
Abstract
Nitric oxide (NO) has been suspected to mediate brain damage during ischemia. Here the authors studied the effects of an antisense oligodeoxynucleotide (ODN) directed against the inducible isoform of NO synthase (iNOS) in a model of transient focal cerebral ischemia in rats. Treatment consisted of seven intracerebroventricular injections of a phosphodiester/phosphorothioate chimera ODN (3 nmol each) at 12-hour intervals, and was initiated 12 hours before a 2-hour occlusion of the left middle cerebral artery and common carotid artery. Outcomes were measured three days after ischemia. When compared with animals treated with vehicle or an appropriate random non-sense control ODN sequence, the antisense treatment reduced the lesion volume by 30% and significantly improved recovery of sensorimotor functions, as assessed on a neuroscore. This effect was associated with a decrease in iNOS expression, as assessed by Western blot, a 39% reduction in iNOS enzymatic activity evaluated as Ca2+-independent NOS activity, and a 37% reduction in nitrotyrosine formation, reflecting protein nitration by NO-derived peroxynitrite. These findings provide new evidence that inhibition of iNOS may be of interest for the treatment of stroke.
Keywords
Nitric oxide (NO) is a pleiotropic effector that is a mediator of major functions, such as vasodilatation (Ignarro et al., 1987), neurotransmission (Garthwaite et al., 1988), and immunoinflammatory cytotoxicity (Knowles and Moncada, 1994). The biosynthesis of NO is mediated through the oxidation of arginine by a family of NO synthases (NOS) comprising constitutive calcium- and calmodulin-dependent neuronal (nNOS) and endothelial (eNOS) isoforms (Bredt et al., 1991; Lamas et al., 1992) and the inducible calcium-independent isoform (iNOS) (Xie et al., 1992; Geller et al., 1993; Charles et al., 1993). In the central nervous system, under pathologic conditions such as ischemia, there is an enhanced production of NO that can react with the radical superoxide anion (O2−) to form peroxynitrite, a highly cytotoxic substance (Kumura et al., 1996; Sakurai et al., 1998). This overproduction of NO may originate from the various isoforms of NOS. However, several lines of evidence have revealed a prominent role of iNOS-derived NO in the pathogenesis of focal cerebral ischemia.
First, both iNOS mRNA levels and iNOS enzymatic activity have been shown to increase during the days after transient or permanent focal ischemia (Iadecola et al., 1995a, 1996; Grandati et al., 1997). Second, high levels of peroxynitrites have been observed at the time of iNOS activation, between 24 and 72 hours after ischemia (Takizawa et al., 1999). Third, iNOS immunoreactivity has been located in invading neutrophils and in vascular endothelial cells within the ischemic territory of rat brains after focal ischemia (Iadecola et al., 1996), as well as in autopsied brains of patients displaying neuropathologic evidence of acute cerebral infarction (Forster et al., 1999).
Inducible NOS therefore may constitute a new attractive target for the treatment of stroke. Accordingly, iNOS contribution to postischemic lesion has been shown by two different strategies—pharmacologic inhibitors and knockout mice. However, these strategies raise some problems of pharmacokinetic and specificity for the former, and of animals lacking iNOS gene from birth for the latter. Thus, in the current study, the authors used another strategy with an antisense oligodeoxynucleotide (ODN) sequence directed towards iNOS to evaluate iNOS contribution to ischemic brain damage. An antisense ODN is a sequence of artificial DNA derived from the gene encoding a given protein that is short enough to be taken up by cells, and long enough to specifically hybridize with the nucleic acids governing its biosynthesis. The hybridization results in a selective decrease in the intracellular levels of the targeted protein (Schlingensiepen et al., 1997). In the current study, the authors investigated the effects of direct injections of an antisense ODN to iNOS into the brain on the neuronal damage produced by transient cerebral ischemia, as well as on iNOS protein levels, iNOS enzymatic activity, and peroxynitrite formation.
MATERIALS AND METHODS
Animals
All experiments were performed with male Sprague–Dawley rats (weighing 300 to 350 g) in accordance with National Institutes of Health and French Department of Agriculture guidelines (license N° 01352).
Oligodeoxynucleotide sequences
The antisense oligonucleotide directed to rat iNOS and the random non-sense control sequence have been designed and manufactured by Biognostik, Germany. To improve the half-life in biologic fluids, both were phosphodiester/phosphorothioate chimera ODN. The sequences were as follows: antisense, GGCAAGCCATGTCTG; random non-sense control, ACCGACCGACGTGT. The ODNs were dissolved in artificial sterile cerebrospinal fluid (Dulbecco; Sigma, Saint-Quentin Fallavier, France) immediately before administration.
