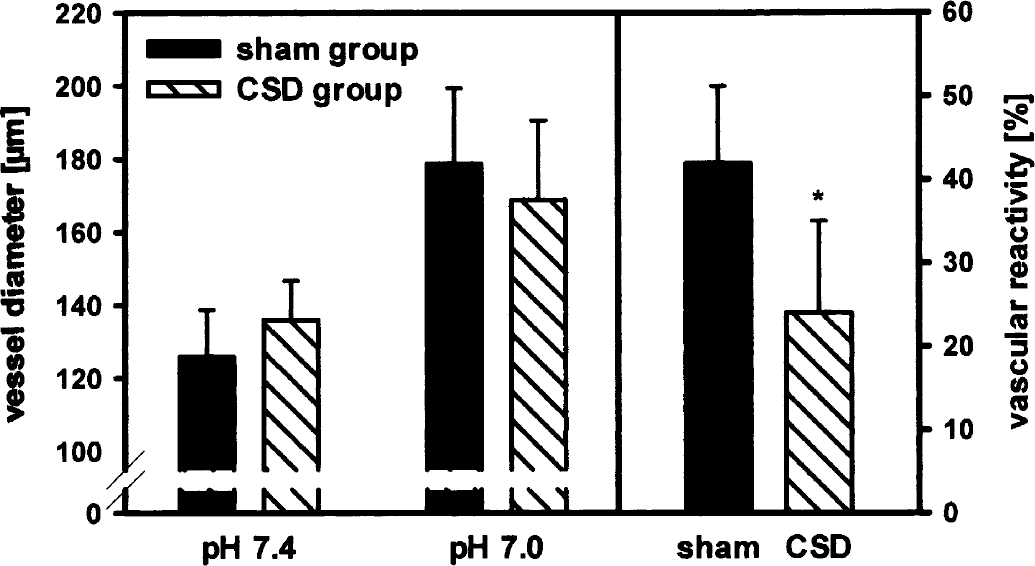
Reactivity of the isolated rat MCA to extraluminal acidosis (change in pH from 7.4 to 7.0): The diameters reached at pH 7.0 are slightly but not significantly reduced in the CSD group compared with the sham-operated group (P > 0.05). However, compared with sham-operated control experiments (n = 7), the percentage reactivity is significantly reduced (*P = 0.006) after elicitation of a single episode of CSD in vivo (n = 7) because of slightly higher baseline diameters in the CSD group at pH 7.4.
During moderately enhanced extraluminal K+ concentration to 12 mmol/L, vasodilation was significantly lower in vessels of CSD-treated animals (25 ± 36 μm) than in sham-operated controls (63 ± 40 μm, P = 0.04) (Fig. 2A). After application of 20-mmol/L extraluminal K+, vessels showed a moderate trend toward smaller values after CSD compared with MCAs from sham-operated animals (vasodilation of 49 ± 38 μm and 64 ± 32 μm, respectively, P > 0.05) (Fig. 2B). Further increase in extraluminal K+ concentration to 40 mmol/L induced a significantly smaller vasodilation in MCAs from CSD-treated animals (74 ± 29 μm) compared with the control group (107 ± 9 μm, P = 0.04) (Fig. 2C). An increase of extraluminal K+ concentrations to 80 mmol/L slightly reduced vessel diameters of MCAs from sham-operated control animals (resting diameter of 135 ± 15 μm to 119 ± 18 μm at 80-mmol/L K+, P = 0.24). In CSD-treated animals, a strong vasoconstriction from a resting diameter of 134 ± 14 μm to 91 ± 2 μm occurred at 80-mmol/L K+ (P = 0.002), which results in a significantly enhanced percentage constriction after CSD compared with arteries of sham-operated controls (P = 0.04) (Fig 2D).
