
Abstract
In the rat, 60 minutes of transient ischemia to the middle cerebral artery results in infarction of the caudate putamen. Ischemic preconditioning with 20 minutes of transient focal ischemia produced tolerance (attenuated infarction volume) to 60 minutes of subsequent focal ischemia administered three days, five days, or seven days later. Western blots from tolerant caudate putamen demonstrated increased bcl-2 expression, maximum at 3 days and persisting through 7 days. Immunocytochemical examination found that bcl-2 was expressed in cells with both neuronal and nonneuronal morphology in striatum after preconditioning ischemia. bcl-2 antisense oligodeoxynucleotides (ODNs), bcl-2 sense ODNs, or artificial cerebrospinal fluid (CSF, vehicle) was infused into the lateral ventricle for the 72 hours between the 20-minute ischemic preconditioning and the 60-minute period of ischemia. Antisense ODN treatment reduced expression of bcl-2 in the striatum and blocked the induction of tolerance by preconditioning ischemia. Sense and CSF treatments had no effect on either bcl-2 expression or tolerance. In this model of induced tolerance to focal ischemia, bcl-2 appears to be a major determinant.
Ischemic tolerance is the phenomenon by which a brief period of “preconditioning” ischemia, which in itself does not result in cell death, attenuates injury from subsequent severe ischemia. The phenomenon was first recognized in the myocardium where reduced infarction size and improved myocardial function occurred in ischemic myocardium that had been subjected to preconditioning ischemia (Swain et al., 1984). Ischemic tolerance in the heart permitted the myocardium to endure twice the duration of ischemia usually producing infarction. Dog and rabbit myocardium, usually able to tolerate 20 minutes of ischemia before infarction, could now tolerate 40 minutes (Lawson and Downey, 1993).
Ischemic tolerance also has been demonstrated in the brain (Kitagawa et al., 1990). Brief periods of transient global ischemia protect the selectively vulnerable neurons of the hippocampus in a dose-dependent fashion. Two minutes, but not 1 minute, of preconditioning produced tolerance and persisted for at least 2 days (Kitagawa et al., 1997). Tolerance may also be induced by focal ischemia. Once induced by either method, tolerance protects the brain against a subsequent challenge by either focal ischemia or global ischemia (Chen et al., 1996; Glazier et al., 1994; Kitagawa et al., 1990; Simon et al., 1993). When focal ischemia is used to induce tolerance against subsequent focal ischemia, more than 24 hours is required for tolerance to become manifest, and once manifested the tolerance persists for approximately 7 days (Chen et al., 1996). The phenomenon of tolerance may not be explained by changes in blood flow. Using transient focal ischemia as the ischemic preconditioning stimulus, Chen et al. (1996) found no change in regional cerebral blood flow induced by preconditioning ischemia in the tolerant regions either before or during the prolonged focal ischemia. When global ischemia was used as the preconditioning ischemia in a model of tolerance, no alteration in regional cerebral blood flow was found in the region subsequently subjected to focal cerebral ischemia (Miyashita et al., 1994). The mechanism that produces ischemic tolerance is unknown. In the myocardium there is evidence suggesting that adenosine is the mediator of tolerance (Belardinelli et al., 1989). Other evidence supports stress protein induction (Yamashita et al., 1994). In the brain, new protein expression is required because ischemic tolerance is blocked by protein synthesis inhibition (Goto et al., 1990). Tolerance also has been induced in vitro by sublethal activation of N-methyl-d-aspartate receptors in cerebellar granule cells. New RNA and protein synthesis is required for tolerance to develop in this model (Marini and Paul, 1992). As in the heart, stress proteins are also candidates, the induction of which could produce tolerance in the brain. For example, expression of the heat shock protein HSP-72 and the glucose regulated proteins GRP-75 and GRP-78 are increased in the brain after ischemia (Lowenstein et al., 1994). The time course of HSP-72 expression induced by preconditioning ischemia is compatible with the time course of stimulation of tolerance, but not with the time course of GRP-75 and GRP-78 expression (Chen et al., 1996). However, if global ischemic preconditioning is carefully quantified by the measurement of ischemic depolarization, tolerance can be generated in the absence of HSP-72 mRNA induction while mRNAs of several transcription factors are produced (Abe and Nowak, 1996). Also, repeated preconditioning ischemic treatment in brain has been demonstrated to enhance tolerance to global ischemia in the absence of enhanced stimulation of HSP-72 (Kitagawa et al., 1996). Thus, proteins other than HSP-72 may be responsible for the induction of tolerance after ischemia.
The authors report here the potential role of bcl-2 expression in the phenomenon of tolerance. bcl-2 inhibits a key step in programed cell death (PCD) by inhibiting egress of cytochrome c from the mitochondria (Kluck et al., 1997). Cytochrome c, when present in the cytoplasm, complexes with Apaf-1, which in turn activates caspase-9 (Zou et al., 1997). Caspase-9 in turn cleaves and activates other caspases, including caspase-3, which executes PCD. bcl-2 expression can also ameliorate necrotic cell death, perhaps through its actions on mitochondrial function (Zhong et al., 1993). bcl-2 expression is induced in the brain by ischemia, suggesting that it could have a role as an endogenous neuroprotectant (Chen et al., 1997; Hara et al., 1996). This hypothesis is supported by the observation that bcl-2 protects neurons against ischemia when it is overexpressed using either viral vector techniques or in transgenic animals (Linnik et al., 1995; Martinou et al., 1994). However, these experiments do not address whether endogenous bcl-2 expression actively inhibits cell death after ischemia, or whether the bcl-2 expression that is induced by ischemia is an epiphenomenon that occurs in neurons already destined to survive ischemia.
To further investigate the potential role of bcl-2 as an effector of ischemic tolerance in brain, the authors used the intraluminal suture technique to precondition the striatum against subsequent focal ischemia and studied the expression of bcl-2 protein in this region of tolerant striatum. The authors then blocked both tolerance and bcl-2 expression with the administration of the bcl-2 antisense oligodeoxynucleotides (ODNs).
MATERIALS AND METHODS
Animal methods
Temporary focal ischemia was induced in male Sprague-Dawley rats (280 to 310 g) using the suture occlusion technique. The rats were anesthetized with 4% isoflurane (70% N2O:30% O2) by face mask. Endotracheal intubation then was performed. The rats were ventilated mechanically with 1.5% isoflurane. The left temporalis muscle temperature was maintained at 37.4°C + 0.1°C with a thermostatically controlled heating lamp until suture removal. One femoral artery was cannulated for continual blood pressure monitoring and measurement of arterial blood gases and blood glucose concentration.
Under the operating microscope, the bifurcation of the common carotid artery was exposed and the external carotid artery ligated with a 6-0 silk suture. The internal carotid artery (ICA) was isolated and separated from the vagus nerve. The extracranial branch of the ICA was ligated close to its origin with a 6-0 silk suture. The origin of the middle cerebral artery (MCA) was occluded by introducing a 3-0 monofilament nylon suture (its tip rounded by heating) into the ICA lumen, through the stump of the external carotid artery. The suture was gently advanced 20 to 22 mm past the carotid artery bifurcation. Preconditioning ischemia was induced, and the suture was gently withdrawn to permit reperfusion. The external carotid was ligated and the wound closed. Sham-operated rats underwent identical surgery except the suture was not inserted.