Induction of transient focal cerebral ischemia
Artery occlusions were performed according to procedures described previously (Parmentier et al., 1999). Rats were anesthetized with chloral hydrate (400 mg kg−1, IP) and allowed to breathe spontaneously. The tail artery was cannulated to monitor mean arterial blood pressure, arterial blood gases, and pH. The left middle cerebral artery (MCA) was exposed through a temporal craniotomy and occluded with a microclip (15 times 0.4 mm; Ohwa Tsuho, Tokyo, Japan), and the left common carotid artery was concomitantly clamped. The microclip was placed on the MCA at a site proximal to the lenticulostriate arteries to induce cortical and striatal infarction. The animals recovered their righting reflex approximately 90 minutes after the onset of ischemia. Thus, animals had to be reanesthetized 2 hours after the induction of ischemia to remove the clips and allow the recirculation. Reperfusion in the two arteries was checked under a microscope. The same surgery was performed in sham-operated rats but the MCA and the common carotid artery were not occluded. During the surgical procedure, and until recovery from anesthesia (approximately 2 hours after the second anesthesia), body temperature was maintained at 37°C to 38°C by means of a heating blanket. Physiologic variables were measured just before ischemia, 30 minutes after the onset of ischemia, and 30 minutes after the onset of reperfusion. After surgery, rats were returned to their home cages. Animals showing feeding difficulties were fed with mashed lab chow.
Intracerebroventricular injection of ODNs and preparation of tissue samples
Animals were anesthetized with chloral hydrate (400 mg kg−1, IP) and placed in a stereotaxic frame (David Kopf). The calvarium was exposed and a hole approximately 1 mm in diameter was drilled over the left hemisphere 1.5 mm lateral to the sagittal suture and 0.8 mm caudal to the bregma. To target the lateral ventricle, a microinjection guide cannula was placed on the surface of the cortex and secured to the skull with dental cement. After a 5-day recovery period, injections were made through this guide cannula using a needle of calibrated length so that its tip was placed 2.5 mm below the surface of the brain. Each injection consisted of 3 nmol of the antisense sequence or of the corresponding random non-sense control (both in 3 μL vehicle), or vehicle alone (artificial sterile cerebrospinal fluid, Dulbecco). Treatments were started 12 hours before ischemia and repeated at 12-hour intervals for 3 days after artery occlusion, representing a total of 7 injections. At this time, rats were killed with an overdose of sodium pentobarbitone and used for iNOS Western blot analysis and activity assay, nitrotyrosine immunoreactivity, and measurement of infarct size (see below).
All biochemical analysis was performed on striatal and cortical infarcted tissues dissected out from a 2-mm-thick coronal brain slice cut at the level of the optic chiasma. A similar segment was isolated in sham-operated and non-operated rats. The tissue samples were homogenized in ice-cold buffer (20 mmol/L HEPES, 1 mmol/L EGTA, 1 mmol/L DTT, 0.32 mol/L sucrose, 1 mg/L leupeptin, 10 mg/L pepstatin, and 1 mg/L PMSF), centrifuged at 20000 g for 20 minutes at 0°C. The supernatant were isolated, frozen, and stored at −40°C for later analysis. Protein concentration in the supernatant was determined by the Bradford Coomassie brilliant blue method (Biorad, Richmond, CA, U.S.A.).
Western blot analysis for iNOS protein
The tissue lysates were diluted and subjected to Western blot analysis. Aliquots (100 μg protein each) were suspended in sodium dodecyl sulfate buffer and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis on a 7.5% polyacrylamide gel. Proteins were transferred to nitrocellulose membranes by electroblotting. The loading and transfer of equal amounts of proteins in each lane were checked by staining the nitrocellulose membrane with Ponceau red and the gel with Coomassie blue. Membranes were probed with mouse monoclonal anti-iNOS antibody (Sigma) at 1/2000 for 2 hours. Detection of bound antibodies was visualized by the enhanced chemiluminescence detection system (Amersham, Orsay, France) after secondary probing of the membrane with peroxidase-conjugated goat anti-mouse antibody (Dako, Trappes, France).
Determination of iNOS enzymatic activity
The catalytic activities of constitutive and inducible NOS activities were measured by monitoring the conversion of L-[14C]arginine to L-[14C]citrulline according to procedures initially described by Bredt and Snyder (1989) and modified as described previously (Grandati et al., 1997). Calcium-independent NOS activity was taken as an index for iNOS activity. Data are expressed as pmol of L-[14C]citrulline formed per mg protein, per minute (pmol/mg protein/min).
Determination of nitrotyrosine immunoreactivity
Nitrotyrosine immunoreactivity of brain samples (10 μg protein each) was analyzed by dot blots on nitrocellulose membranes, as described previously (Mesenge et al., 1998). Membranes were incubated overnight (4°C) with a rabbit nitrotyrosine polyclonal antibody (Upstate Biotechnology, Euromedex, France) at 1/2000, washed, and incubated with a secondary peroxidase-conjugated goat anti-rabbit antibody (Dako, France) at 1/4000 for 30 minutes.