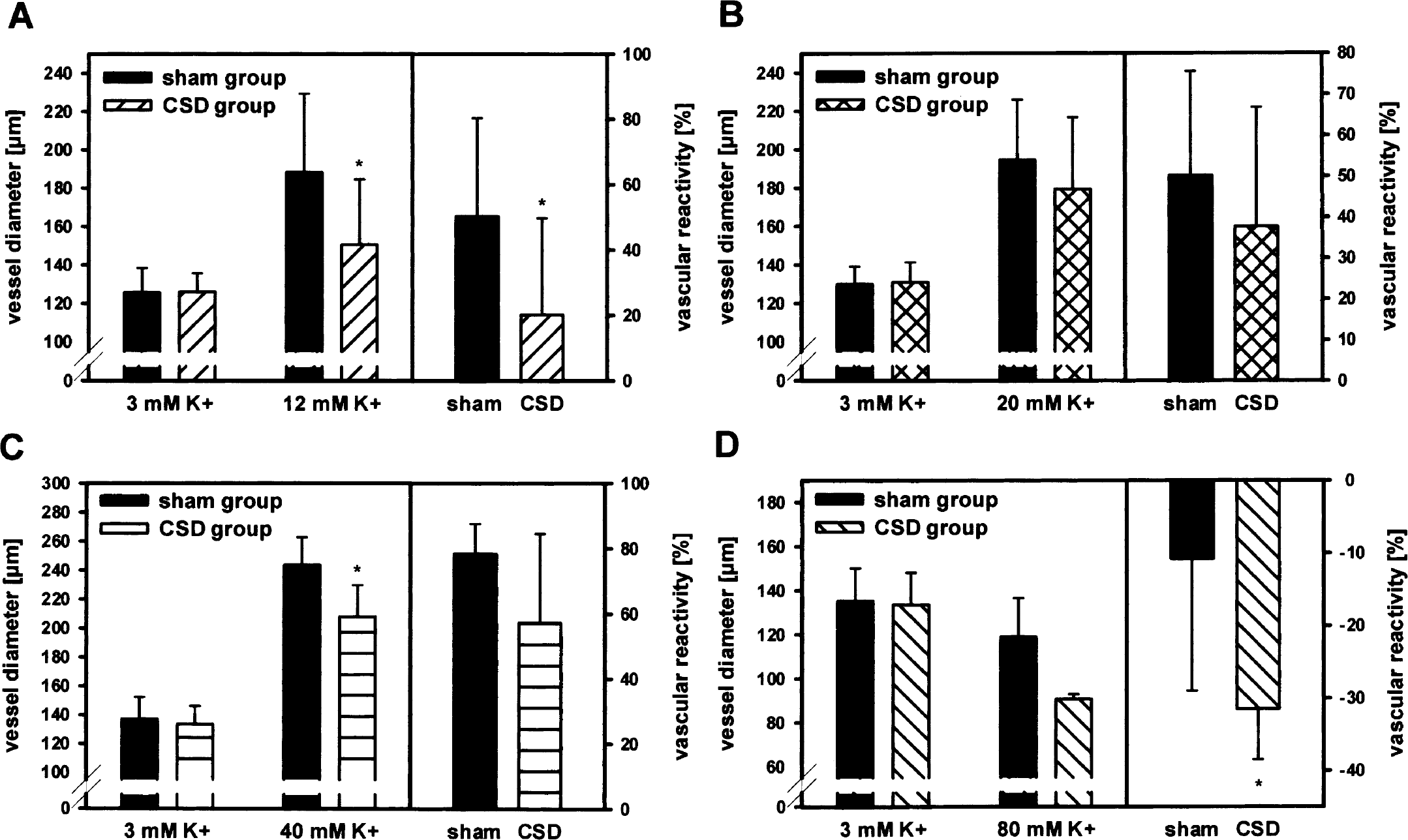
Reactivity of isolated rat MCA to increased extraluminal K+ concentrations of 12 mmol/L (
DISCUSSION
In the present study, we show that the vascular response of isolated rat MCA to extraluminal acidosis and moderately increased extraluminal K+ concentrations was reduced after CSD in vivo compared with sham-operated controls. Increasing the potassium concentration to 80 mmol/L induced strong vasoconstriction after CSD.
In vivo experiments have shown that resting cerebral blood flow is reduced by 20% to 30% after CSD and recovers within the second hour after CSD (Fabricius and Lauritzen, 1993). Because of methodologic constraints, the reactivity of the isolated and cannulated MCA in our model was tested not until 1 hour after elicitation of CSD in vivo. Therefore, we did not find a reduction of resting diameters of MCAs from CSD-treated rats compared with sham-operated controls. However, vascular reactivity to increased H+ or K+ concentrations was reduced 1 hour after CSD. The present in vitro results are in line with the observed impairment of the cerebral blood flow responses to hypercapnia and functional activation in vivo, at least up to 1 hour after CSD (Busch et al., 1999; Lauritzen, 1984). Functional brain activation results in the release of potassium ions from active neurons, reaching extracellular potassium concentration up to 12 mmol/L (Sykova, 1983). After CSD, vasodilation to moderately enhanced extraluminal K+ in the physiologic range of 12 mmol/L was strongly reduced, whereas only a slight impairment of vasodilation to 20 mmol/L occurred. Vasodilation to 12- to 20-mmol/L K+ is mediated by inward rectifier K+-channel (Kir) activity (Johnson et al., 1998). From the present data it can be suggested that CSD induces a reduction in Kir function that is most effective at moderate stimuli in the physiologic range, and increasing stimulus strength can overcome CSD induced channel impairment. Under pathophysiologic conditions like stroke or brain injury, the extracellular K+-concentration increases above 60 mmol/L (Astrup et al., 1980; Katayama et al., 1990). To the authors' knowledge, the strong vasoconstriction of isolated rat MCA induced by pathophysiologically high K+ concentrations of 80 mmol/L after CSD in vivo has not been shown so far. These findings may have important relevance for the understanding of the pathophysiologic cascade of focal cerebral ischemia (Back et al., 1994; Marrelli et al., 1998), of cortical spreading ischemia (Dreier et al., 1998), and of vasospasm after subarachnoid hemorrhage (Beaulieu et al., 2000; Hubschmann and Kornhauser, 1980), where CSD is speculated to participate in the development of damage.