At a later time, the suture was reinserted to induce prolonged MCA ischemia. Rats were anesthetized in the same manner as noted above. Rectal and temporalis muscle temperature monitoring and femoral artery cannulation then were performed and the cervical incision reopened. Microvascular clips were applied to both proximal and distal sides of the common carotid artery bifurcation. The stump ligation was undone, and the same 3-0 monofilament suture was introduced again into the ICA through the stump. After ischemia was induced, the suture was again gently withdrawn to permit reperfusion. The external carotid stump was ligated and the wound was closed.
Transient focal ischemia 20 minutes in duration was used to induce tolerance. Preliminary experiments with 5 and 10 minutes of transient focal ischemia produced variable tolerance, and with 30 minutes of transient focal ischemia, incomplete infarction occurred in the striatum (data not shown). Accordingly, 20 minutes of middle cerebral artery occlusion was used to induce tolerance in subsequent studies. After 20 minutes of focal ischemia, animals then were challenged with 60 minutes of ischemia, which produces striatal infarction in the absence of tolerance. The interval between the two periods of ischemia was 1, 3, 5, or 7 days. A sham-operated group in which the initial surgery was performed (the carotid artery exposed, but no suture inserted) was studied as well (n = 7 per group).
Quantification of infarction volume
An overdose of 8% chloral hydrate was used to kill animals 72 hours after the second MCA occlusion. Brains were rapidly removed and sectioned coronally at 2-mm intervals. The sections were immersed in 2% 2,3,5-triphenyltetrazolium hydrochloride (TTC) in saline for 20 minutes at 37°C and then transferred to 4% paraformaldehyde for 15 minutes. Six sections were analyzed for infarction size by a blinded observer using a computerized imaging analysis system. The infarction area in each section was calculated by subtracting the normal area that stained with TTC in the right (ischemic) hemisphere from the area of the left (nonischemic) hemisphere. Infarction volume was calculated by summing the infarction areas of all sections and multiplying by the slice thickness.
For confirmation of the TTC determinations of infarct volume, an additional group of animals (n = 7) was subjected to 20 minutes of transient focal ischemia or sham surgery (n = 7) and 3 days later to 60 minutes of ischemia. Animals were killed with 8% chloral hydrate 72 hours later and perfused transcardially with saline (100 mL) and 4% paraformaldehyde (400 mg, 0.1 N phosphate buffer, pH 7.4). Brains were removed and postfixed in the same fixative for 7 days, then were embedded in paraffin, and 6-mm-thick coronal sections were taken at −0.3 mm from the bregma. Sections were stained with cresyl violet and the infarction volume was determined using the method above.
Western blotting
Brain tissue was dissected from the striatum at various times after the 20-minute preconditioning period of ischemia. Tissue was homogenized and lysed in 0.1 mol/L NaCl, 0.01 mol/L Tris-Cl (pH 7.6), 0.001 mol/L MEDTA (pH 8.0), 1 μg/mL aprotinin, and 100 μg/mL PMSF.
Lysates were cleared by centrifugation at 14,000 g for 30 minutes at 4°C and boiled at 100°C in sodium dodecyl sulfate (SDS) gel loading buffer (100 mmol/L Tris-Cl, 200 mmol/L dithiothreitol, 4% SDS, 0.2% bromophenol blue, and 20% glycerol) for 6 minutes. The samples were then run on a 12% SDS-polyacrylamide gel. Western blots were performed as previously described (Chen et al., 1997). The transferred PVDF membrane was incubated with the primary antibody (monoclonal antibodies against bcl-2, bcl-x, or a polyclonal antibody against Bax; Santa Cruz, CA) in a dilution range of 1:500-1000 according to the manufacturer's instructions at 4°C overnight. This was followed by 3 washings in buffer (0.1% Tween-20, 0.5% bovine serum albumin, and 1% nonfat dry milk in 1x phosphate-buffered saline (PBS) buffer) and then by incubation at room temperature for 60 minutes at different relative alkaline phosphatase (AP)-conjugated secondary antibodies according to the requirements for the various primary antibodies. The membrane was incubated at room temperature for 60 minutes with conjugated secondary antibodies appropriate for the primary antibody and detected with chemiluminescent substrate (CSPD 25 mmol/L). The blot was wrapped in plastic wrap and exposed to a Kodak X-OMAT film. The film was developed and the band optical density was measured by the MCID image system (St. Catherine's, Ontario, Canada).
Immunocytochemistry and TUNEL
bcl-2 immunocytochemistry was performed using the methods previously described (Clark et al., 1997). Briefly, rats were anesthetized as described above, then perfused transcardially with 200 mL heparinized saline (8 units/mL) followed by 500 mL 2% paraformaldehyde. Brains were removed and further immersion-fixed in 2% paraformaldehyde for 30 minutes, rinsed 3 times in PBS, and then placed in 30% sucrose at 4°C before snap freezing in 2-methylbutane in liquid nitrogen. Brains were cut in 5-μm sections using a cryostat, washed 3 times with PBS, and then washed 3 times in PBS containing 0.5% bovine serum albuminute and 0.15% glycine (buffer A). Nonspecific activity was blocked with 5% normal goat serum in buffer A. Brain sections then were incubated at room temperature for 1 hour in a 1:100 dilution of mouse monoclonal antibody against human bcl-2 (Dako, Cupertino, CA, U.S.A.). Sections were washed 3 times in buffer A and then incubated for 1 hour in a 1:3000 dilution of goat anti-mouse Cy3.18 immunoconjugate (Jackson Immunochemicals, West Grove, PA, U.S.A.). Sections then were washed 6 times (5 min/wash) in buffer A, mounted in gelvatol, and coverslipped for light microscopy. For double- and triple-label experiments, terminal deoxynucleotidyl transferase-mediated biotin-dUTP nick end labeling (TUNEL) was performed on additional sections using a modification of the technique (Gavrieli et al., 1992). Sections were washed twice with PBS and then incubated at 37°C for 1 hour in buffer containing TdT and fluorescein-conjugated 12-dUTP (Boeringer Mannheim, Indianapolis, IN, U.S.A.). Sections then were processed for bcl-2 immunocytochemistry as described above. To assess nuclear morphology, bisbenzimide (Sigma, St. Louis, MO, U.S.A.; similar to Hoechst 33258) diluted in sterile water was applied to some sections for 30 seconds before coverslipping. A light microscope equipped for epifluorescent illumination and differential interference contrast was used for observation, and fluorescent images were collected using an integrating 3-chip color video camera. Excitation and emission wavelengths of 550/565 (red), 494/520 (green), and 346/460 (blue) λ were used for bcl-2, TUNEL, and bisbenzimide, respectively. In sections from each specimen, the primary antibody was omitted to assess for nonspecific binding of the secondary antibody.