Detection of bound antibody was performed by the use of a chemiluminescence Blotting Kit (Amersham, France). Quantification was performed by densitometric measurement using an image analyzer (Imstar, Paris, France).
Determination of infarct volume
Damage to brain tissue was determined on 7 consecutive 2-mm-thick coronal sections spanning the entire damaged area, as previously described (Parmentier et al., 1999). Sections were immersed in 2% 2,3,5-triphenyltetrazolium chloride, incubated for 20 minutes at 20°C, and then placed overnight in formalin in the dark. The striatal and cortical areas of infarction (unstained tissue) were measured on each section using an image analyzer (Imstar, Paris, France). Striatal and cortical necrotic volumes were determined by integrating the areas of infarction corrected for edema according to Golanov and Reis (1995).
Determination of neurologic deficits
Three days after ischemia, sensorimotor neurologic functions were assessed in a blinded fashion using the scoring system described previously by Wahl and coworkers (1992). The following parameters were scored for right and left paws: grasping reflex, righting reflex, visual placing reaction, and leg hanging placing reaction. For each side, a maximum neurologic score of 5 corresponds to a normal sensorimotor function in non-operated rats. The lower the neurologic score is the more severe are the deficits.
Data analysis
Data in text and figures are expressed as mean ± SD. Multiple comparisons were statistically evaluated by the analysis of variance and Fisher's PLSD test. Nonparametric Kruskall and Wallis rank analysis evaluated neurologic score with subsequent group comparisons by Mann–Whitney U test. Differences were considered statistically significant for P < 0.05.
RESULTS
Effect of the antisense oligodeoxynucleotide on iNOS expression
Three days after ischemia, vehicle-treated rats showed marked induction of iNOS protein expression (Fig. 1A) and calcium-independent enzymatic activity (5.1 ± 2.9 vs. 0.5 ± 0.4 pmol/mg protein/min in sham-operated rats; P < 0.001) (Fig. 1B, upper graph) in the area of infarction. Treatment with the antisense ODN notably reduced iNOS protein levels and significantly attenuated iNOS activity by 39% in the infarcted tissue (3.1 ± 1.6 pmol/mg protein/min, P < 0.05 with respect to vehicle-treated rats). In contrast, treatment with the random non-sense control ODN did not alter iNOS activity (4.2 ± 1.8 pmol/mg protein/min).
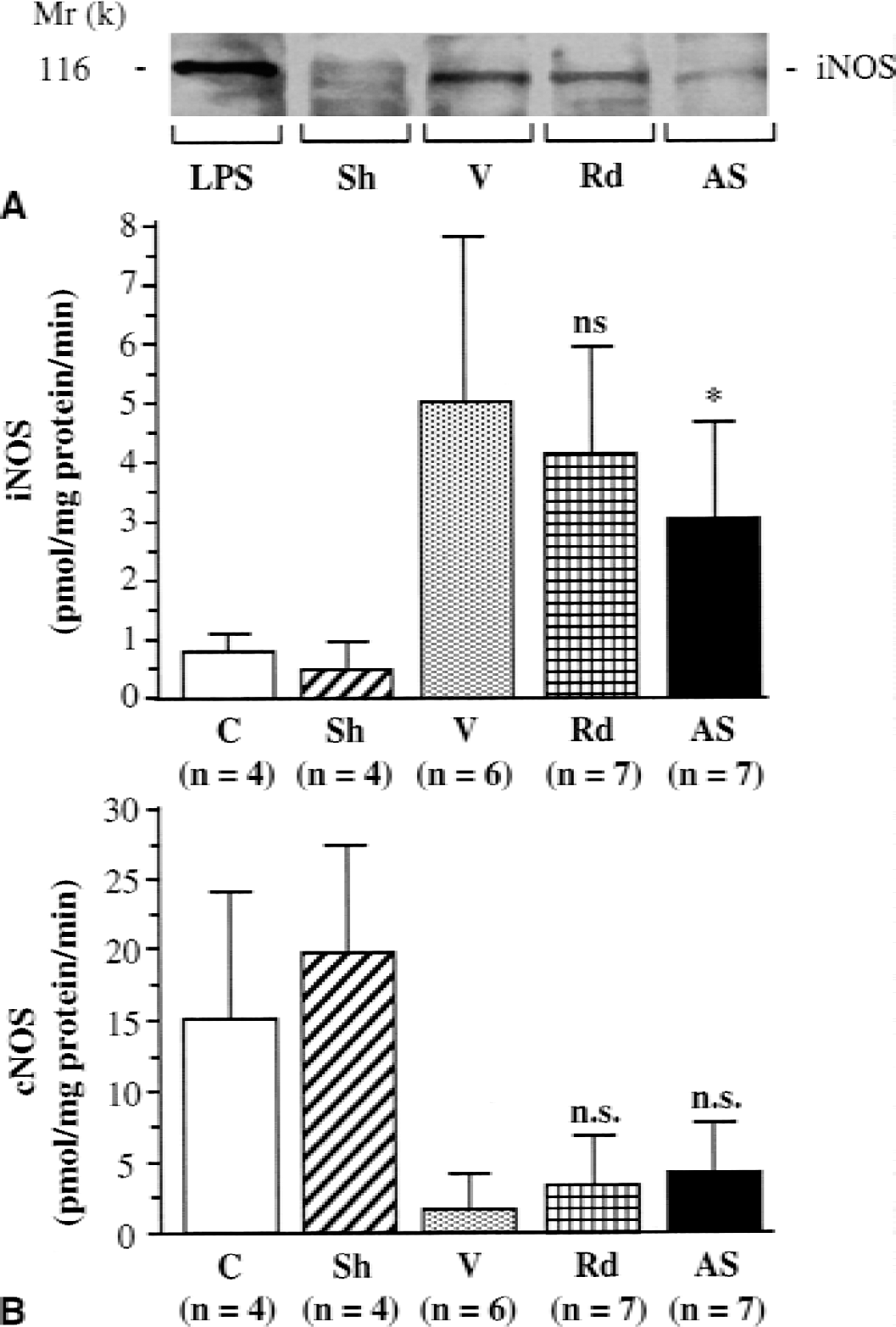
Expression of iNOS protein and nitric oxide synthase (NOS) enzymatic activities in the cerebral infarction area three days after experimental transient focal ischemia in rats. iNOS protein and NOS enzymatic activities were evaluated in sham-operated rats (Sh) and in ischemic rats treated with an antisense oligodeoxynucleotide (ODN) (AS) directed towards iNOS, a random non-sense control ODN (Rd), or vehicle (V).