In the present study, an impairment of the reactivity of the distal part of the main trunk of MCA was shown after CSD. The question of why reduced cerebrovascular responses occur only in the cerebral cortex after CSD and not in deeper structures, as shown by previous hemodynamic studies in vivo (Lauritzen, 1984), is therefore raised. Local cerebral blood flow is mainly regulated by small arteries and arterioles as the main resistance vessels in cerebral tissue. Impairment of local tissue blood flow regulation exclusively induced by disturbances of the function of the main trunk of MCA will only occur under severe reductions of the diameter of the main trunk of MCA as for example during severe vasospasm after brain hemorrhage. In parallel to changes of the main trunk of MCA after CSD, it can be suggested that vascular reactivity of cortical pial arteries and penetrating arterioles is comparably reduced because they are identically affected by tissue changes during CSD. This will lead to reduced corticovascular responses within the microcirculation. However, propagation of spreading depression elicited within the cerebral cortex is mainly restricted to cortical layers. Therefore, resistance vessels of deeper brain areas will not be struck by CSD. Because of the preserved or only slightly reduced resting diameter of the MCA trunk after CSD, no significant changes in local vascular reactivity seem to occur in deeper brain areas not reached by the CSD wave.
The mechanisms impairing cerebrovascular smooth muscle cell function after CSD are not known. The results of the present study after CSD are reminiscent of published studies after pharmacologic NO synthase inhibition, in which reduced reactivity to acidosis and 40-mmol/L K+ and vasoconstriction to 80-mmol/L K+ occurred (Golding et al., 2001; Lindauer et al., 2001; Schuh-Hofer et al., 2001). During CSD, NO concentrations in the tissue transiently increase (Read and Parsons, 1998). Therefore, a shortage of L-arginine, the substrate for NO production, has been suggested, and recovery of hypercapnic flow rise was enhanced in L-arginine–treated animals (Fabricius et al., 1995). Therefore, one might consider the possibility of a CSD-induced reduction in the physiologic basal NO concentration surrounding cerebral blood vessels. However, no reduction of basal NO concentration was detected after CSD (Read and Parsons, 1998). Other mechanisms may also lead to vasoconstriction and reduced vascular reactivity after CSD. A rapid activation of protein kinase C in the cerebral cortex has been shown within the first 30 minutes after CSD (Kurkinen et al., 2001). Increased protein kinase C activity induces inhibition of Kir, ATP-sensitive, and Ca2+-activated K+ channels by phosphorylation (Chrissobolis and Sobey, 2002; Standen and Quayle, 1998; Taguchi et al., 2000). These K+-channel families are involved in vasodilation to increased K+ concentrations or acidosis (Johnson et al., 1998; Lindauer et al., 2003). Furthermore, mechanisms leading to a reduction in vascular sensitivity to vasodilators and amplification to vasoconstrictors by sensitization of myofilaments to Ca2+ can be suggested, leading to reduced vasodilation to acidosis and moderately enhanced K+ and strong vasoconstriction to 80-mmol/L K+ after CSD. Further experiments have to be performed to resolve the mechanisms of reduced vascular reactivity after CSD.
While investigating vascular reactivity of isolated MCA from CSD treated rats, the present study combines the advantage of CSD elicitation as a pathophysiologic model of migraine aura in vivo and the investigation of vascular reactivity of cerebral arteries without parenchymal influences. It can be concluded that the impairment of vascular reactivity after CSD in vivo occurs, at least in part, at the vascular level. These findings point toward a functional impairment of the vasculature of the cerebral cortex itself within the pathophysiology during migraine.