
Abstract
Albeit controversely discussed, it has been suggested by several authors that nitric oxide (NO) serves as a permissive factor in the cerebral blood flow response to systemic hypercapnia. Potassium channels are important regulators of cerebrovascular tone and may be modulated by a basal perivascular NO level. To elucidate the functional targets of the proposed NO modulation during hypercapnia-induced vasodilation, the authors performed experiments in isolated, cannulated, and pressurized rat middle cerebral arteries (MCA). Extracellular pH was reduced from 7.4 to 7.0 in the extraluminal bath to induce NO dependent vasdilation. Acidosis increased vessel diameter by 35 ± 10%. In separate experiments, ATP-sensitive potassium channels (KATP) were blocked by extraluminal application of glibenclamide (Glib), Ca2+-activated potassium channels (KCa) by tetraethylammonium (TEA), voltage-gated potassium channels (Kv) by 4-aminopyridine, and inward rectifier potassium channels (KIR) by BaCl2. Na+-K+-ATP-ase was inhibited by ouabain. Application of TEA slightly constricted the arteries at pH 7.4 and slightly but significantly attenuated the vasodilation to acidosis. Inhibition of the other potassium channels or Na+-K+-ATP-ase had no effect. Combined blockade of KATP and KCa channels further reduced resting diameter, and abolished acidosis induced vasodilation. The authors conclude that mainly KCa channels are active under resting conditions. KATP and KCa channels are responsible for vasodilation to acidosis. Activity of one of these potassium channel families is sufficient for vasodilation to acidosis, and only combined inhibition completely abolishes vasodilation. During NO synthase inhibition, dilation to the KATP channel opener pinacidil or the KCa channel opener NS1619 was attenuated or abolished, respectively. The authors suggest that a basal perivascular NO level is necessary for physiologic KATP and KCa channel function in rat MCA. Future studies have to elucidate whether this NO dependent effect on KATP and KCa channel function is a principle mechanism of NO induced modulation of cerebrovascular reactivity and whether the variability of findings in the literature concerning a modulatory role of NO can be explained by different levels of vascular NO/cGMP concentrations within the cerebrovascular tree.
The diameter of cerebral blood vessels and consequently cerebral blood flow (CBF) are highly dependent on tissue proton (H+) concentration (Busija and Heistad, 1984; Kontos et al., 1977). Hypercapnic, as well as normocapnic, acidosis has been shown to cause vasodilation in cerebral blood vessels (Aalkjaer and Poston, 1996; Dacey and Duling, 1982; Dietrich and Dacey, 1994). Changes in extracellular pH have been discussed to mediate coupling of CBF to brain function (Lou et al., 1987), and tissue acidification is one of the major early events during focal cerebral ischemia (Kohno et al., 1995; Mabe et al., 1983). The means by which extra- or intracellular H+ dilates cerebral vessels is, however, still unknown.
The bioradical nitric oxide (NO) is not only a potent vasodilator but may also be an important modulator of cerebrovascular reactivity (Lindauer et al., 1999; Iadecola et al., 1994). It is suggested that a basal NO level in perivascular brain tissue is necessary for proper vascular smooth muscle cell function. It has been shown that the cerebrovascular responses to functional activation, hypercapnia, or tissue acidosis are dependent on NO in both in vivo and in vitro studies (Dirnagl et al., 1993; Lindauer et al., 2001; Niwa et al., 1993; You et al., 1994). Thereby, the role of NO is suggested to be modulatory; a basal vascular NO/cGMP concentration may be necessary for other, still unknown mechanisms, which mediate vasodilation (Iadecola et al., 1994; Lindauer et al., 1999, 2001; Okamoto et al., 1997; Tuettenberg et al., 2001). In vitro evidence additionally suggests that extracellular acidosis does not induce NO or cGMP production (You et al., 1994).
The molecular mechanism of acidosis-induced vasodilation and the role of the basal NO level in the vicinity of cerebral arteries, providing physiologic smooth muscle cell function and pH-dependent as well as functional activation-induced vascular reactivity, remain enigmatic. Hypercapnic and normocapnic acidosis (Tian et al., 1995), as well as acidosis in HCO3−/CO2 free systems (Austin and Wray, 1993), are associated with intracellular acidosis in vascular tissues. Nevertheless, there is evidence that changes in intracellular pH are not responsible for smooth muscle cell relaxation during extracellular pH reduction (Aalkjaer and Poston, 1996). Thus, the question arises how the information of increased extracellular proton concentration is transduced into vasodilation. Relaxation or constriction of cerebrovascular smooth muscle is mainly dependent on the intracellular Ca2+ concentration, which is in part regulated by voltage-gated Ca2+ channels (for review, see Faraci and Sobey, 1998). Extracellular acidosis is associated with increased potassium conductance causing hyperpolarisation that, in turn, leads to closure of voltage-gated Ca2+ channels and smooth muscle relaxation (Bonnet et al., 1991; Dietrich and Dacey, 1994; Harder, 1982). Potassium channel activity has been shown to be modulated by NO or cGMP and by intracellular proton concentration (Bolotina et al., 1994; Bonnet et al., 1991; Robertson et al., 1993; Santa et al., 2003; Xu et al., 2001).
In cerebral vessels, the expression of at least four different types of potassium (K+) channel families has been shown: ATP-sensitive K+ channels (KATP), calcium (Ca2+)-activated K+ channels (KCa), voltage-gated K+ channels (Kv), and classical inwardly rectifying K+ channels (KIR) (Faraci and Sobey, 1998; Johnson et al., 1998; Nelson and Quayle, 1995). In addition, the enzyme Na+-K+-ATP-ase is involved in the regulation of extra-/intracellular K+ concentration, and NO has been suggested to modulate the vascular smooth muscle Na+-K+-ATP-ase (Gupta et al., 1994).
Recent findings provide evidence for a contribution of KATP channels in the vasodilation of cerebral arteries to acidosis (Faraci et al., 1994; Horiuchi et al., 2002; Kinoshita and Katusic, 1997; Kontos and Wei, 1996). However, the results concerning a role for K+ channels in acidosis-induced cerebrovasodilation are inconclusive, as others did not find evidence for a role of K+ channels in hypercapnia-induced cerebrovasodilation under physiologic conditions (Bayerle-Eder et al., 2000; Reid et al., 1993; Wang et al., 1998; for review, see Faraci and Sobey, 1998).
The present study is the first to systematically investigate the role of the four known K+ channel families and of Na+-K+-ATP-ase of vascular smooth muscle cells in vasodilation to extraluminal acidosis in rat middle cerebral artery (MCA). The authors show that a combined activation of KATP and KCa channels is responsible for smooth muscle relaxation to extraluminal acidosis of the isolated MCA. In addition, the authors provide strong evidence for a role of NO regulating the activity of KATP and KCa channels.
METHODS
Animal preparation
All animal experiments were approved by the Governmental Animal Care and Use Committee. Male Wistar rats (250 to 350 g; n = 124) were anesthetized with halothane and decapitated. The brain was rapidly removed from the skull and placed in cold (4°C) 3-(N-morpholino)propanesulfonic acid (MOPS) buffered saline solution (with 1% dialyzed bovine serum albumin) at pH 7.40 ± 0.02 (Duling and Rivers, 1986).
Isolation and cannulation of rat MCA
A detailed description of isolation and cannulation of rat MCA was provided in a recent publication (Lindauer et al., 2001). Briefly, approximately 1 cm of MCA was carefully dissected from the brain and cannulated on glass micropipettes. The vessel was continuously perfused with MOPS buffered saline solution at a transmural pressure of 80 mm Hg and heated to 37°C. The extraluminal bath contained MOPS buffered saline solution without bovine serum albumine; it was heated to 37°C and continuously exchanged at a rate of 20 mL/min. The vessel chamber was placed on an inverted microscope equipped with a videocamera and monitor for online analysis of luminal diameters of the arteries. The camera was connected to a PC to store video images after each manipulation for later off-line analysis.
