
Abstract
The current study determined whether short durations of ischemia that produce ischemia-induced tolerance stimulate glial proliferation in brain. Adult male gerbils were injected with BrdU (50 mg/kg) and dividing cells were detected using immunocytochemistry after sham operations, 2.5 or 5 minutes of global ischemia, or ischemia-induced tolerance. The 2.5-minute ischemia and the ischemia-induced tolerance did not kill hippocampal CA1 pyramidal neurons, whereas the 5-minute ischemia did kill the neurons. At 4 days after 2.5-minute global ischemia, when cell proliferation was maximal, BrdU-labeled cells increased in striatum and in neocortex, but not in hippocampus. The majority of the BrdU-labeled cells were double-labeled with isolectin B4, showing that these dividing cells were primarily microglia or macrophages, or both. Similarly, BrdU-labeled microglia/macrophages were found in striatum and neocortex but not in hippocampus of most animals 4 days after ischemia-induced tolerance (2.5 minutes of global ischemia followed 3 days later by 5 minutes of global ischemia). No detectable neuronal cell death existed in striatal and cortical regions where the microglia/macrophage proliferation occurred. Though 3 of 7 animals subjected to 2.5 minutes of ischemia showed decreased myelin-associated glycoprotein (MAG) immunostaining and increased numbers of adenomatous polyposis coli-stained oligodendrocytes in lateral striatum, this did not explain the microglia/macrophage proliferation. Data show that ischemia-induced tolerance in the gerbil is associated with proliferation of microglia/macrophages in striatum and cortex but not in hippocampus. Because there is no apparent neuronal death, it is postulated that the microglia/ macrophage proliferation occurs in response to an unknown nonlethal injury to neurons or glia and may be beneficial.
Keywords
Markers for astrocytes (Chen et al., 1993; Petito et al., 1990), microglia (Giordana et al., 1994; Soriano et al., 1994), and oligodendrocytes (Hattori et al., 1992; Irving et al., 1997; Yam et al., 1997) provide useful indices of the glial responses to ischemia and other types of injury. One characteristic response that has been intensively studied is the activation of glia to form reactive microglia and reactive astrocytes after global and focal ischemia and many other types of brain injury. Reactive glia demonstrate changes in size and shape that can occur rapidly after injury (Bruce et al., 1996; Chen et al., 1993; Fukuda et al., 1996; Gehrmann et al., 1992a, 1992b , 1993, 1995a,1995b; Ivacko et al., 1996; Morioka et al., 1992, 1993) and can persist for days, weeks, or longer (Banati et al., 1995; Kato et al., 1994, 1995; Kumar and Evans, 1997; McRae et al., 1995). Glial activation also can occur when there is no apparent injury, because spreading depression stimulates the appearance of reactive microglia (Gehrmann et al., 1993) and reactive astrocytes (Kraig et al., 1991).
Glia around an injury change their cytoskeleton and often appear to increase in number, suggesting proliferation of cells in response to the injury (Del Bigio and Becker, 1994; Giordana et al., 1994; Hailer et al., 1999). All types of glia can proliferate in response to brain injury, including ischemia and trauma (Alliot et al., 1991; Amat et al., 1996; Eglitis and Mezey, 1997; Elkabes et al., 1996; Giordana et al., 1994; Giulian et al., 1991; Kondo et al., 1995; Mandai et al., 1997; Kreutzberg, 1996; Streit, 1993; Zielasek and Hartung, 1996). This results in increased numbers of microglia and, to a lesser extent, increased numbers of astrocytes and oligodendroglia at various times after ischemic and traumatic injury (Amat et al., 1996; Ivacko et al., 1996, 1997; Del Bigio and Becker, 1994). Although the signals for the glial proliferation are still being examined, damaged cells may release factors that trigger glial cell division (Ivacko et al., 1997; Kreutzberg, 1996; Raivich et al., 1994).