Antisense treatment
A miniosmotic infusion pump was implanted 30 minutes before the 20-minute period of preconditioning ischemia. Rats were anesthetized with isoflurane in the same manner as described above and placed in a stereotactic frame. An access hole was drilled through the skull to permit placement of a cannula into the right cerebral ventricle. Brain infusion cannulas (Alzet brain infusion kit; Alza, Palo Alto, CA, U.S.A.) were stereotactically placed following the manufacturer's recommendation at the following coordinates: 0.8 mm posterior to bregma, 1.5 mm lateral to midline, and 3.8 mm below the dura with the incisor bar positioned 5 mm above the interaural line. Cannulas were fixed in place with dental cement and anchored to a screw placed 5 mm away. Cerebrospinal fluid (CSF), bcl-2 antisense ODN, or bcl-2 sense ODN were continuously infused into the right cerebral ventricle for 72 hours by Alzet mini pumps (1.0 μL/h flow rate). The bcl-2 antisense phosphorothioate ODNs used for these experiments are those that straddle the predicted translation initiation site of rat bcl-2 mRNA and are completely complementary to this region. The rat bcl-2 antisense ODN was based on the prior development of human antisense ODNs to bcl-2 (Kitada et al., 1993) by the availability of the rat bcl-2 sequence (Sato et al., 1994). The antisense sequence used was TGT TCT CCC GGC TTG CGC CAT. The complimentary sense sequence used was ATG GCG CAA GCC GGG AGA ACA. The scrambled sequence used was GTC ATC CCC TGC TGC CGT. Sixty-minute cerebral ischemia was induced and the rat brains were killed after 3 days for Western blots and determination of infarction volume by TTC. Rats were killed at 24 hours after ischemia for immunocytochemistry because previous studies had found more robust expression at this time (Chen et al, 1997).
RESULTS
Characterization of the effect of preconditioning ischemia on infarction volume
The effect of preconditioning ischemia on the volume of striatal infarction is summarized in Fig. 1. Control animals received only the 60-minute period of focal ischemia; their infarct volume was the same as animals receiving 60 minutes of ischemia but previously subjected to a sham operation (sham) 3 days before. Animals with an interval of 1 day between 20 minutes of preconditioning ischemia and 60 minutes of focal ischemia had infarct volumes in the striatum not different from those of control and sham animals. However, the 3-day interval between the preconditioning ischemia and the 60 minutes of focal ischemia resulted in a reduction of the striatal infarct volume by nearly half. A significant reduction in infarct volume continued to be demonstrated even when the interval between the preconditioning ischemia and the 60 minutes of focal ischemia was extended to 5 and 7 days. There were no statistically significant differences in the physiologic variables among any of the experimental groups (Table 1).
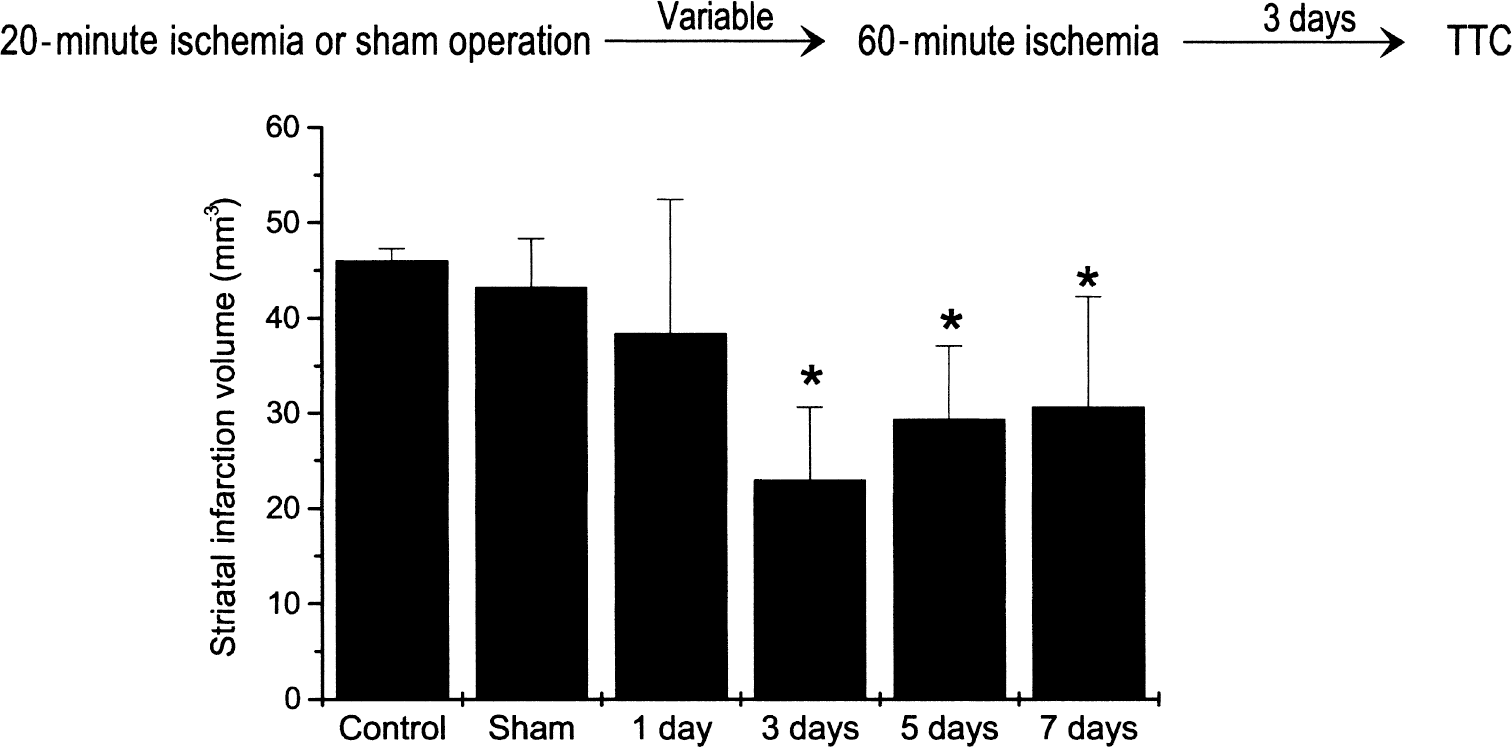
Effect of preconditioning ischemia on infarction volume determined by 2,3,5-triphenyltetrazolium hydrochloride (TTC). Graph illustrating mean infarction volume determined by TTC-staining 72 hours after 60-minute ischemia. Rats were subjected to variable intervals between 20-minute preconditioning ischemia and 60-minute ischemia. Control: rats had no surgery or preconditioning ischemia before 60 minutes of ischemia. Sham: rats underwent sham surgery 3 days before 60 minutes of ischemia. Data expressed as mean ± standard deviation. *P < 0.05 analysis of variance, post hoc testing by Fischer's PLSD multiple comparison test.