After preparation, the vessels were allowed to equilibrate for 1 hour. Within this time period, spontaneous tone developed, reducing resting diameter to approximately 60%, compared with diameters measured directly after start of inflow and pressurizing at room temperature. During the entire experiment, temperature, perfusion inflow pressure, and flow rate were kept constant. All pharmacologically active substances were added to the extraluminal bath. Acidotic vasodilation was induced by changing the pH of the extraluminal bath by adding HCl to the MOPS buffered saline solution. Luminal diameters were registered after an equilibration period of 10 minutes.
Chemicals
The following drugs were used: Tetraethylammonium ion (TEA, 10−3 mol/L); BaCl2 (4 × 10−5 or 10−4 mol/L); 4-aminopyridine (4-AP, 10−3 mol/L), ouabain (10−3 mol/L), charybdotoxin (CHTX, 10−7 mol/L), NS1619 (10−5 mol/L dissolved in ethanol reaching final concentration of 1.0%), and Nω-nitro-L-arginine (L-NNA, 10−5 mol/L); purchased from Sigma Chemicals. Glibenclamide (Glib, 10−5 mol/L dissolved in ethanol reaching final concentration of 0.2%) and pinacidil (10−5 mol/L, dissolved in ethanol reaching final concentration of 0.1%) were purchased from Research Biochemicals International. The drugs were dissolved in MOPS buffer if not further mentioned.
Experimental design
Group I: Effect of NOS inhibition on vasodilation to increased extraluminal proton concentrations
Vascular responses to a stepwise decrease in extraluminal pH (7.4, 7.3, 7.2, 7.1, 7.0, 6.8, 6.5, and 6.0) were recorded under normal conditions and after NOS inhibition by L-NNA (30 minutes extraluminal application, n = 4). In all further experiments, vasodilation was recorded after change of extraluminal pH from 7.4 (referred to as “physiologic”) to 7.0 (referred to as “acidosis”).
Groups II–VII: Influence of blockade of single potassium channel types on vessel response to extraluminal acidosis in the isolated cerebral artery
In control group II (n = 6), three consecutive responses to pH 7.0 acidosis were recorded. In experimental group III to VII, after recording a baseline response to extraluminal acidosis, specific potassium channel inhibitors were applied to the extraluminal bath for 20 minutes (group III, n = 19, TEA; group IV, n = 7, Glib; group V, n = 5 each concentration, BaCl2; group VI, n = 6, 4-AP; group VII, n = 4, ouabain). The vessel response to acidosis was tested again.
Groups VIII–XII: Influence of combined blockade of potassium channel types on vessel response to extraluminal acidosis in the isolated cerebral artery
In experimental group VIII to XII, after recording a baseline response to extraluminal acidosis, combinations of potassium channel inhibitors were applied to the extraluminal bath for 20 minutes (group VIII, n = 5, Glib + BaCl2 10−4 mol/L; group IX, n = 6, TEA + Glib; group X, n = 4, CHTX + Glib; group XI, n = 6, TEA + BaCl2 10−4 mol/L, followed by outwash of BaCl2 and followed by application of TEA + 4-AP; group XII, n = 5, 4-AP + BaCl2 10−4 mol/L, followed by outwash of BaCl2 and followed by application of 4-AP + Glib). The vessel response to acidosis was tested again.
Group XIII: Vasodilation to acidosis during increasing concentrations of the KATP channel inhibitor Glib
After recording a baseline response to acidosis, the KATP channel inhibitor Glib was added to the extraluminal bath in increasing concentrations of 10−6, 10−5, 10−4, and 10−3 mol/L. Vasodilation to acidosis was tested at each concentration (n = 5).
Groups XIV and XV: Effect of NOS inhibition during response to KATP opener pinacidil and KCa opener NS1619
The vasodilation to the KATP channel opener pinacidil (group XIV, n = 10) or the KCa channel opener NS1619 (group XV, n = 13) was recorded before and after NOS inhibition by L-NNA, which was applied for 30 minutes.
Statistical analysis
From each rat brain, only one vessel was studied. Values are mean ± SD. Comparisons were made using repeated-measures ANOVA followed by multiple comparison procedures (Bonferroni's method). When normality test failed, comparisons were made using Friedmann repeated measures ANOVA on ranks with a subsequent Student-Newman-Keuls test to perform multiple comparisons. P values < 0.05 were considered statistically significant.
RESULTS
After developing spontaneous tone, smooth muscle cell function was tested at the beginning of each experiment by increasing extraluminal potassium concentration to 2 × 10−2 mol/L. Only vessels were used for the experiments that showed a potassium-induced dilation of more than 25%.
Vasodilation to increased extraluminal proton concentration
Stepwise increases of extraluminal proton concentration from pH 7.4 to pH 7.0 led to a linear increase in vessel diameter. The maximum of dilation was reached at pH 7.0 with no further diameter increase on lowering pH to 6.0. After NOS inhibition by L-NNA, the vascular response to acidosis down to pH 6.5 was abolished. However, vasodilation in response to further acidification to pH 6.0 was preserved despite NOS inhibition (Fig. 1). In control experiments, repetitive stepwise reductions of pH from 7.4 to 6.0 led to identical dose-response curves (n = 3, data not shown).
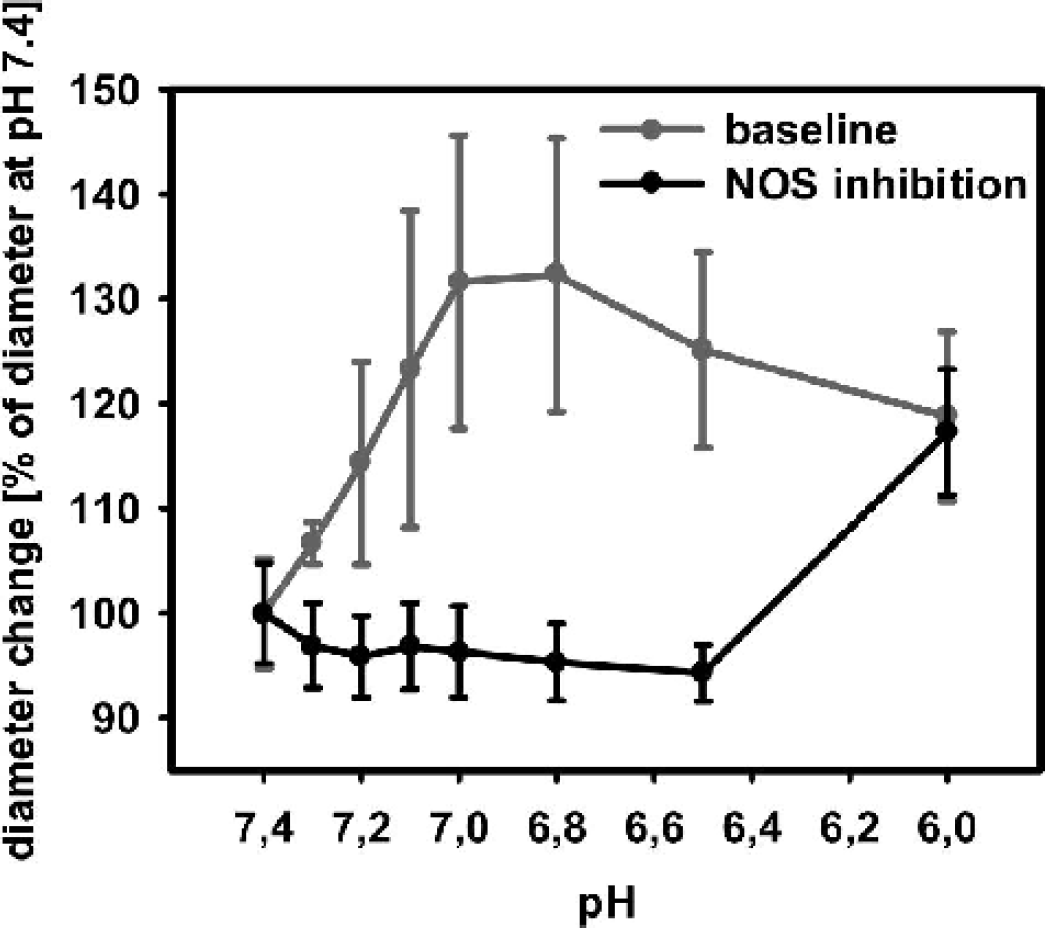
Significant change of vascular diameter during stepwise increase of extraluminal proton concentration from pH 7.4 to pH 6.0. After NOS inhibition by L-NNA, vascular response to acidosis up to pH 6.5 was abolished, whereas vasodilation to pH 6.0 was preserved. NOS, nitric oxide synthase; L-NNA, Nω-nitro-L-arginine.