A recent ischemia study showed that 10 minutes of global ischemia has no effect on the proliferation of stem cells located in the hippocampal subgranular zone for the first week (Liu et al., 1998). However, between 7 and 14 days after ischemia, a 10-fold or greater increase in the proliferation of cells in the subgranular zone of the hippocampus appears. Approximately 40% of the newborn cells migrate into the dentate hilus where approximately one third of these cells differentiate into glial fibrillary acidic protein (GFAP)-labeled astrocytes (Liu et al., 1998).
During the study of astrocyte proliferation, marked cellular proliferation was noted 3 to 4 days after 5 minutes of global ischemia that occurred throughout the forebrain including the hippocampus, cortex, and striatum. This was surprising because 5 minutes of global ischemia in the gerbil was believed to cause neuronal death mainly in CA1 and the hilus of the hippocampus (Ito et al., 1975; Kirino, 1982). To investigate this further, the current study determined whether cellular proliferation occurred in gerbil brain after 2.5 minutes of global ischemia alone and after ischemia-induced tolerance, in which a 2.5-minute conditioning ischemia protects against 5 minutes of global ischemia 3 days later (Kitagawa et al., 1990, 1991; Kirino et al., 1991). The brief 2.5-minute period of global ischemia is not believed to cause neuronal death in gerbil brain, and the gerbil ischemic tolerance model protects CA1 pyramidal neurons (Kirino et al., 1991). The results of the current study demonstrate that 2.5 minutes of global ischemia and ischemia-induced tolerance stimulate proliferation of BrdU-labeled cells in striatum and cortex but not hippocampus. Because most of the dividing cells appear to be microglia, or macrophages, or both, it is postulated that ischemia-induced tolerance leads to sublethal injury in striatum and cortex, the nature and the cause of the injury as yet unknown.
MATERIALS AND METHODS
Animals and treatment
Adult male Mongolian gerbils (11 to 13 weeks, Harlan, Indianapolis, IN, U.S.A.) were used for these studies according to an animal protocol approved by the San Francisco VA. Animals were anesthetized with 3% isoflurane in 20% O2:77% N2. After bilateral neck incisions, both common carotid arteries (CCAs) were exposed and occluded with aneurysm clips for either 0 (sham), 2.5, or 5 minutes. Rectal temperature was maintained at 37†C ± 0.5†C with a heating blanket until the animals recovered from surgery.
Four groups of gerbils were examined in this study. Sham-operated gerbils served as controls, with both CCAs isolated but not occluded (n = 10). The second group had bilateral CCA occlusions performed for 2.5 minutes (n = 26). The third group had bilateral CCA occlusions performed for 5 minutes (n = 30). A 5-minute bilateral CCA occlusion lead to the death of most CA1 pyramidal neurons in the authors' laboratory (Liu et al., 1998) and in other published studies (Kirino, 1982; Colbourne et al., 1999). The fourth group had a 2.5 minute bilateral CCA occlusion performed and a 5-minute bilateral CCA occlusion was performed 3 days later (n = 16). This paradigm produces ischemia-induced tolerance (Kirino et al., 1991; Kitagawa et al., 1990, 1991). As reported by Kitagawa et al. (1990, 1991) and Kirino et al. (1991), the authors also found that the initial brief period of ischemia protects CA1 neurons against death produced by the second lethal duration of ischemia 3 days later (Liu et al., 1998).