Physiologic variables in tolerance experiment
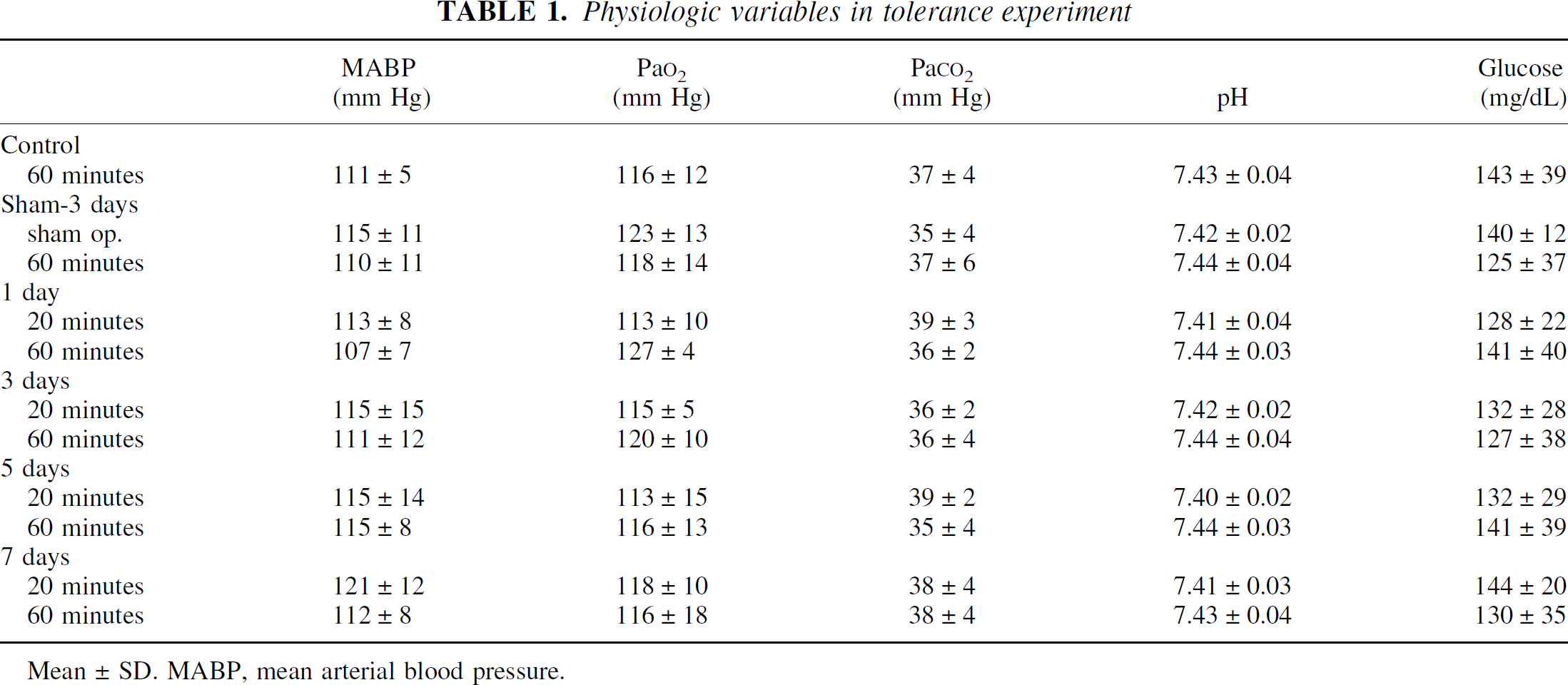
Mean±SD. MABP, mean arterial blood pressure.
To confirm the above findings, infarction volume was also assessed in cresyl violet-stained sections from rats killed 3 days after the second (severe) ischemia. These rats were subjected to either sham operation or preconditioning ischemia. Using this method, there also was a significant reduction in infarction volume in those rats that received preconditioning ischemia as compared with the sham-operated rats (Fig. 2). The degree of protection induced by preconditioning ischemia observed with cresyl violet histology is smaller than the TTC method. This difference may be due to several reasons, including detection of inflammatory cells by the TTC method or the lack of sensitivity of TTC to selective neuronal loss compared with cresyl violet histology.
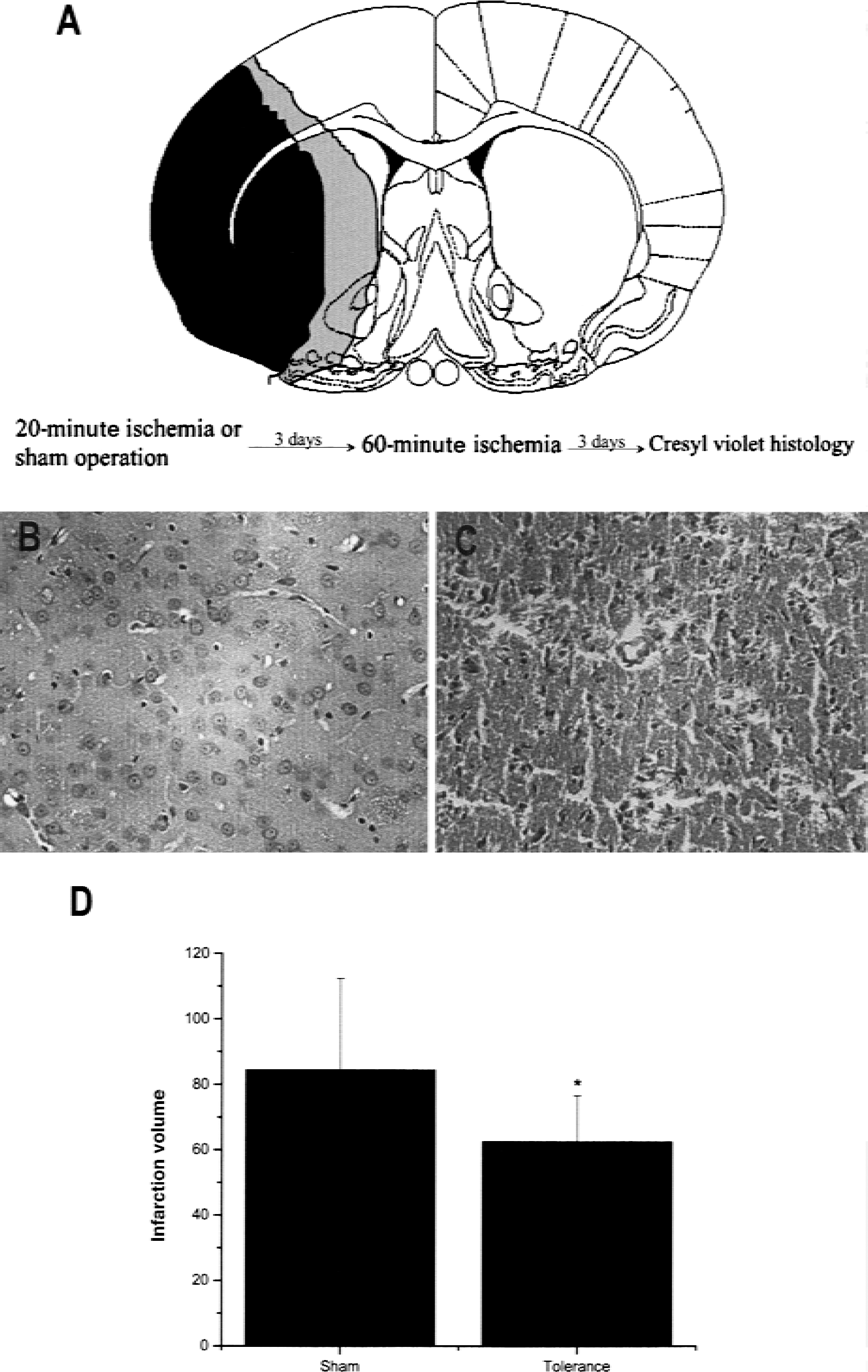
Effect of preconditioning ischemia on infarction volume determined by cresyl violet histology.