Constitutive NOS activity was reduced by a similar extent in both vehicle and treated groups of ischemic rats, indicating that this activity was not affected by any ODN treatments (Fig. 1B, lower graph). The reduction in constitutive NOS activity at 3 days after ischemia probably reflects a loss of nNOS-containing neurons.
Effect of the antisense oligodeoxynucleotide on nitrotyrosine immunoreactivity
Nitrotyrosine levels reflect protein nitration by NO-derived peroxynitrites (Szabo, 1996). As determined by the dot blots of brain proteins using a nitrotyrosine specific antibody, nitrotyrosine levels were doubled 3 days after ischemia (0.98 ± 0.46 vs. 0.48 ± 0.16 in sham-operated rats, P < 0.05) (Fig. 2). The iNOS antisense ODN treatment significantly reduced this ischemia-induced nitrotyrosine production by 37% (0.62 ± 0.18, P < 0.05) whereas random non-sense control ODN had no significant effect (0.80 ± 0.23).
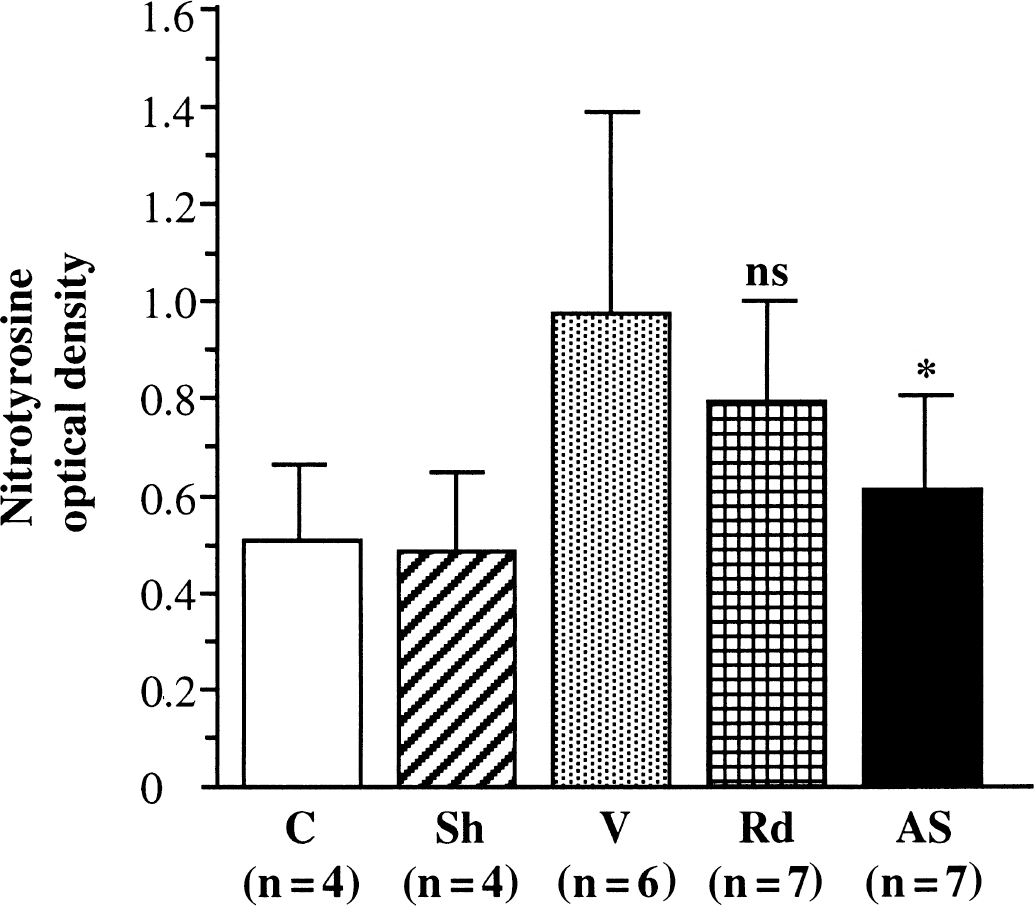
Immunoreactivity of nitrotyrosine in cerebral infarction area at three days after ischemia. Nitrotyrosine was detected by dot blots in sample tissues in nonoperated (C) and sham-operated (Sh) rats and in ischemic rats treated with antisense oligodeoxynucleotide (ODN) (AS), random non-sense control ODN (Rd), or vehicle (V). Values are expressed as mean ± SD. The antisense ODN treatment significantly reduced nitrotyrosine formation (*P < 0.05).
Effect of the antisense ODN on infarct volume