Labeling cell turnover and histology
Three days after global ischemia, all animals were injected intraperitoneally with a thymidine analog, 5-bromo-2′-deoxyuridine-5′-monophosphate (BrdU, 50 mg/kg; Sigma, St. Louis, MO, U.S.A.). Sham animals were injected 3 days after sham surgery and ischemia-tolerant animals were injected 3 days after the 5-minute ischemia. Twenty-four hours after the BrdU injections, animals were anesthetized with ketamine (80 mg/kg) and xylazine (20 mg/kg) and perfused transcardially with 300 cc of 0.9% saline followed by 400 cc of 4% paraformaldehyde in (pH 7.4, 0.1 mol/L) phosphate buffer (PB). The brains were removed, postfixed for 6 hours in 4% paraformaldehyde, and placed in 30% sucrose for 2 days. Coronal sections (50-μm-thick) were cut on a vibratome, stored in PB, and processed for BrdU immunocytochemistry as described below. Therefore, the number of BrdU-labeled cells was assessed on day 4 after the last ischemia in all of the animals, the BrdU labeling represented the number of cells born on days 3 to 4 after the last ischemia.
Sections for histology were floated onto gelatin-subbed slides and Nissl stained with cresyl violet. To detect isolated neuronal loss, selected sections were immunostained using a neuron specific antibody to NeuN as described below. The authors previously have shown that more than 90% of the NeuN-stained neuronal nuclei disappear in the CA1 region of the hippocampus after 5 minutes of global ischemia in the gerbil model used in the current study (Liu et al., 1998), whereas 2.5 minutes of global ischemia and ischemia-induced tolerance (2.5 minutes of global ischemia followed 3 days later by 5 minutes of global ischemia) do not result in loss of any NeuN-stained neuronal nuclei in CA1 of hippocampus (Liu et al., 1998).
DNA denaturation
For the immunocytochemical detection of BrdU-labeled nuclei, DNA was denatured before incubation with the primary antibody (Liu et al., 1997, 1998). Free floating brain sections were pretreated in 50% formamide/2 × SSC at 65†C for 2 hours, followed by a 30-minute incubation in 2 N HCl at 37†C. Sections were rinsed for 10 minutes in 0.1 mol/L boric acid, pH 8.5 at room temperature followed by PB for 20 minutes at room temperature.
Immunohistochemistry
The following antibodies and reagents were used: mouse anti-BrdU (0.25 μg/mL; Roche, Indianapolis, IN, U.S.A.); mouse anti–myelin-associated glycoprotein (MAG; 1 μg/mL; Roche/Boehringer); mouse anti-vimentin (1 μg/mL; Chemicon, Temecula, CA, U.S.A.); mouse anti-GFAP (1 μg/mL, DAKO); mouse anti-adenomatous polyposis coli (APC; 1 μg/ mL; CalBiochem); and mouse anti-NeuN (1 μg/mL; Chemicon). Biotinylated sheep anti-mouse IgG (5 μg/mL; Amersham) was used as the second antibody for all mouse monoclonal antibodies. Biotinylated isolectin B4 (Sigma) was used to stain microglia/macrophages. Avidin-peroxidase complex was diluted 1:100 in PB (Vector laboratories, Burlingame, CA, U.S.A.). ExtrAvidin-Fluorescein 5′-isothiocyanate conjugate (15 μg/mL) and Texas Red-avidin conjugate (15 μg/mL, Sigma) were used as fluorescent labels.
Sections were incubated overnight at room temperature in primary antibody diluted in PB with 1% serum, 0.1% bovine serum albumin, and 0.3% TritonX100. After washing in 0.1 mol/L PB (pH 7.4), sections were incubated in biotinylated secondary antibody for 2 hours at room temperature. After 3 × 5-minute rinse in PB, sections were placed in avidin-peroxidase complex solution containing avidin-peroxidase conjugate for 2 hours followed by a 5-minute peroxidase reaction (0.25 mg/mL diaminobenzidine, 0.01% H2O2, 0.04% NiCl2).