Effect of preconditioning ischemia on expression of bcl-2 and related genes
Preconditioning ischemia produces a sublethal ischemic stress to brain. One would therefore expect induction of expression of genes such as bcl-2 in regions of brain that are ischemic yet survive. To test whether preconditioning ischemia induces expression of bcl-2 in regions of brain that are spared by preconditioning ischemia, the authors studied expression of bcl-2 and related genes in striatum after 20 minutes of preconditioning ischemia or sham operation, by Western blotting and immunocytochemistry. Figure 3A shows representative Western blots from striatal tissue obtained from rats killed at various times after the 20-minute preconditioning ischemia or naïve (nonischemic) controls. These data are quantified in the graph in Fig. 3B. Rats subjected to 20 minutes of ischemia evidenced up to a 15-fold increase in bcl-2 expression as compared with nonischemic controls. Interestingly, the peak increase in bcl-2 expression occurred at 3 days, the interval after preconditioning ischemia when there is maximal reduction of infarction volume (Fig. 1A).
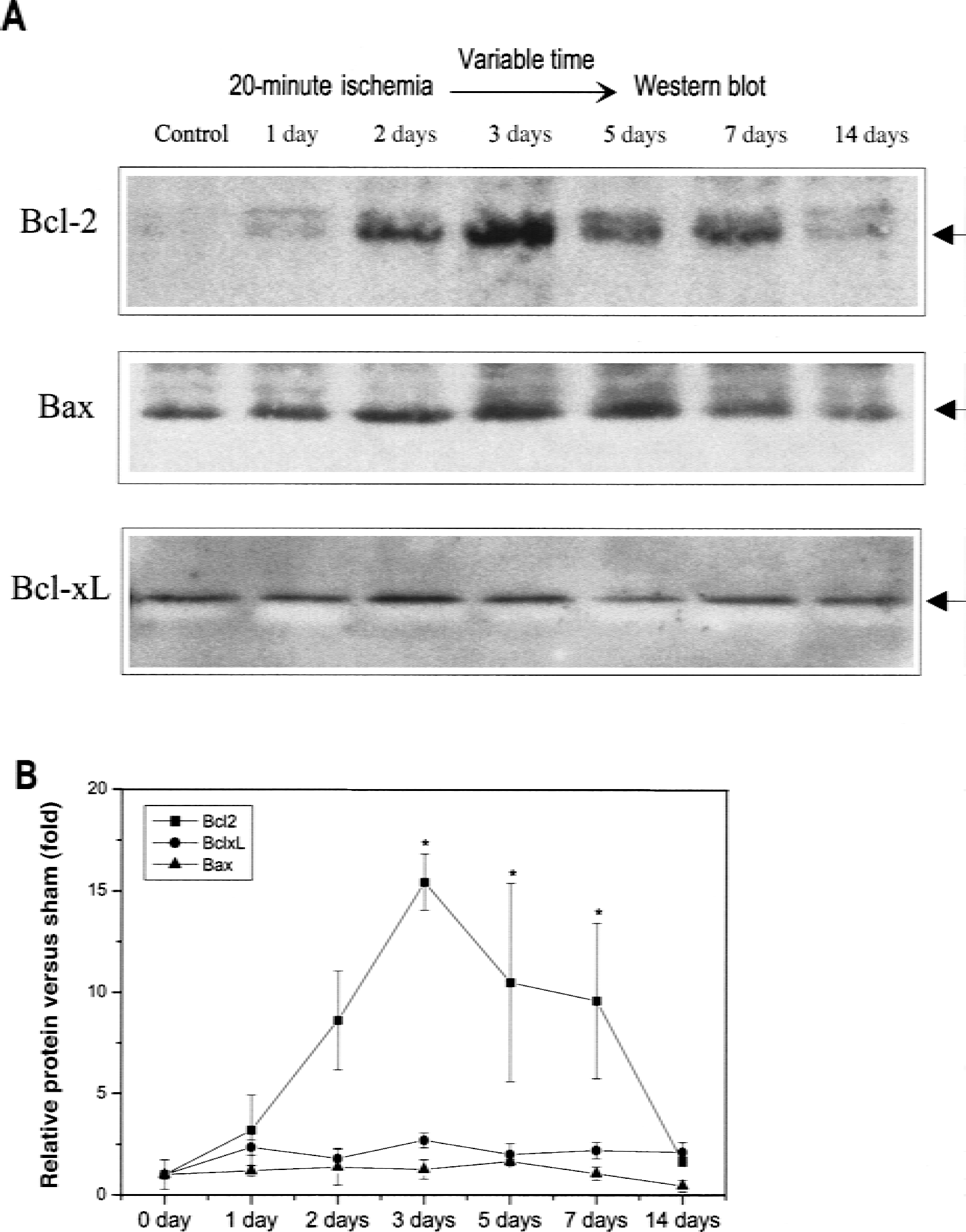
Western blots after 20 minutes of preconditioning ischemia.
To determine whether bcl-2 was expressed in regions that were spared by preconditioning ischemia, 20 minutes of ischemia was induced in rats that were killed 24 hours later for immunocytochemical analysis. Increased expression of bcl-2 in medial caudate occurred in cells with both neuronal and nonneuronal morphology (Fig. 4). Bisbenzamide double staining revealed that bcl-2 was expressed primarily in the cytoplasmic compartment in cells with normal-appearing nuclear morphology. Almost no cells TUNEL-labeled after 20-minute ischemia, which is consistent with the previous results that this interval of ischemia stresses but does not kill neurons (Chen et al., 1996). There was extensive TUNEL labeling in the ischemic cortex and striatum of brains subjected to 60 minutes of ischemia, and these were used as positive controls (not shown). Interestingly, the region where bcl-2 reactive cells are present corresponds closely to the region that is spared from infarction by preconditioning ischemia (Fig. 2A). There was no identifiable cellular expression of bcl-2 in cells in nonischemic regions; thus, in animals subjected to preconditioning ischemia, bcl-2 expression is induced in regions that are spared.