In the following experiments, reduction of pH to 7.0 was chosen to investigate the role of potassium channels during the NO dependent vasodilation to acidosis.
Vessel diameter at pH 7.4 or pH 7.0
After equilibration, MCA diameter stabilized at 130 ± 11 μm at pH 7.4, and extraluminal pH reduction to 7.0 led to a consistent and statistically significant (P < 0.01) diameter increase of 46 ± 13 μm (35 ± 10%), reaching vessel diameters of 176 ± 18 μm. Repetitive reductions of extraluminal pH led to comparable vasodilations, as shown in the first panel of Fig. 2.
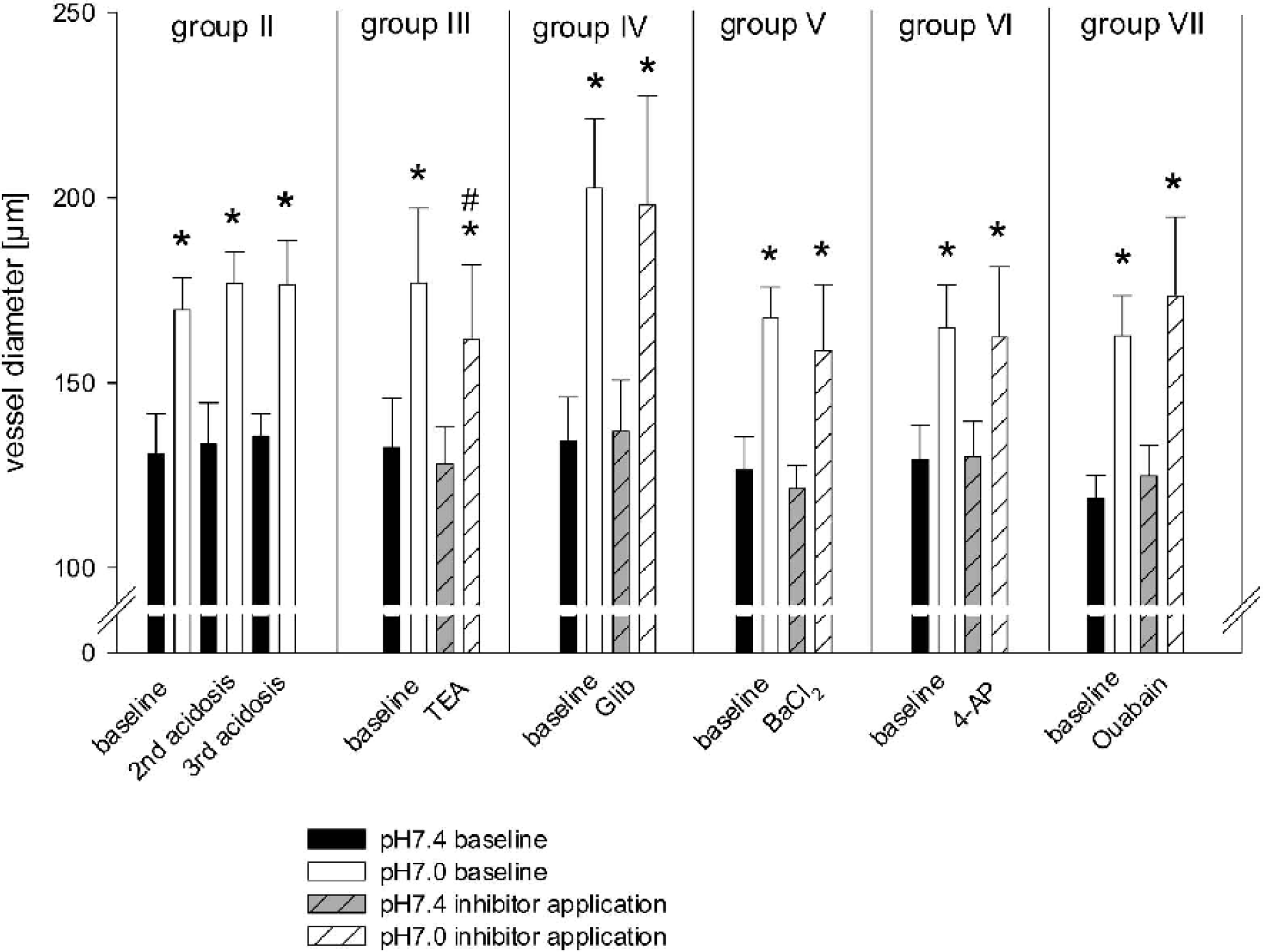
Effect of single potassium channel inhibition on vasodilation to extraluminal acidosis (pH 7.0). During KCa channel inhibition by TEA, vasodilation to extraluminal acidosis was significantly reduced to approximately 77%, compared with baseline dilation before KCa channel inhibition. ∗ P < 0.05 significantly different to vessel diameter at pH 7.4 under same condition; #P < 0.05 significantly different to vessel diameter at pH 7.0 under baseline condition. TEA, tetraethylammonium; Glib, glibenclamide.
Effect of inhibition of single K+ channel types on resting diameter at pH 7.4 and on reactivity to acidosis
Extraluminal application of the KCa channel inhibitor TEA (group III) slightly constricted the vessels at pH 7.4 (P = 0.05), whereas neither Glib (KATP channel inhibitor) nor BaCl2 (KIR channel inhibitor), 4-AP (KV channel inhibitor), or ouabain (Na+-K+-ATPase inhibitor) changed resting diameter. The acidosis induced dilation was significantly reduced by the KCa channel inhibitor TEA to approximately 77%, compared with baseline dilation before KCa channel inhibition (P < 0.01). In contrast, the dilation to acidosis was not altered by the other inhibitors Glib, BaCl2, 4-AP, or ouabain (Fig. 2).
Efficacy of channel inhibition by TEA (KCa channel), Glib (KATP channel), or BaCl2 (10−4 mol/L; KIR channel) was confirmed by the ability of blocking the dilation to the specific channel openers NS1619 (n = 3), pinacidil (n = 3), or K+ (2 × 10−2 mol/L; n = 5), respectively (Fig. 3).