For double immunofluorescent labeling, sections were reacted overnight with anti-BrdU followed by a 2-hour incubation in biotinylated sheep anti-mouse antibody. Texas Red-avidin conjugate was added for 2 hours and then washed with PB to detect the first antibody. Sections next were incubated with biotinylated isolectin B4 followed by ExtrAvidin-Fluorescein 5′-isothiocyanate conjugate to visualize the microglia, or macrophages, or both. Fluorescent labeling was detected using a Leitz microscope and the resulting images collected and processed with Adobe Photoshop (Adobe Systems, Mountain View, CA, U.S.A.). A few double-labeling experiments were performed using primary antibodies to BrdU and GFAP to determine if some of the newborn cells were astrocytes.
RESULTS
BrdU labeling of proliferating cells in subgranular and subventricular zones
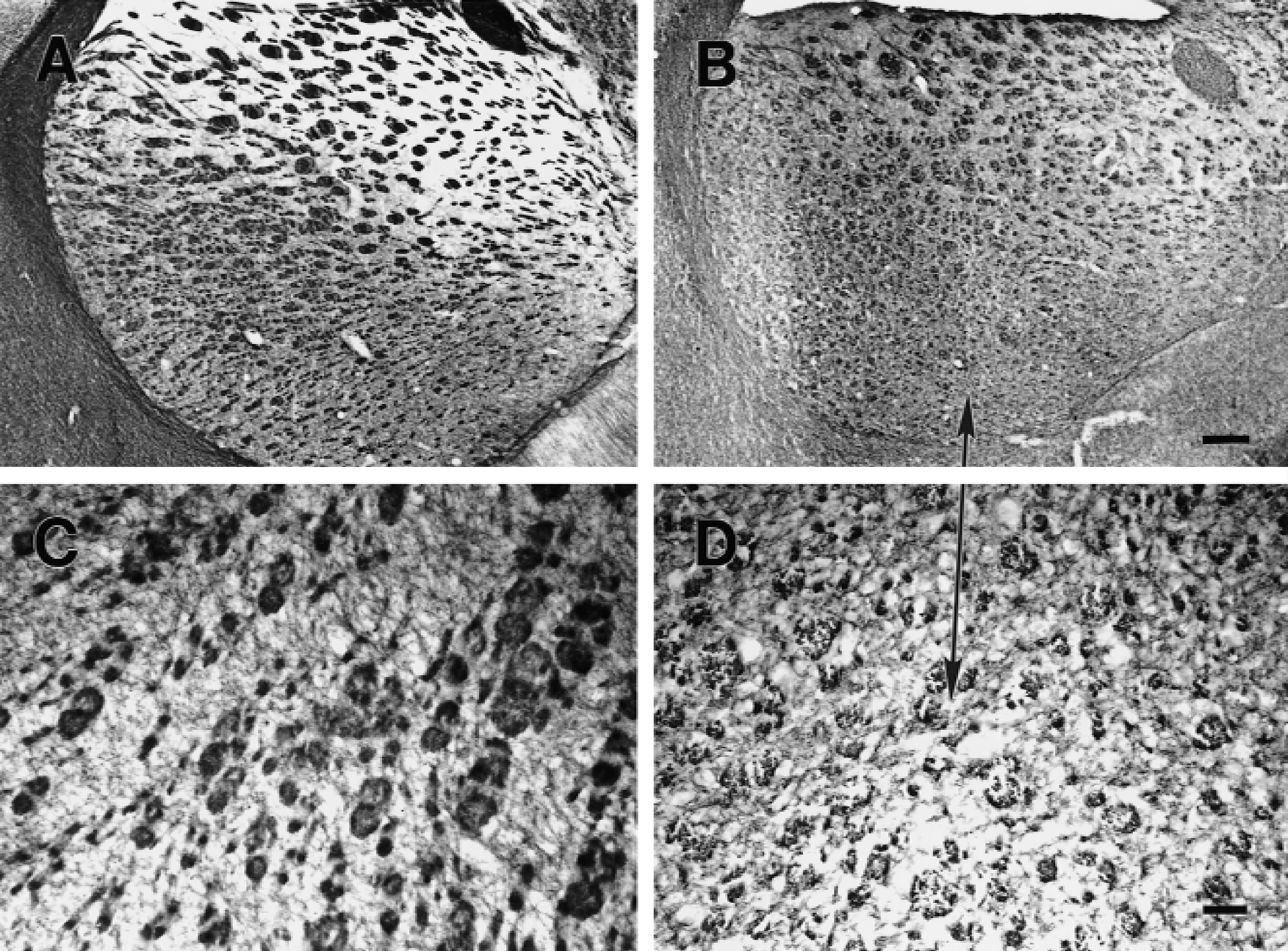
BrdU labeling was detected as black staining in the nucleus of cells. In sham-operated and ischemic animals, there were always BrdU-stained progenitor cells in the subventricular (subependymal) region of the medial striatum, adjacent to the walls of the lateral ventricle (Fig. 1A to 1D, dark cells at top of each figure). The number of cells in this region did not appear to change related to whether animals had been ischemic. There were also scattered cells in hippocampus, with clusters of BrdU-labeled cells in the subgranular zone of the dentate gyrus of sham-operated (Fig. 1E) and ischemic animals (Fig. 1F and 1H). As reported previously, the number of BrdU-stained cells in the dentate subgranular zone did not change for the first seven days after ischemia (Liu et al., 1998). In sham-operated animals, there were rare BrdU-stained nuclei scattered throughout every region in the brain, including the striatum, hippocampus, and neocortex (Fig. 1A, 1E, and 1I).
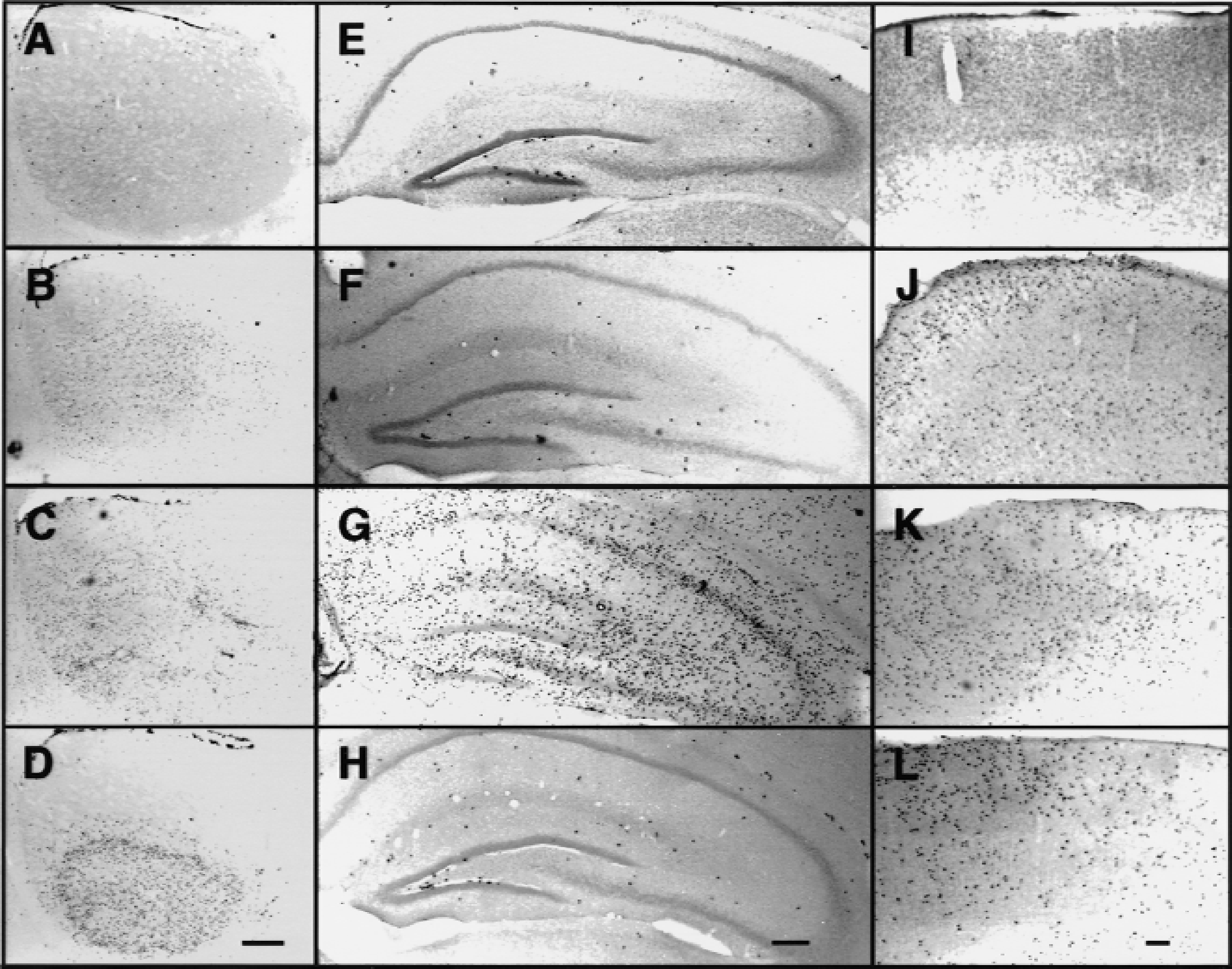
Dividing cells in brain four days after global ischemia. BrdU-labeled, dividing cells in striatum (
Cell proliferation in striatum, cortex, and hippocampus after 5 minutes of ischemia