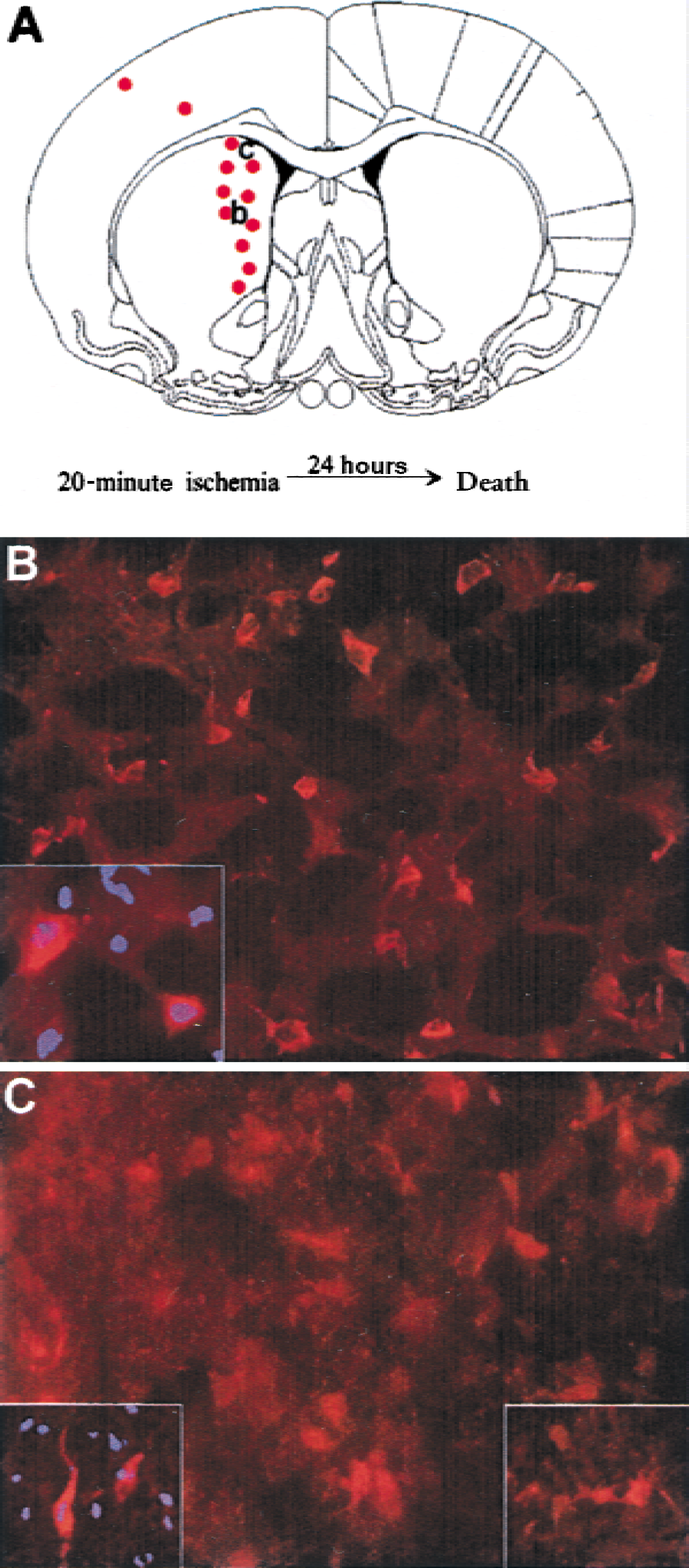
bcl-2 immunocytochemistry after 20 minutes of preconditioning ischemia.
Effect of bcl-2 antisense treatment on expression of bcl-2 and related genes
To test whether bcl-2 expression induced by preconditioning ischemia affords protection against severe ischemia, expression of bcl-2 induced by preconditioning ischemia was inhibited by intraventricular infusion of antisense ODN, with brains removed 72 hours after treatment with either antisense, sense, or vehicle treatment (artificial CSF). Sham-operated animals that had the pump and catheter implanted and infused with mock CSF and carotid dissection, but no ischemia, were used as additional controls. Animals with CSF or sense infusion demonstrated a fivefold increase in bcl-2 expression in the striatum compared with the sham-operated rats (Fig. 5). bcl-2 expression in the antisense-treated rat brain was similar to bcl-2 expression in sham-operated controls. Animals treated with antisense showed a trend toward increased bcl-x-l expression. These results indicate that antisense treatment suppressed translation of bcl-2 protein in a sequence-specific fashion.
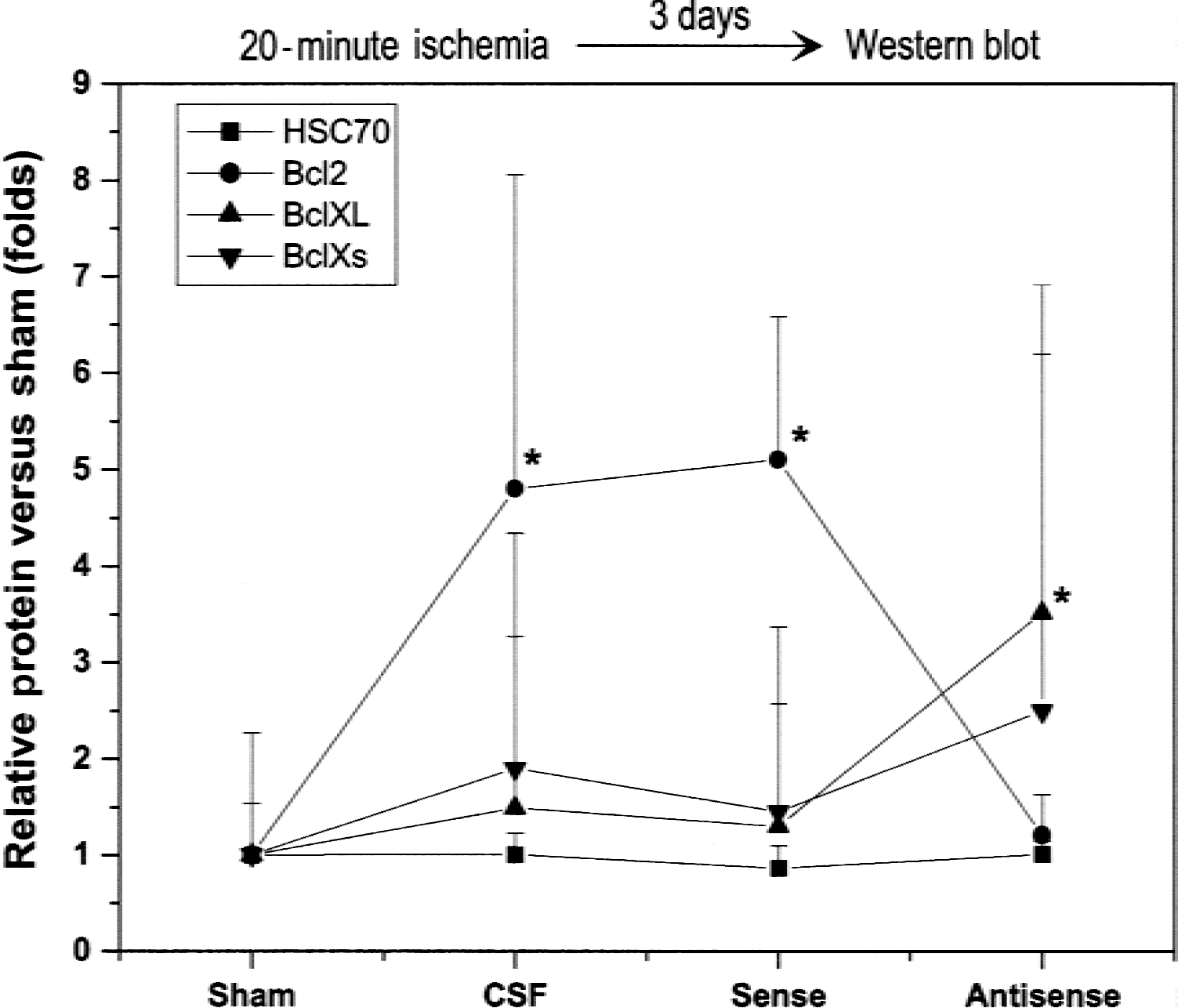
Effect of oligodeoxynucleotides (ODN) treatment on bcl-2 expression after 20 minutes of preconditioning ischemia. Graph summarizes mean changes in bcl-2, Bax, and bcl-x-l expression in sham-operated animals (sham) and rats subjected to 20-minute ischemia and infused with antisense, sense, or cerebrospinal fluid controls. Three days after sham surgery or 20-minute ischemia, brains were removed for determination of protein content in the ipsilateral cortex by Western blot. Data presented as mean ± SD of the ratio of mean of two sham samples run on the same gel. *P < 0.05 different from sham-operated group. Analysis of variance with post hoc testing by Fischer's PLSD multiple comparison test.