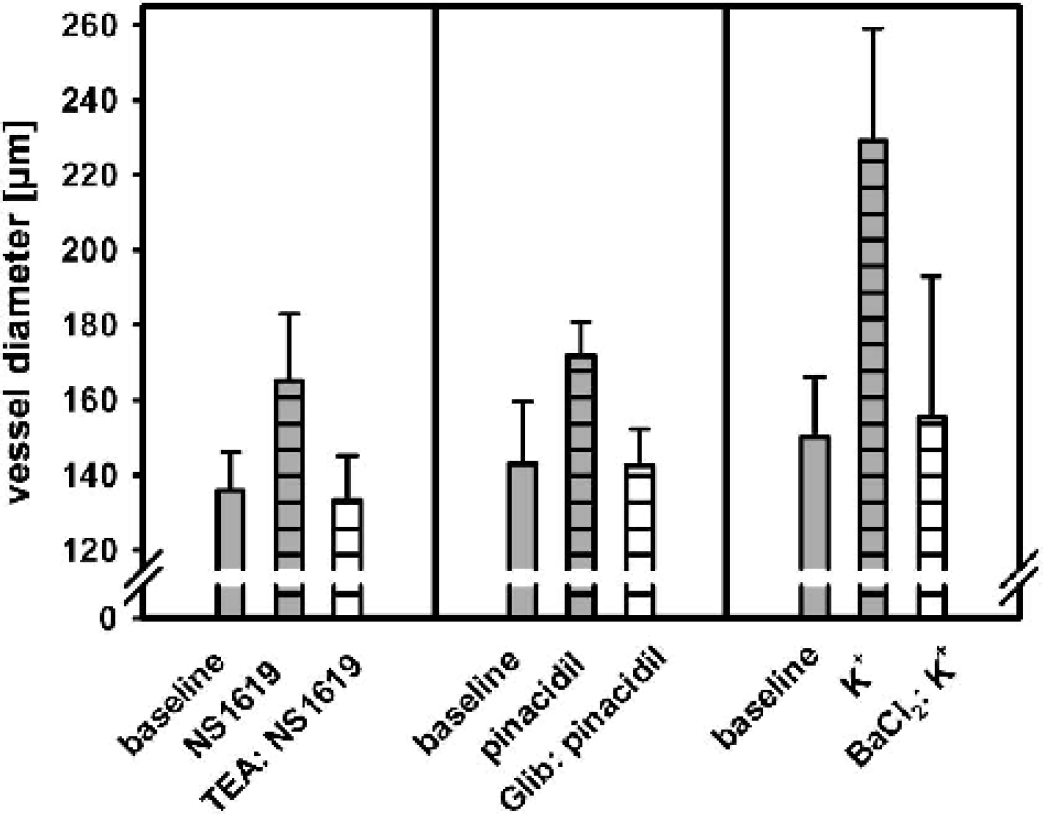
Potassium channel inhibition by TEA, Glib, or BaCl2 in the concentrations used during vasodilation to acidosis completely blocked the dilation to the specific channel openers NS1619, pinacidil or K+ (2 × 10−2 mol/L), respectively. TEA, tetraethylammonium; Glib, glibenclamide.
Effect of combined inhibition of K+ channel types on resting diameter at pH 7.4 and on reactivity to acidosis
Combined inhibition of KCa channels with TEA and KATP channels with Glib (group IX) or KIR channels with BaCl2 (group XI) significantly reduced vessel diameter at pH 7.4, whereas the combination of TEA and 4-AP (inhibition of KCa channels and KV channels, group XI, 2nd combination) did not change resting diameter. Specific inhibition of large conductance KCa channels with CHTX combined with KATP channel blockade with Glib (group X) also significantly constricted the vessels at resting conditions. Constriction at pH 7.4 during combined inhibition of KCa and KATP or KCa and KIR channels was more pronounced than reduction of resting diameter during KCa channel blockade alone. All other combinations tested had no effect on vessel diameter at pH 7.4 (Fig. 4).
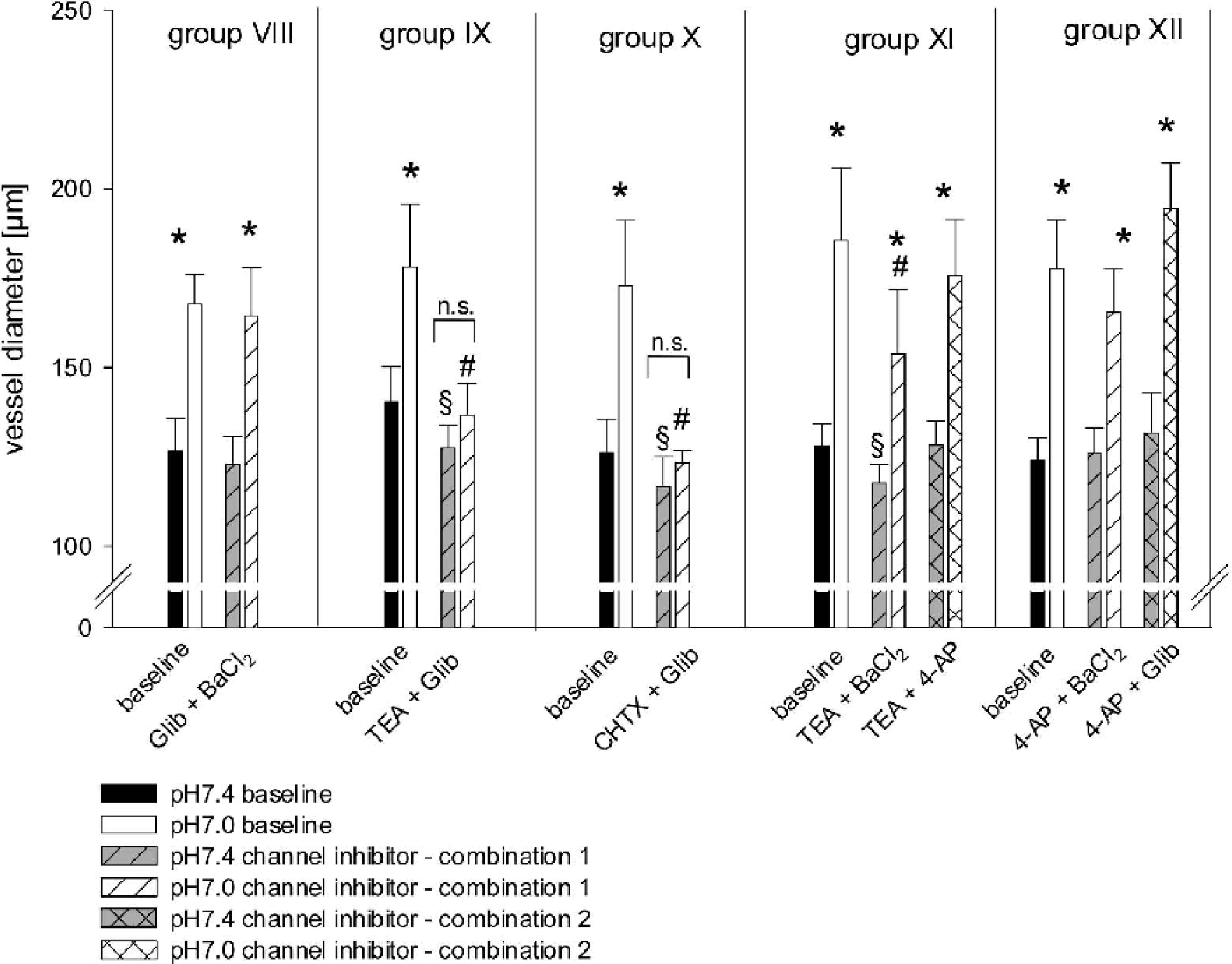
Effect of combined potassium channel inhibition on vasodilation to extraluminal acidosis (pH 7.0). During combined inhibition of KCa and KATP (TEA + Glib, TEA + CHTX) or KCa and KIR (TEA + BaCl2) channels constriction at pH 7.4 occurred, which was more pronounced than reduction of resting diameter during KCa channel blockade alone (see Fig. 2). Combined inhibition of KCa and KATP (TEA + Glib, TEA + CHTX) abolished the dilation response to acidosis. ∗ P < 0.05 significantly different to vessel diameter at pH 7.4 under same condition; #P < 0.05 significantly different to vessel diameter at pH 7.0 under baseline condition; ∗ P < 0.05 significantly different to vessel diameter at pH 7.4 under baseline condition. TEA, tetraethylammonium; Glib, glibenclamide; CHTX, charybdotoxin.
Combined extraluminal application of TEA and Glib (group IX) or CHTX and Glib (group X) abolished the dilation response to acidosis. Inhibition of KCa channels with TEA together with blockade of KIR channels with BaCl2 (group XI) significantly attenuated but did not abolish the dilation to pH 7.0. The effect of TEA + BaCl2 on the vessel dilation to acidosis was not significantly different to the effect of KCa channel inhibition by TEA alone. All other combinations tested had no influence on vascular reactivity to extraluminally applied acidosis (Fig. 4).