Four days after 5 minutes of global ischemia (group 3), the number of BrdU-labeled cells increased in striatum (Fig. 1C), parietal cortex (Fig. 1K), and throughout all layers of the hippocampus (Fig. 1G). Labeled cells increased by 1 day after ischemia, were maximal at 4 days after ischemia, and decreased by 6 to 8 days.; therefore, this report focused on the findings at 4 days after ischemia (Figs. 1 to 5; Table 1). The 5-minute period of ischemia resulted in loss of most CA1 pyramidal neurons in hippocampus by 4 days after ischemia (Fig. 2E), as previously described from the authors' laboratory using Nissl and NeuN immunostaining (Liu et al., 1998). All 30 animals subjected to 5 minutes of global ischemia showed marked increases in the number of BrdU-labeled cells in the hippocampus, striatum, and neocortex (Table 1, 5 minutes).
Number of gerbils that demonstrated 1 of 5 different patterns of BrdU labeling in ST, CTX, and HP 4 days after global ischemia compared with controls
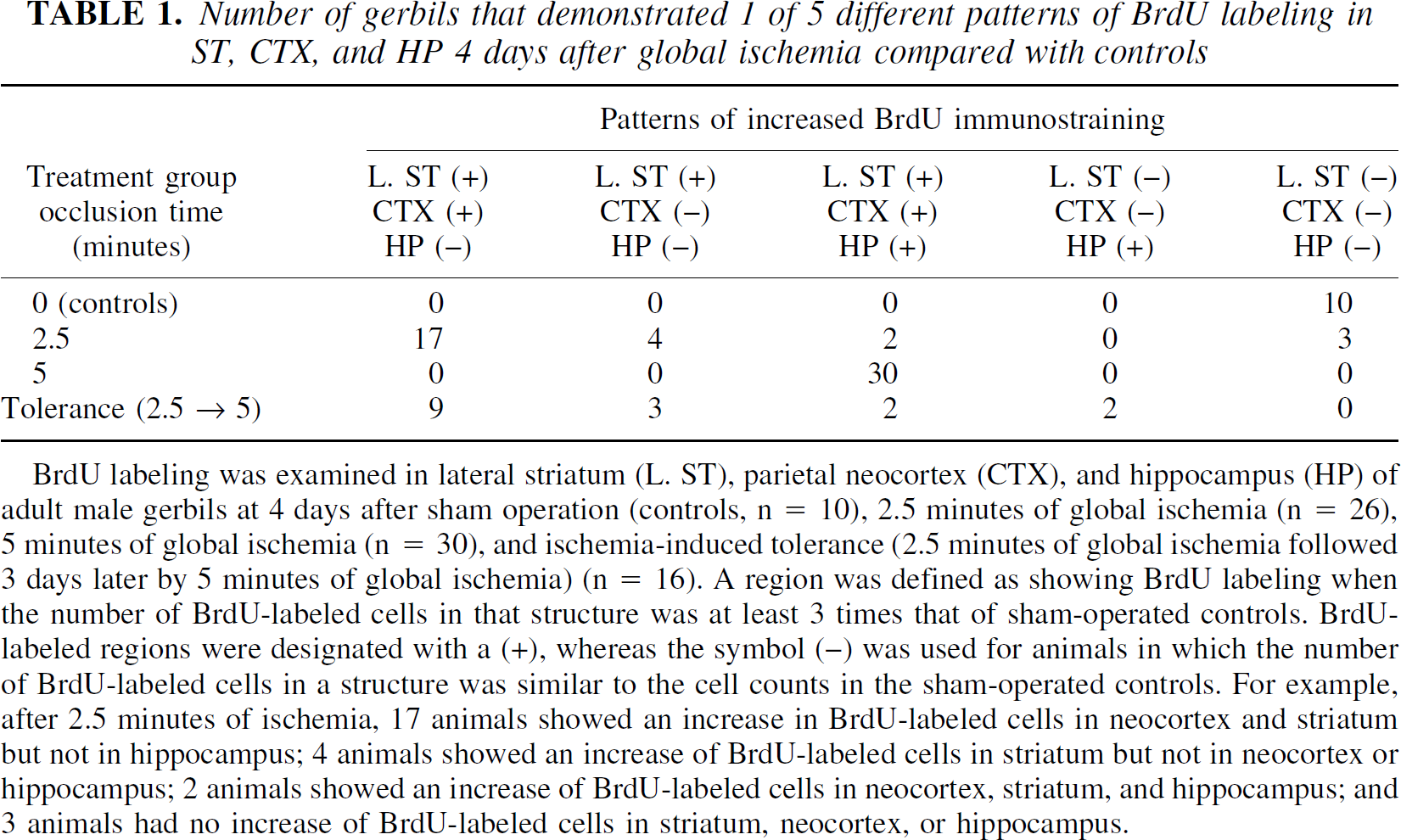
BrdU labeling was examined in lateral striatum (L. ST), parietal neocortex (CTX), and hippocampus (HP) of adult male gerbils at 4 days after sham operation (controls, n = 10), 2.5 minutes of global ischemia (n = 26), 5 minutes of global ischemia (n = 30), and ischemia-induced tolerance (2.5 minutes of global ischemia followed 3 days later by 5 minutes of global ischemia) (n = 16). A region was defined as showing BrdU labeling when the number of BrdU-labeled cells in that structure was at least 3 times that of sham-operated controls. BrdU-labeled regions were designated with a (+), whereas the symbol (−) was used for animals in which the number of BrdU-labeled cells in a structure was similar to the cell counts in the sham-operated controls. For example, after 2.5 minutes of ischemia, 17 animals showed an increase in BrdU-labeled cells in neocortex and striatum but not in hippocampus; 4 animals showed an increase of BrdU-labeled cells in striatum but not in neocortex or hippocampus; 2 animals showed an increase of BrdU-labeled cells in neocortex, striatum, and hippocampus; and 3 animals had no increase of BrdU-labeled cells in striatum, neocortex, or hippocampus.