Effect of bcl-2 antisense treatment on infarction volume
The effect of bcl-2 antisense treatment on the induction of ischemic tolerance by preconditioning ischemia also was studied (Fig. 6). All animals were subjected to preconditioning ischemia or sham operation, and the lateral ventricles were perfused with either antisense or sense ODN, or vehicle (CSF). The infarction volume of antisense-treated animals is not different from sham-operated animals, so antisense treatment prevented the induction of tolerance. However, CSF and sense-infused animals have an attenuated striatal infarct volume compared with sham controls, so tolerance is not affected by these treatments. There were no significant differences in physiologic parameters among groups (Table 2).
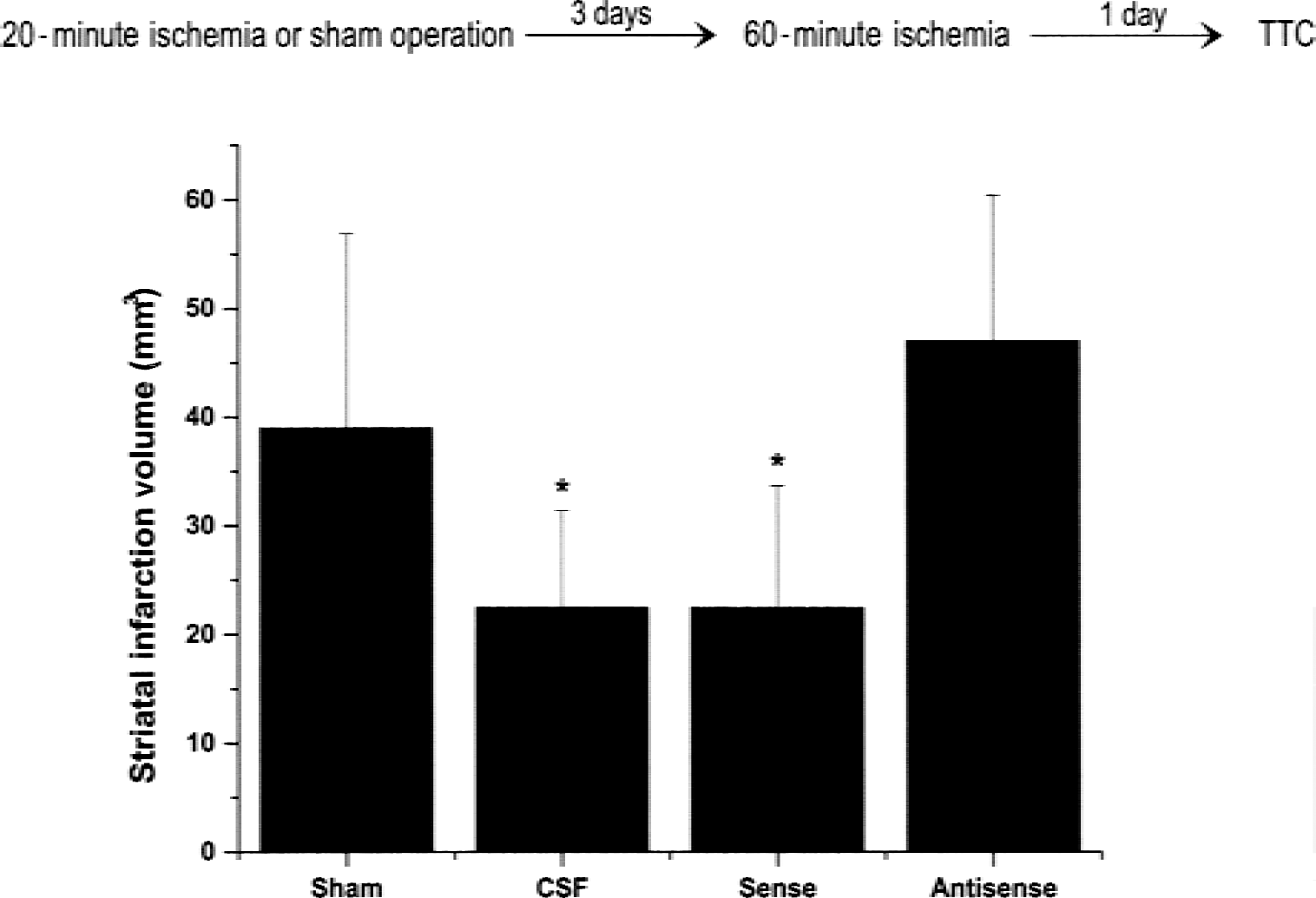
Effect of oligodeoxynucleotides (ODN) treatment on tolerance. Graph summarizes mean changes in infarction volume in sham-operated animals (sham) and rats subjected to 20 minutes of ischemia and infused with either antisense, sense, or cerebrospinal fluid controls. Three days after sham surgery or 20-minute ischemia, rats were subjected to 60-minute ischemia. Three days after 60-minute ischemia, brains were removed for histologic analysis. Data presented as mean ± SD. *P < 0.05 different from sham-operated group determined by analysis of variance with post hoc testing by Fischer's PLSD multiple comparison test.
Physiologic variables in antisense experiment
Mean±SD. MABP, mean arterial blood pressure; CSF, cerebrospinal fluid.
DISCUSSION
The major findings of this study include the following:
brief sublethal ischemia protects the striatum against subsequent lethal ischemia; thus, ischemic tolerance can be induced in the striatum by preconditioning ischemia; Preconditioning ischemia induces expression of bcl-2 protein in the striatum at times that correlate with the induction of ischemic tolerance; 3) bcl-2 antisense treatment prevents increased expression of bcl-2 protein in the striatum induced by preconditioning ischemia; 4) bcl-2 antisense treatment blocked the induction of tolerance in the striatum by preconditioning ischemia.