Vasodilation to acidosis during increased concentration of the KATP channel inhibitor Glib
A dose-response study was undertaken to investigate whether vasodilation to acidosis in the present study was inhibited by higher, more unspecific concentrations of Glib. At concentrations of 10−6 or 10−5 mol/L, no change in vasodilation to acidosis was observed. As shown previously (Fig. 3), Glib at a concentration of 10−5 mol/L effectively blocked the dilation to the specific KATP channel opener pinacidil. By increasing Glib to concentrations of 10−4 or 10−3 mol/L, which have been shown to block not only KATP but also KCa channels (Gelband and McCullough, 1993), vasodilation to acidosis was significantly reduced or abolished, respectively (Fig. 5). Resting diameter at pH 7.4 did not change during glibenclamide application at concentrations of 10−6 and 10−5 mol/L. At the concentration of 10−4 mol/L, a significant reduction of approximately 15% (117 ± 13 μm compared with 139 ± 12 μm before Glib application) of resting diameter was observed, whereas Glib at the concentration of 10−3 mol/L induced a significant increase in resting diameter of approximately 55%.
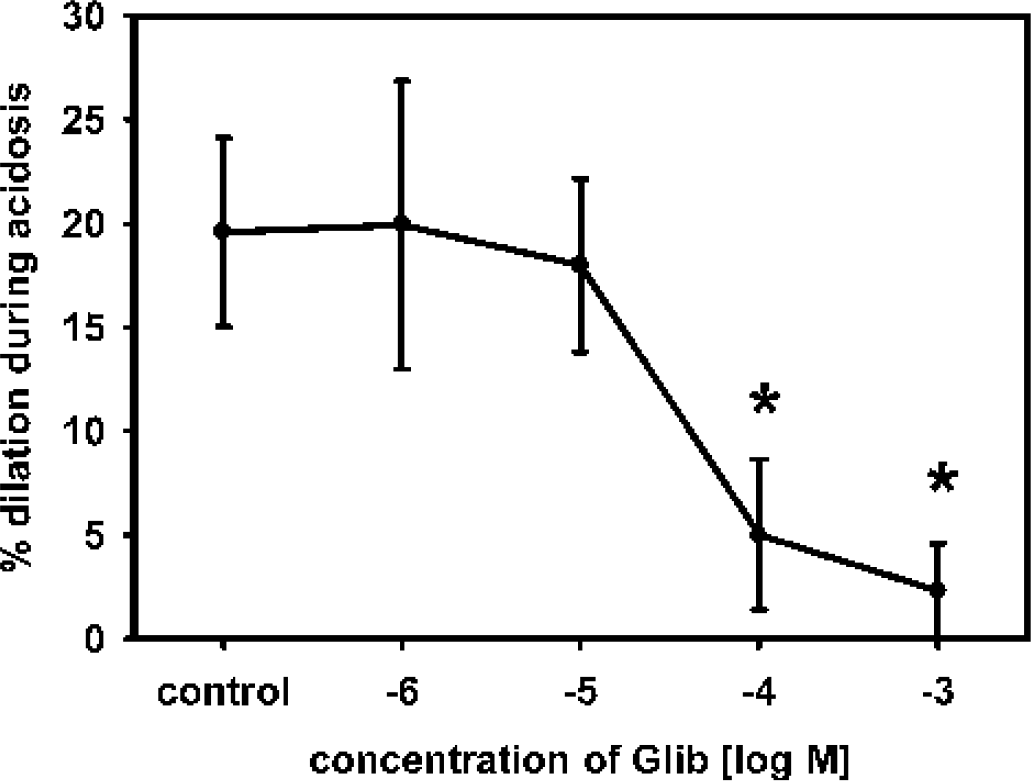
Dose response relationship of Glib during vasodilation to acidosis. At concentrations of 10−6 or 10−5 mol/L, no significant change in vasodilation to acidosis was observed. Whereas increasing Glib to concentrations of greater than 10−5 mol/L, vasodilation to acidosis was significantly reduced or abolished, respectively. ∗ P < 0.05 significantly different to control response. Glib, glibenclamide.
Vasodilation to KATP channel opener pinacidil or KCa channel opener NS1619: effect of L-NNA application
The KATP channel opener pinacidil significantly dilated MCAs by 33 ± 12 μm under baseline conditions. Repetitive application of pinacidil induced comparable dilations (time control experiments, n = 3, data not shown). Extraluminal application of L-NNA significantly attenuated resting diameter to 78 ± 5% of baseline value. During L-NNA application pinacidil dilated the vessels by 17 ± 7 μm (P < 0.01 compared with baseline dilation).
The KCa channel opener NS1619 significantly dilated MCAs by 17 ± 11 μm under baseline conditions. Repetitive application of NS1619 induced comparable dilations (time control experiments, n = 3, data not shown). Extraluminal application of L-NNA significantly attenuated resting diameter to 79 ± 6% of baseline value. During L-NNA application, dilation to NS1619 was abolished (P < 0.01 compared with baseline dilation) (Fig. 6).
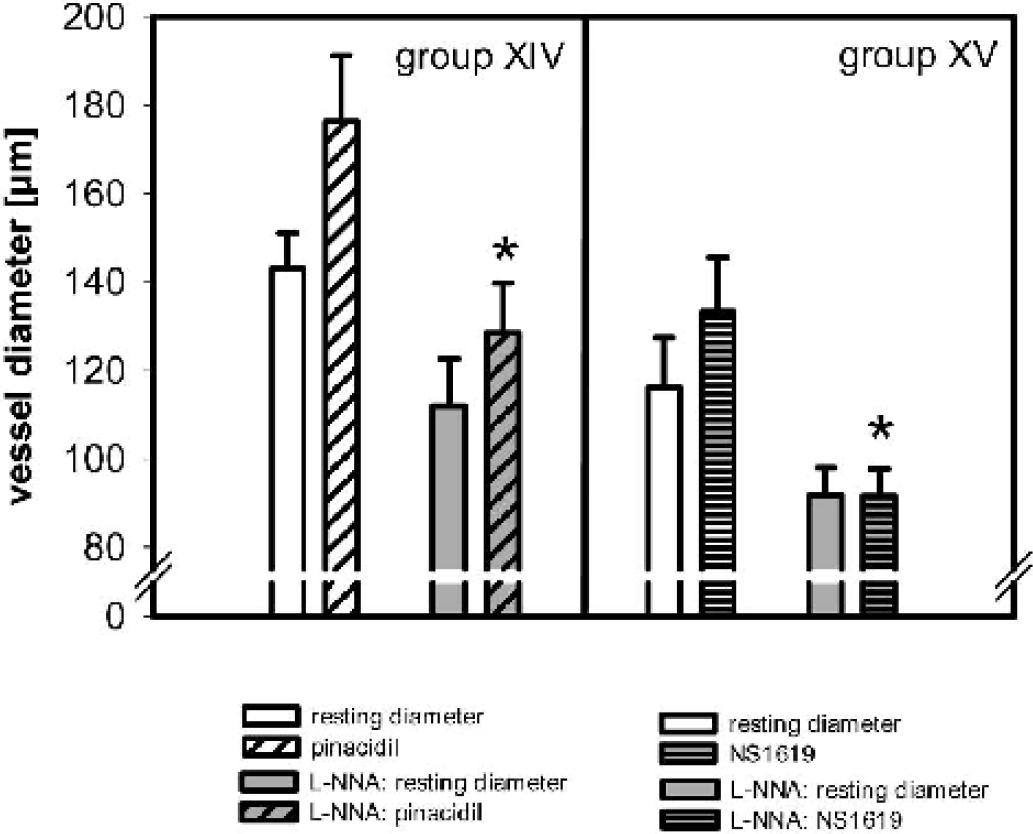
Effect of NOS inhibition (L-NNA application) on vasodilation to the specific KATP channel opener pinacidil or to the specific KCa channel opener NS1619. During NOS inhibition, vasodilation to pinacidil was significantly reduced, whereas vasodilation to NS1619 was abolished. ∗ P < 0.05 significantly different to vessel diameter during pinacidil/NS1619 application under resting conditions. NOS, nitric oxide synthase; L-NNA, Nω-nitro-L-arginine.