The mean arterial blood pressure, arterial blood gases, and pH data obtained from rats treated with the antisense ODN, with the random non-sense control ODN or with vehicle, remained within the physiologic range before and during ischemia, as well as during reperfusion (Table 1). Three days after ischemia, vehicle-treated rats presented a wide infarction involving the cerebral cortex and the striatum (cortex: 112 ± 26 mm3, striatum: 42 ± 7 mm3) (Fig. 3).
Effect of ischemia on physiologic variables and rectal temperature in vehicle-, random non-sense ODN- and iNOS antisense ODN-treated rats
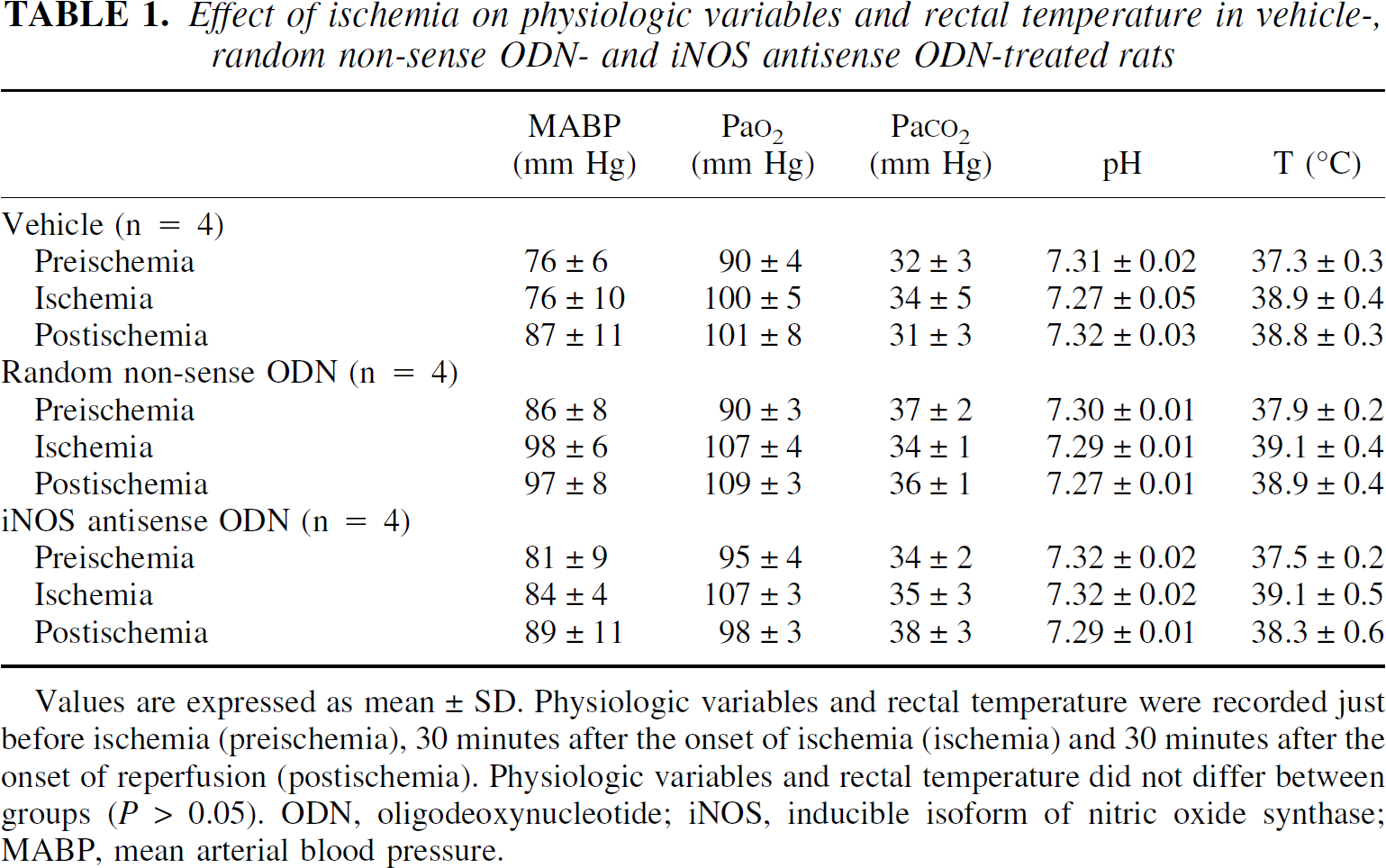
Values are expressed as mean ± SD. Physiologic variables and rectal temperature were recorded just before ischemia (preischemia), 30 minutes after the onset of ischemia (ischemia) and 30 minutes after the onset of reperfusion (postischemia). Physiologic variables and rectal temperature did not differ between groups (P > 0.05). ODN, oligodeoxynucleotide; iNOS, inducible isoform of nitric oxide synthase; MABP, mean arterial blood pressure.
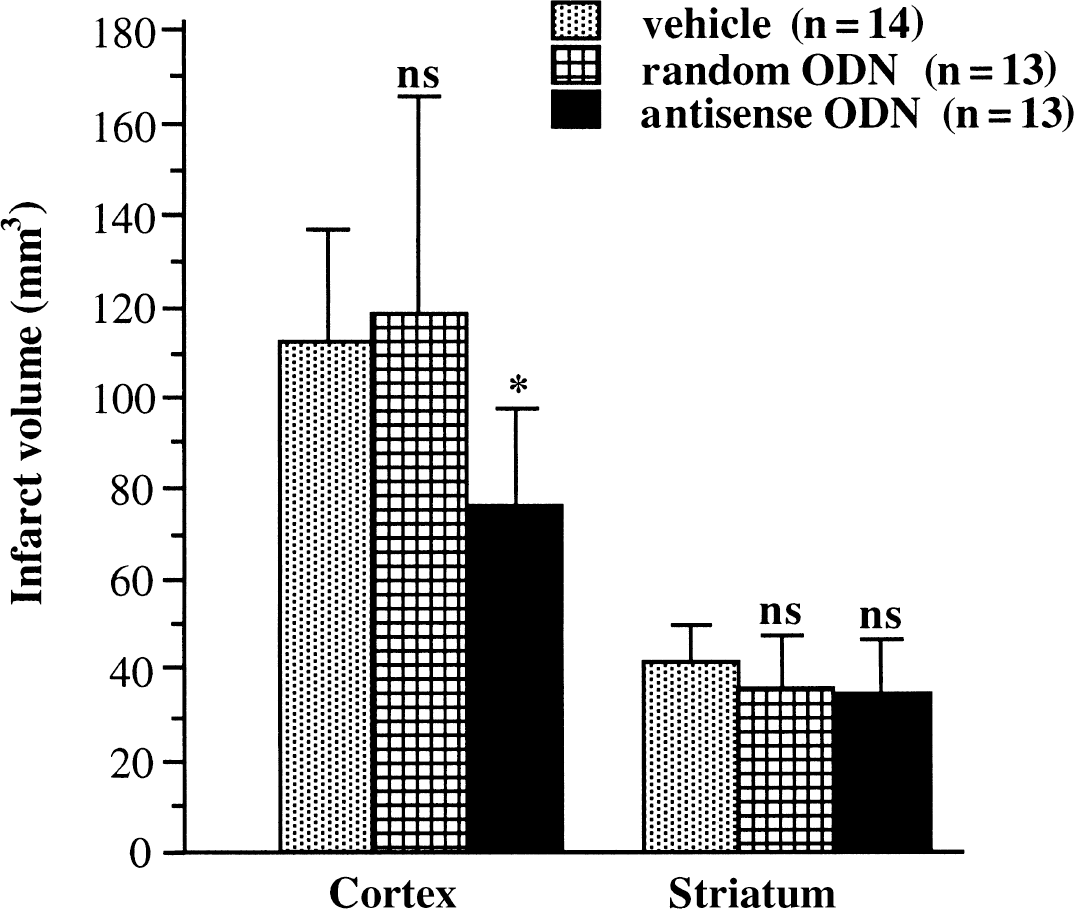
Infarct volume in rats treated with antisense oligodeoxynucleotide (ODN), random non-sense ODN, or vehicle three days after transient focal cerebral ischemia. Values are expressed as mean ± SD. Cortical infarct size is smaller in antisense ODN-treated rats (*P < 0.01). Striatal infarct size is not significantly different between the groups.