The phenomenon of ischemic tolerance in brain was first demonstrated in vivo in hippocampal pyramidal neurons in the setting of global ischemia-reperfusion (Kitagawa et al., 1990). The phenomenon of ischemic tolerance was subsequently demonstrated in cortex after focal ischemia (Chen et al., 1996; Miyashita et al., 1994; Simon et al., 1993). In the current study, the authors found that brief focal ischemia can induce tolerance in the caudate as well. Previously, however, their group found the site of tolerance to be in the cortex. This may be because of the differences in duration of MCA occlusion. In a previous study (Chen et al., 1996), 100 minutes of ischemia, which results in extensive cortical infarction, was used as the second severe ischemia. In the current study, 60 minutes of ischemia, which produces primarily striatal infarction, was used. Thus, the phenomenon of ischemic tolerance can be induced in a variety of brain regions depending on the duration and type of ischemia.
These data strongly suggest that bcl-2 is a major mechanism by which ischemic tolerance is produced, at least in the striatum. The time window of the up-regulation of bcl-2 in this experiment mirrors the time window of the development of tolerance. In this experiment and in the authors' previous studies (Chen et al., 1996), tolerance is not found at 1 day after ischemic preconditioning and is maximal at 3 days; the duration of tolerance in focal ischemia is approximately 7 days.
This is also the temporal profile of bcl-2 expression after temporary focal ischemia. There is very little constitutive expression of bcl-2 in the nonischemic rat brain (Merry et al., 1994). Its maximal expression in this experiment is at 3 days, when tolerance is maximal, and it persists to 7 days, as does tolerance in this model. Furthermore, preconditioning ischemia induces expression of bcl-2 in the medial caudate, the region that is spared by preconditioning ischemia.
Administration of antisense ODNs to bcl-2 blocked its expression and also blocked tolerance. The phosphorothioate ODN used for these studies straddled the predicted translation-initiation site of rat bcl-2 mRNA. An antisense ODN complimentary to this region for human bcl-2 has been shown to inhibit bcl-2 translation in several in vitro systems (Kitada et al., 1993). Controls for the antisense ODN treatment included the sense version of this 21-mer ODN and infusion of mock CSF. The antisense treatment significantly reduced bcl-2 protein expression compared with sense or CSF controls. The antisense-treated animals had a volume of striatal infarction not different from the sham animals, whereas CSF and sense animals continued to demonstrate the phenomenon of tolerance (Fig. 5). Suppressing bcl-2 expression with antisense ODN almost completely abrogates the effect of preconditioning ischemia in inducing ischemic tolerance. This observation suggests that bcl-2 expression may be the major mechanism by which ischemic tolerance is induced in brain.
Bcl-x-l is also an antiapoptotic gene product that is up-regulated in ischemic brain (Chen et al., 1997). In contrast to bcl-2, bcl-x protein is expressed in normal rodent brain (Merry et al., 1994). In the current study, there were no significant changes in bcl-x-l expression after preconditioning ischemia. Also, there were no decreases in Bax expression found that could also explain this finding. Therefore, the time course and localization of bcl-2 expression after preconditioning ischemia is consistent with its role as an effector of ischemic tolerance.
Interestingly, there was a slight but significant increase in expression of bcl-x-l in rats treated with bcl-2 antisense ODNs compared with CSF controls after preconditioning ischemia. As has been observed when gene expression is disrupted during development, proteins with similar functions were induced in an attempt to replace the function of the missing protein. In this case, the attempt was not successful because tolerance was abolished. There are at least three possible reasons for this: 1) the increase in bcl-x-l expression is quite modest in comparison with the effects on bcl-2 expression; 2) there is also a slight increase in the proapoptotic bcl-x-s splice variant; and 3) bcl-x-l may have no protective effect. There was no effect of antisense treatment on expression of the constitutive 70 kDa heat shock protein hsc-70, suggesting that antisense treatment is not non-specifically suppressing synthesis of constitutively expressed proteins.
There are several mechanisms by which bcl-2 expression may produce ischemic tolerance. Ischemic cell injury involves accumulation of intracellular calcium entering the cell through excitatory amino acid modulated channels; intracellularly, such calcium is sequestered mainly in mitochondria and, to a lesser degree, in endoplasmic reticulum and Golgi bodies (Simon et al., 1984). bcl-2 protein is primarily found in the mitochondrial outer membrane and produces homo- and heterodimers with other bcl-2 family members that form membrane channels (Schlesinger et al., 1997). These channels may flux calcium (Lam et al., 1994). bcl-2 further modifies the calcium uptake of mitochondria in neuronal cells (Murphy et al., 1996). bcl-2-expressing cells have increased capacity for calcium uptake and an increased resistance to calcium-induced mitochondrial injury (Murphy et al., 1996). Furthermore, Shiraiwa et al. (1996), using the oxalate pryoantimonate technique, observed intracellular calcium deposition in organelles of CA1 pyramidal neurons exposed to a period of ischemic preconditioning adequate to induce tolerance. Although the mitochondria of the tolerant neurons were able to sequester increased amounts of calcium during an ischemic challenge, they also were able to decrease their calcium load more rapidly during the reperfusion. The authors noted that the plasma membrane calcium-ATPase activity of CA1 neurons was enhanced by the ischemic preconditioning.
bcl-2 expression also serves as a key control step that inhibits PCD. In the presence of bcl-2 protein, the egress of cytochrome c from the mitochondria is inhibited (Yang et al., 1997). Cytochrome c, when released into the cytosol, complexes with Apaf-1 and activates caspase-9 (Zou et al., 1997), a key step in the activation of PCD. bcl-2 expression in the mitochondria reduces the formation of reactive oxygen species in neurons in response to oxidative stress (Kane et al., 1993). Thus, bcl-2 may exert its protective effects through several mechanisms.
There are a number of possible limitations to these findings. Antisense ODN, as with any drug treatment, may have effects other than those intended on message translation. They may interact with cell surface glycoproteins, their nucleotide degradation products may affect the cell cycle, or they may have a variety of other nonsequence-specific effects. However, the authors used controls to help exclude these nonspecific effects. As in any in vivo experiment, there are innumerable complex and interdependent biochemical and physiologic changes that result in infarction after cerebral ischemia. Thus, the effect of antisense ODN treatment on bcl-2 expression may not be the sole mechanism by which the antisense treatment exerts its effects on ischemic tolerance.
Despite these limitations, the authors have shown that there is spatial and temporal correlation between the induction of bcl-2 expression after preconditioning ischemia and the induction of tolerance. bcl-2 antisense ODN almost completely blocks the induction of bcl-2 expression after ischemia and the induction of ischemic tolerance. These data support the hypothesis that bcl-2 expression is a major mechanism by which ischemic tolerance is induced in the striatum by focal ischemic preconditioning.
Footnotes
Acknowledgments
The authors thank Jingyu Luan and Jennifer Sinclair Sergi for technical support and Pat Strickler for secretarial support.