Effect of combined blockade of KCa and KATP channels or NOS inhibition on vasodilation to 1.2 and/or 2 × 10−2 mol/L extraluminal K+
To ascertain that combined blockade of KCa and KATP channels by TEA + Glib did not unspecifically abolish vasodilation of the isolated arteries, the authors tested the reactivity of the MCA to moderately elevated extraluminal K+ concentrations of 1.2 and 2 × 10−2 mol/L (n = 4). Vasodilation in this concentration range has been shown to be exclusively mediated by KIR channels at vascular smooth muscle cells independently of NO/cGMP. Vasodilation to 1.2 and 2 × 10−2 mol/L extraluminal K+ was unchanged during TEA + Glib application (Fig. 7A).
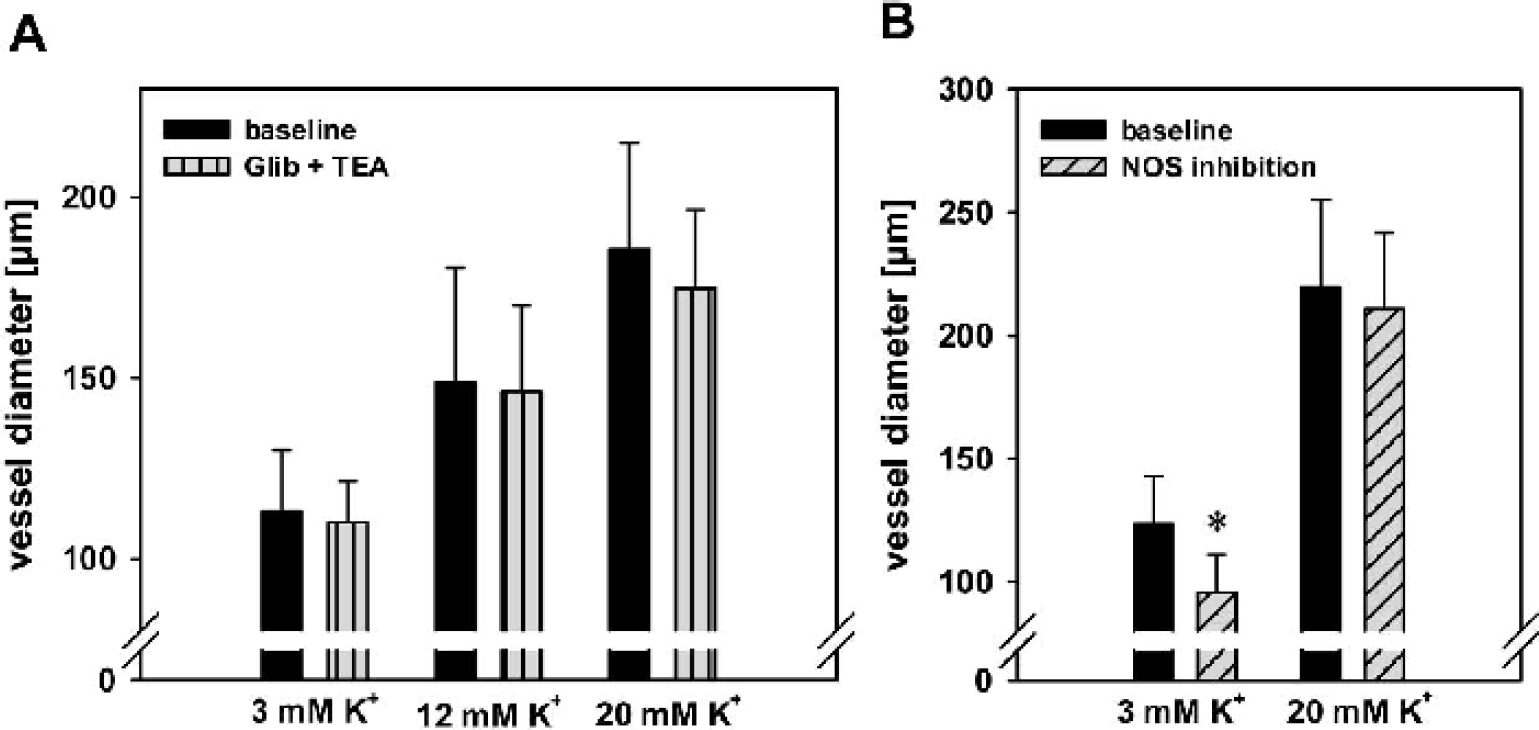
To rule out that NOS inhibition by L-NNA (10−5 M) is exhibiting nonselective effects in this model, the authors tested the reactivity of the MCA to moderately elevated extraluminal K+ concentrations of 2 × 10−2 mol/L (n = 4), which was unchanged during L-NNA application compared with baseline reactivity (Fig. 7B). This finding is in agreement with the authors' previous results on the role of NOS inhibition on cerebrovascular potassium reactivity using the same model of rat MCA preparation (Schuh-Hofer et al., 2001).
DISCUSSION
In the present study, the authors systematically characterized the basal and extracellular acidosis induced activity of K+ channels on isolated rat brain MCA. The key findings of this study are as follows:
Under resting conditions at pH 7.4, KCa channels are active, and significantly contribute to resting tone of rat MCA. KATP, KIR, KV channels, or Na+-K+-ATPase are silent at rest. KCa channels significantly contribute to acidosis induced vasodilation, and their blockade leads to reduced vasodilation at pH 7.0. Combined inhibition of KCa and KATP channels completely prevented vasodilation to acidosis. KCa and KATP channels seem to function in a redundant manner and, to a large degree, can compensate each other. Since NO has been suggested to modulate cerebrovascular vasodilation to acidosis in a permissive way, the authors tested the involvement of NO in physiologic KCa and KATP channel function. Vasodilation to specific KCa channel activation is abolished by L-NNA application, and vasodilation to specific KATP channel activation is significantly reduced by L-NNA application. The authors suggest that KCa and KATP channels of rat MCA are modulated by NO.
Technical considerations
In the present study, the authors used the well-known model of the cannulated and perfused isolated cerebral artery, which allows us to investigate cerebrovascular reactivity without parenchymal influences. The artery develops spontaneous tone in response to physiologic intraluminal pressure without pharmacologic intervention (Duling and Rivers, 1986; Johnson et al., 1998), and the intact adventitial layer provides a physiologic neurotransmitter and neuromodulator tonus from perivascular nerve endings (Ignacio et al., 1997; Lindauer et al., 2001). MOPS buffered physiologic salt solution was used for the extraluminal bath as well as the intraluminal perfusion, and intraluminally applied albumin (1%) maintained the integrity of the endothelial cells and luminal matrix (Duling and Rivers, 1986). MOPS and other organic buffers have been shown to prevent development of spontaneous mechanical activity and attenuate responses to vasoconstrictors when applied at concentrations of 5 mM (Altura et al., 1980). In the present study, a lower concentration of MOPS was used (2 mM); compared with rat MCA investigated using bicarbonate buffer in the same model of the cannulated and perfused isolated cerebral artery (Golding et al., 1998, 2001), the vessels develop comparable spontaneous tone when pressurized, and identically react to changes in extraluminal potassium concentrations (Golding et al., 2001; Schuh-Hofer et al., 2001). The authors therefore have no evidence of an adverse effect of MOPS buffering.