Administration of the antisense ODN significantly reduced infarct volume in the cerebral cortex by 30% (78 ± 22 mm3, P < 0.01). Treatment with the random non-sense control ODN had no effect on the size of the cortical infarct (120 ± 50 mm3). None of the ODN treatments affected the striatal infarction volume.
Effect of the antisense ODN on sensorimotor deficits
In vehicle-treated rats, the ischemic episode induced in the left hemisphere produced a profound neurologic deficit on the right side of the body. At 3 days after injury, the neuroscore was decreased to 0.5 ± 0.7 in vehicle-treated rats, whereas it was 5 ± 0 in sham-operated rats (P < 0.001). The scoring of sensorimotor functions on the left part of the body was not significantly altered (Fig. 4). Treatment with the antisense ODN significantly increased the neurologic scores on the right side (1.7 ± 0.9, P < 0.001), whereas the random non-sense control ODN had no effect (0.5 ± 0.6).
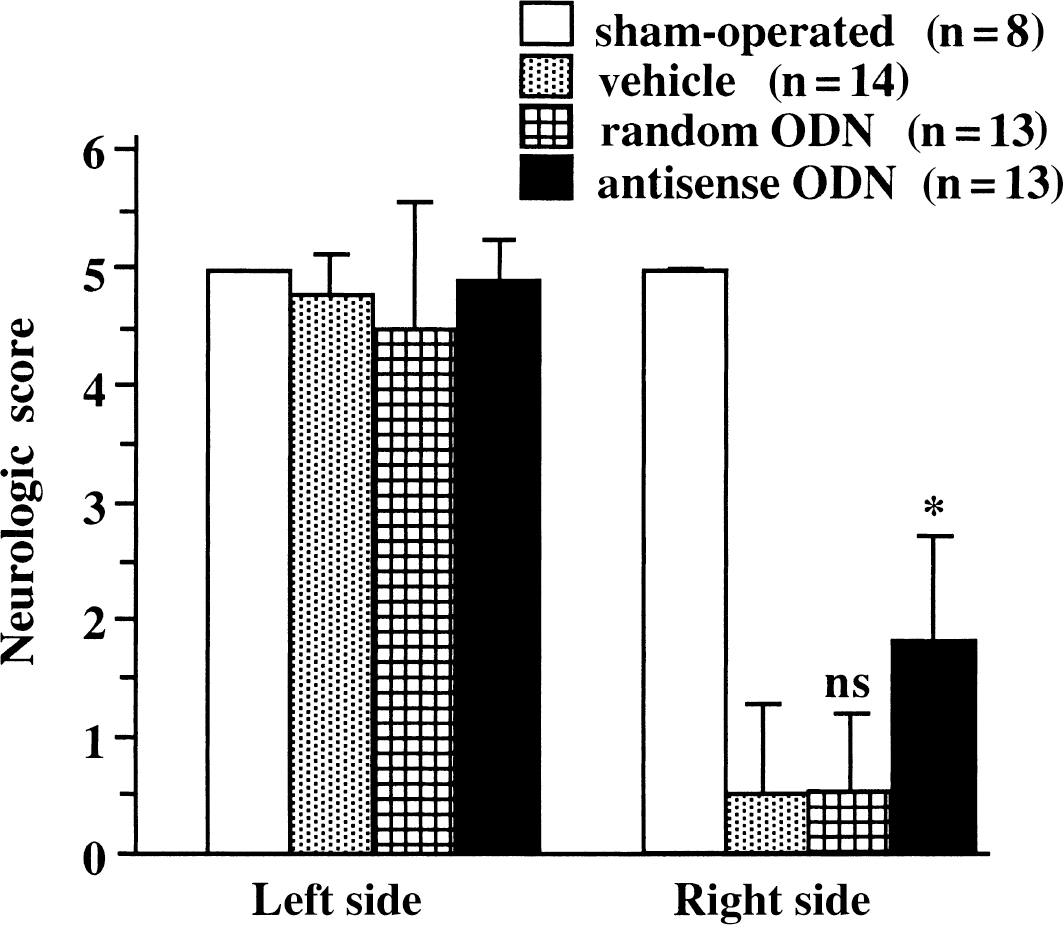
Neurologic score in rats treated with antisense oligodeoxynucleotide (ODN), random non-sense ODN, or vehicle at three days after ischemia. Values are expressed as mean ± SD. Ischemia induces a neurologic deficit on the right side of the body. The antisense ODN-treated rats present a significant increase in neurologic score (*P < 0.001).