Changes in pH were induced by addition of small amounts of HCl to the hypocarbic buffer solution. As already discussed in a recent paper (Lindauer et al., 2001), identical mechanisms of hypo- and hyperpolarisation during changes of diameters of cerebral arteries induced by extracellular pH reduction have been shown during physiologic hypercapnic acidosis and acidosis induced in in vitro systems using HCO3−/CO2 free buffered saline solutions (Austin and Wray, 1993; Dietrich and Dacey, 1994; Kontos et al., 1977; Niwa et al., 1993). In addition, there is strong evidence that it is mainly the reduction of extracellular pH that explains the vasodilation to hypercapnic acidosis, whereas the concomitant reduction of intracellular smooth muscle pH is of little or no importance for the effect (Tian et al., 1995). Whereas vasodilation caused by extracellular acidosis seems to be dependent on NO (Horiuchi et al., 2002; Lindauer et al., 2001), vasodilation during intracellular acidification, which may be an early event during ischemia, was not affected by NOS inhibition but was significantly reduced during KATP channel blockade by glibenclamide (Santa et al., 2003). Therefore, it can be suggested that selective intracellular acidification activates KATP channels of vascular smooth muscle cells by a direct, proton induced activation of an intracellular domain followed by increased open probability of the channels (Xu et al., 2001), which occurs NO-independently. During vasodilation to hypercapnia or extracellular acidification, however, NO-dependent mechansims involving potassium channels have to be considered, which are not identified so far. Therefore the findings of the present study investigating the role of potassium channels during extracellular acidosis may have a strong impact upon the understanding of the mechanisms leading to vasodilation during hypercapnia but less to mechanisms of vasodilation during intracellular acidification mainly occuring during hypoxia or ischemia.
In the present study, the authors investigated the role of potassium channels under resting conditions and during vasodilation to acidosis. As shown above, several chemicals have been diluted by using ethanol reaching final concentration of at least 0.1%. Recently, Rosenblum et al. have shown that KATP channels were inhibited by low concentrations of ethanol (0.01 to 0.075%), but above this dose the blockade disappeared (Rosenblum et al., 2001b). In the authors' experiments, an impairment of KATP channel function by ethanol can be ruled out since higher concentrations of ethanol were used and vasodilation to pinacidil, a specific opener of KATP channels, was comparable with other studies (Zimmermann et al., 1997),
Nitric oxide dependence of vasodilation to increased proton concentration
Vasodilation to extraluminal acidosis was nearly abolished during NOS inhibition down to pH values of 6.5 in the present study. However at lower pH, vasodilation became independent of NO as shown by the preserved dilation during NOS inhibition at pH 6.0. These findings are in agreement with earlier in vivo studies (Iadecola and Zhang, 1994): the CBF response to hypercapnia is not uniformly dependent on NO, and vascular reactivity to high arterial partial pressure of CO2 is completely independent of the bioradical NO. NOS dependent vasodilation to acidosis has also been shown for the reactivity of the basilar artery (You et al., 1994). In contrast, dilation of intracerebral arterioles to acidic buffer solution at pH 6.8 was independent of NO (Ngai and Winn, 1995). These findings from the literature and these results suggest that vasodilation of cerebral arteries during moderate elevation of proton concentration is highly dependent on the basal perivascular NO level (Lindauer et al., 2001), whereas at lower extraluminal pH vasodilation becomes independent of NO and is probably mediated by different mechanisms. In the present study, the authors were mainly interested in the NO dependent range of vasodilation during acidosis. The authors therefore used the reactivity to pH 7.0 in all further experiments to investigate the potassium channel dependent mechanism leading to vasodilation modulated by NO. Whether vasodilation to extraluminal/extracellular acidosis can serve as a model reaction for other NO dependent changes of vascular tone such as functional activation induced increases of CBF (Lindauer et al., 1999) has to be investigated in future studies.
Potassium channels: Contribution to resting tone and to vasodilation during acidosis
Several studies have shown that in cerebral arteries other types of potassium channels are active under resting conditions than are in cerebral arterioles. In larger arteries, inhibition of KCa channels induces vasoconstriction, as shown for rat basilar (Fujii et al., 1991; Sobey and Faraci, 1997), rat middle cerebral (Geary et al., 1998), and human pial arteries (Gokina et al., 1996; for review, see Faraci and Heistad, 1998; Faraci and Sobey, 1998). These findings are consistent with the authors' results of single or combined KCa channel inhibition during TEA or CHTX application. Other potassium channels seem not to be involved in the maintenance of resting tone in the middle cerebral artery. In contrast, in basilar artery constriction has been found during 4-AP application (Sobey and Faraci, 1999). In cerebral arterioles there is strong evidence that KV and KIR channels are active under resting conditions (Horiuchi et al., 2001, 2002). In this vascular bed KCa channel inhibition induces only a slight and non significant constriction in vitro (Horiuchi et al., 2002), and no change of resting diameter in vivo (Wang et al., 1998). Despite the wide distribution of KATP channels in both larger cerebral arteries and arterioles (Faraci and Heistad, 1998; Faraci and Sobey, 1998; Lang et al., 1997), it is generally accepted that these channels are not active under resting conditions (Fujii et al., 1991; Lang et al., 1997; Wang et al., 1998), as shown also in the present study.
At present, the role of potassium channels during dilation of cerebral arteries and arterioles to hypercapnia or acidosis is discussed controversly. Recent findings provide evidence for a contribution of KATP channels in the vasodilation of cerebral arteries to acidosis (Faraci et al., 1994; Horiuchi et al., 2002; Kontos and Wei, 1996; Kinoshita and Katusic, 1997; Rosenblum et al., 2001a; Wei et al., 1999). However, others did not support a role of KCa or KATP channels in hypercapnia induced cerebrovasodilation under physiologic conditions (Bayerle-Eder et al., 2000; Reid et al., 1993; Wang et al., 1998; for review, see Faraci and Heistad, 1998; Faraci and Sobey, 1998). The authors' finding of a redundant action of KCa or KATP channels in acidosis induced vasodilation of rat MCA may at least partially explain the controversial results of previous studies. These results lead to the hypothesis that during specific inhibition of KCa channels, activation of KCa channels can be substituted by KATP channel activity during acidosis. In turn, KATP channel activity during acidosis can completely be replaced by KCa channel activity, when KATP channels are inhibited. A similar pattern of substitution of specific potassium channel function has recently been shown for the vasodilation to NO in rat basilar artery (Hempelmann et al., 2001).
In addition, the specificity of several potassium channel blockers has been shown to be dose-dependent. The glibenclamide dose-response relationship in the present study showed that vasodilation to acidosis was inhibited by higher, more unspecific concentrations of Glib. Glib at concentrations of greater than 10−5 mol/L has been shown to block not only KATP channels but also KCa channels (Gelband and McCullough, 1993). The significant reduction of acidosis induced vasodilation at higher Glib concentrations may therefore be caused by combined KATP and KCa channel inhibition, which supports these findings of blocking vasodilation to extracellular acidosis during combined application of Glib and TEA in specific concentrations. The narrow concentration range for efficacy and specifity of glibenclamide may at least in some studies in part explain the differences of the results achieved by KATP channel inhibition by glibenclamide concerning vasodilation to acidosis.
Role of NO/cGMP during vasodilation to acidosis: Modulation of KCa or KATP channels
In a recent study, the authors have shown that acidotic vasodilation is significantly dependent on basal perivascular NO/cGMP concentration in large cerebral arteries. NOS inhibition nearly abolished the vasodilation response to extraluminal acidotic buffer solution of rat MCA. Reestablishing the vascular tone by adding NO donors to the extraluminal bath completely restored pH dependent reactivity of the isolated MCA. This NO effect seems to be mediated by cGMP because cGMP application during NOS inhibition or during soluble guanylyl cyclase inhibition restored pH-reactivity (Lindauer et al., 2001). In contrast, Kontos and Wei failed to find a permissive effect of NO donors under NOS inhibitor application during hypercapnia in cat pial arterioles (Kontos and Wei, 1996). In a later study by Rosenblum, Kontos, and Wei, application of the inhibitor of soluble guanylyl cyclase also failed to reduce vasodilation of rat pial arterioles to hypercapnia (Rosenblum et al., 2002). Other studies, however, provided strong evidence for a permissive role of the basal NO or cGMP concentration in the tissue during hypercapnia- or acetazolamide-induced increase of microcirculatory CBF in rats (Iadecola et al., 1994; Okamoto et al., 1997; Tuettenberg et al., 2001).