DISCUSSION
The results of the current study demonstrate that the administration of an antisense ODN targeted to iNOS gene products significantly reduces the infarct size by approximately one third and ameliorates the neurologic outcomes in rats submitted to transient focal cerebral ischemia.
The intracerebroventricular injection of the antisense ODN also induced a comparable reduction of iNOS protein levels, iNOS enzymatic activity, and peroxynitrite formation. A similar inhibition by approximately one half of iNOS protein expression after ODN treatment led to significant improvement of symptoms in mice with experimental autoimmune encephalomyelitis, a model of multiple sclerosis (Ding et al., 1998). The extent by which a target protein is inhibited by an antisense can vary because of a number of factors, including expression levels of the corresponding gene products, the capacity of the antisense ODN to penetrate cells, the resistance of the antisense to degradation by endo- and exonucleases, and the stability of the protein itself (Schlingensiepen et al., 1997; Gyurko et al., 1997). In addition, the fact that iNOS gene expression after cerebral ischemia is restricted to certain cell types (for example, neutrophils, endothelial cells, and macrophage; Iadecola et al., 1996) also probably contributes to the partial effect observed in the current study, by preventing the iNOS antisense to accumulate in the relevant cell population. An autoradiographic analysis of the radiolabeled antisense would have been useful to quantify and localize the extent of diffusion of the ODN throughout the brain after intracerebroventricular administration. Importantly, the random non-sense control ODN did not produce significant neuroprotection or alteration of any of the markers of iNOS expression. These data indicate that the neuroprotective effect of antisense ODN treatment specifically correlates with an inhibition of iNOS gene products and suggests that iNOS antisense ODN treatment may be a novel method to treat cerebral ischemia. However, it remains to be established whether systemic administration of ODN would be effective, although it has been reported that the blood–brain barrier is opened after cerebral ischemia (Belayev et al., 1996).
The current study did not allow the authors to determine whether iNOS inhibition offers a window of therapeutic intervention as they administered the ODNs before the experimental ischemic episode to allow them to be taken up by the cells. Recent work in the authors' laboratory has demonstrated that the pharmacologic inhibition of iNOS with a selective small molecule inhibitor (1400W) produces neuroprotection in experimental transient focal ischemia with an 18-hour therapeutic window (Parmentier et al., 1999). High doses of the rather weak but relatively selective iNOS inhibitor, aminoguanidine, have been shown previously to reduce infarct size after permanent and transient MCA occlusion with an even greater window of 24 hours (Iadecola et al., 1995b, 1996; Zhang et al., 1996). Although the pharmacokinetic properties of these drugs may not be optimal to target the brain, it is interesting to note that these drugs led to a similar reduction in ischemic tissue damage by one third. iNOS knockout mice have also been used to investigate the role of iNOS expression in ischemic brain damage (Iadecola et al., 1997). Although this approach has limitations because of the possible interaction of background genes in the observed phenotype (Gerlai, 1996) and the engagement of compensatory mechanisms, as the protein is extinguished throughout all development, these mutant mice again showed a partial 30% to 35% reduction in their cortical infarct size after focal ischemia. The consistency of the degree of neuroprotection in three different approaches addressing the regulation of iNOS provides strong evidence that an overproduction of NO from iNOS is responsible for a significant fraction of the brain injury during experimental ischemia.
Peroxynitrite is considered to be a major mediator of NO toxicity (Darley-usmar et al., 1995; Lipton et al., 1993; Szabo, 1996). Nitrotyrosine formation is widely used as an index of peroxynitrite production even if mechanisms other than protein nitration lead to nitrotyrosine (Eiserich et al., 1998). In the current study, nitrotyrosine levels were reduced in the iNOS antisensetreated animals, suggesting that iNOS activity is indeed linked to peroxynitrite formation. This is consistent with recent data from Takizawa et al. (1999) and Hirabayashi et al. (2000) showing that nitrotyrosine levels are decreased after treatment with aminoguanidine and in iNOS knockout mice, respectively. The functional consequences of tyrosine nitration may include abnormal cellular signaling, inhibition of glutamate transporters (Trotti et al., 1996), and changes in the physico-chemical properties of intraneuronal proteins (Crow et al., 1997). Nitrotyrosine immunoreactivity is also found increased in other neurodegenerative diseases such as Alzheimer's disease (Smith et al., 1997) or Parkinson's disease (Liberatore et al., 1999).
In conclusion, the current study provides new evidence that iNOS induction contributes to the mechanism of postischemic neuronal injury and may represent a target for novel neuroprotective drugs.
Footnotes
Acknowledgment
The authors are grateful to Dr. T. Rooney for a critical reading of the manuscript.