The molecular target of the postulated NO/cGMP modulating activity for cerebrovascular reactivity at least in larger cerebral arteries and at the microcirculatory level is not known so far. It has been shown that hyper- or hypopolarizing K+ channel activity of vascular smooth muscle cells can be modulated by NO or cGMP (Bolotina et al., 1994; Bonnet et al., 1991; Robertson et al., 1993). In isolated cerebral arteries, a functional proof of a permissive action of NO/cGMP on potassium channels involved in vasodilation to acidosis is lacking. The authors have shown here that KCa and KATP channels are mediating vasodilation to acidosis in rat MCA. In addition, the authors provide evidence that both KCa and KATP channel function is indeed dependent on basal perivascular NO concentration. NOS inhibition reduced vasodilation to pinacidil, a specific KATP channel opener, by approximately 50%. These results are in agreement with findings by Kontos and Wei (1996) and Rosenblum et al. (2001a), demonstrating that the vasodilation to specific openers of KATP channels is partially reduced by NOS inhibitor application. However, from their data, Kontos and Wei rejected the concept of a permissive role of NO/cGMP (1996). The authors suggested that arginine analogues inhibit hypercapnic vasodilation by blocking KATP channels independently of NO or cGMP and that KATP channels may have an arginine site that influences their function instead of the proposed specific role of NO or cGMP. Further complicating the understanding of the role of basal NO for KATP channel function, Sobey and Faraci did not find an effect of NOS inhibition during vasodilation to specific openers of KATP channels in rat pial arterioles or basilar arteries (Mayhan and Faraci, 1993; Sobey and Faraci, 1997). Further experiments have to clarify the specific role of arginine analogues during inhibition of acidotic/hypercapnic vasodilation concerning a modulatory role of NO/cGMP at the level of potassium channel function of cerebral arteries.
In contrast to KATP channels, the conductivity of KCa channels seems to be completely dependent on basal perivascular NO concentrations, since NOS inhibition abolished vasodilation during NS1619 application. It has recently been shown that the vasodilator response to NO in rat MCA is mediated by activation of KCa channels via a cGMP independent pathway (Yu et al., 2002), a finding that is in accordance with these results of an important role of NO for KCa channel activity. It has been suggested that besides specific KCa channel opening properties, NS1619 may possess Ca2+ channel antagonistic activity (Holland et al., 1996). However, TEA in a concentration specifically blocking KCa channel activity completely abolished dilation to NS1619 in this study, which strongly points against an unselective action of NS1619 in the concentration used in the authors' experiments. In addition, recent findings in the piglet showed that vasoconstriction to a Ca2+ channel agonist was unchanged in the presence of NS1619 (Armstead, 1997). In the present study, NS1619 may therefore be considered to be selective for the activation of KCa channels rather than to block Ca2+ channels.
Wang et al. (1998) have recently criticized the current strategies for investigating permissive actions of cGMP or NO by blocking NOS synthase with subsequent cGMP or NO donor application. They have shown that neither inhibition of KCa channels nor of KATP channels changed vasodilation of rat pial arterioles during hypercapnia under normal conditions. However, during NOS inhibition followed by low concentration cGMP application, hypercapnia induced vasodilation was significantly attenuated by KCa channel or KATP channel blockade (Wang et al., 1998). On the basis of these present results of a redundant action of both channels during acidosis under control conditions, the authors suggest that during application of a subthreshold dose of cGMP, K+ channel conductance may be increased but not completely restored. This may lead to a condition in which the function of both channels during vasodilation to hypercapnia is additive and not redundant as under normal conditions. Further experiments have to be performed to prove this possible explanation.
In general, the variability in the literature concerning the role of specific potassium channels as well as the involvement of the basal NO concentration during resting and hypercapnia- or acidosis-activated conditions may point towards regional as well as species differences. Region-dependent expression of different potassium channel families at vascular smooth muscle cells has been shown for different vascular segments in rats and rabbits (Horiuchi et al., 2001; Nagao et al., 1996).
NOS inhibition reduced the diameter of larger cerebral arteries by 20 to 30% (Lindauer et al., 2001; Schuh-Hofer et al., 2001; You et al., 1999) and significantly reduced CBF-measured autoradiographically (Cholet et al., 1997) or microcirculatory CBF assessed by laser Doppler flowmetry (Iadecola et al., 1994; Lindauer et al., 1999; Okamoto et al., 1997). However, resting pial arteriolar diameters either did not change during NOS inhibition (Kontos and Wei, 1996, 1998; Rosenblum et al., 2001a; Sun et al., 2002; Wang et al., 1998) or were reduced by less than 15% (Santizo et al., 2001; Xu et al., 2002). The present authors and others have shown that not the endothelium but perivascular nerves from extracerebral origin are the main source of basal NO in large cerebral arteries originating from the anterior portion of the circle of Willis (Iadecola et al., 1993; Lindauer et al., 2001). In addition, evidence is strong that perivascular NO-releasing neurons within the parenchyma may play a significant role in maintaining the resting cerebrovascular tone at the level of small intracerebral arterioles and capillaries within the microcirculation (Iadecola et al., 1993; Kashiwagi et al., 2002). In contrast to larger cerebral arteries of the anterior part of the circle of Willis and to the intraparenchymal microcirculation, it can be suggested that significantly lower perivascular NO concentrations may be present at arterioles at the pial surface. They are not surrounded by NO-producing tissue and lack perivascular nerve endings from extracerebral origin. At the basilar artery, the innervation with NOS-positive nerve endings has been shown to be less dense (Iadecola et al., 1993; Tomimoto et al., 1993). The functional impact of perivascular NO on vascular reactivity may therefore significantly differ between larger cerebral arteries of the anterior part of the circle of Willis and the intraparenchymal microcirculation, on one hand, and arterioles at the pial surface or basilar artery preparations on the other hand. This hypothesis is supported by findings of Bryan's and Faraci's groups. Third-order branches and penetrating arterioles originating from rat MCA (You et al., 1999), as well as preparations of rat basilar artery (Faraci, 1991), showed significantly smaller constrictions to NOS inhibition than rat MCA itself (Lindauer et al., 2001; You et al., 1999) (10 to 13% compared with 20 to 30%, respectively). The relevance of this proposed diminished functional role of basal perivascular NO in pial and larger penetrating arterioles and basilar artery compared with larger cerebral arteries of the anterior part of the circle of Willis and the intraparenchymal microcirculation for activated conditions has to be thoroughly investigated in future studies. The authors' hypothesis may explain at least some of the discrepant findings of the authors' studies and of the results by Rosenblum, Kontos, and Wei, as well as Wang et al. (1998), concerning a modulatory role of NO within the cerebral circulation. In addition, it may also provide the basis for further discussion concerning the lack of effect of NOS inhibition on vasodilation of rat pial arterioles and basilar artery to specific openers of KATP channels (Mayhan and Faraci, 1993; Sobey and Faraci, 1997). In vessel segments adapted to lower perivascular NO concentrations, KATP channel function may occur less dependent or even completely independent of NO, whereas in vessels with suggested higher perivascular NO concentration, at least part of the channel conductance may be specifically influenced by NO.
The authors have shown that KCa channels are active under basal conditions in rat middle cerebral artery and that KATP and KCa channels are mainly responsible for the vasodilation because of increased extraluminal proton concentration. Activity of one of these two potassium channels is sufficient for vasodilation to extraluminal acidosis, and only combined inhibition abolishes vasodilation to acidosis in the middle cerebral artery. In addition, from these results, the authors suggest that basal vascular NO is an important modulator of physiologic KATP and KCa channel function of rat MCA. According to these results, KATP and KCa channels can be considered likely targets of NO/cGMP modulation during maintenance of resting tone as well as cerebrovascular reactivity to acidosis of large cerebral arteries. It has to be further investigated whether this NO dependent effect on KATP and KCa channel function is a principle mechanism of NO induced modulation of cerebrovascular reactivity.
Footnotes
Abbreviations:
Acknowledgment
The authors thank Claire Gehlhaar and Marco Foddis for excellent technical assistance.