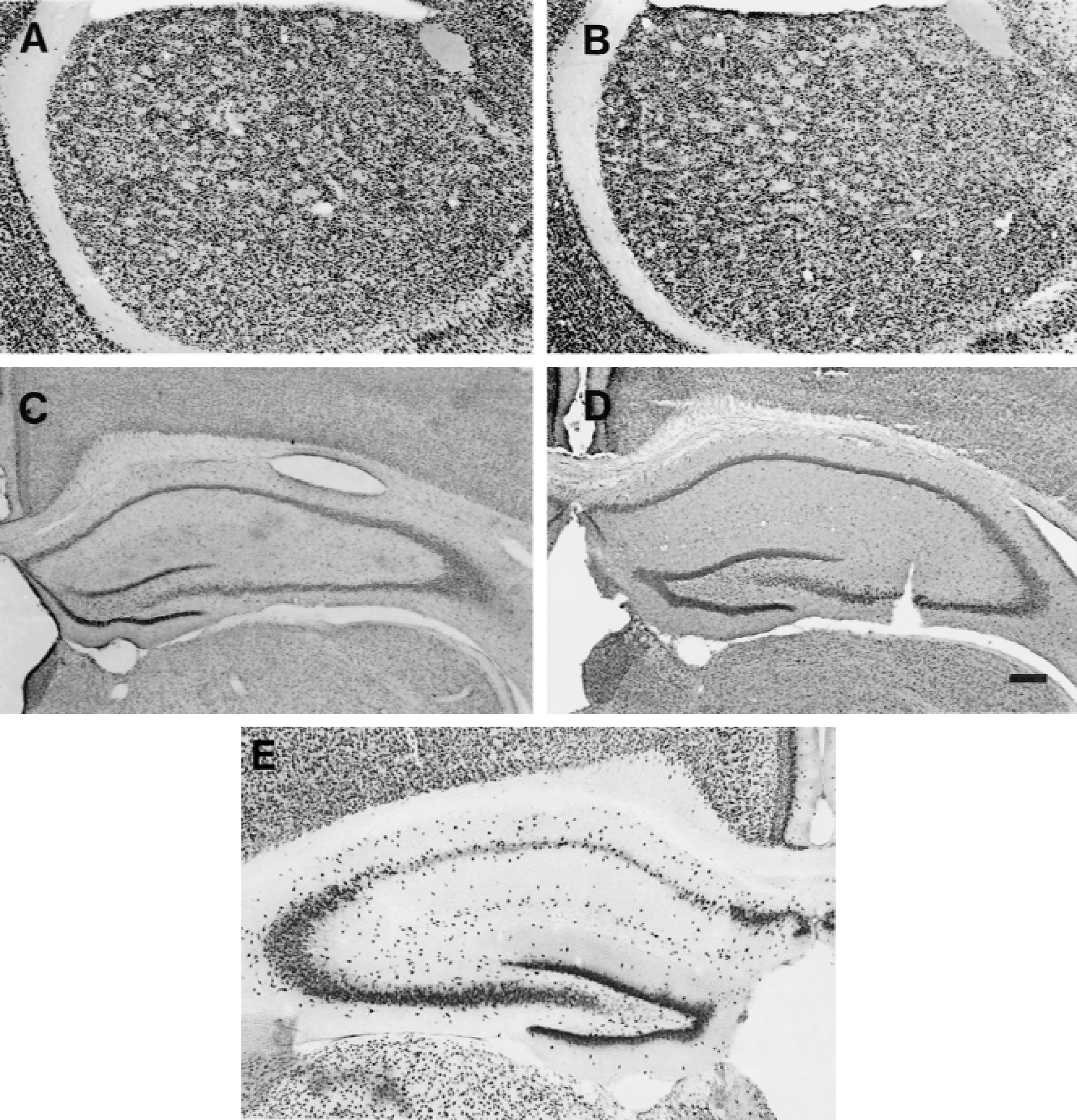
No neuronal loss exists in striatum (
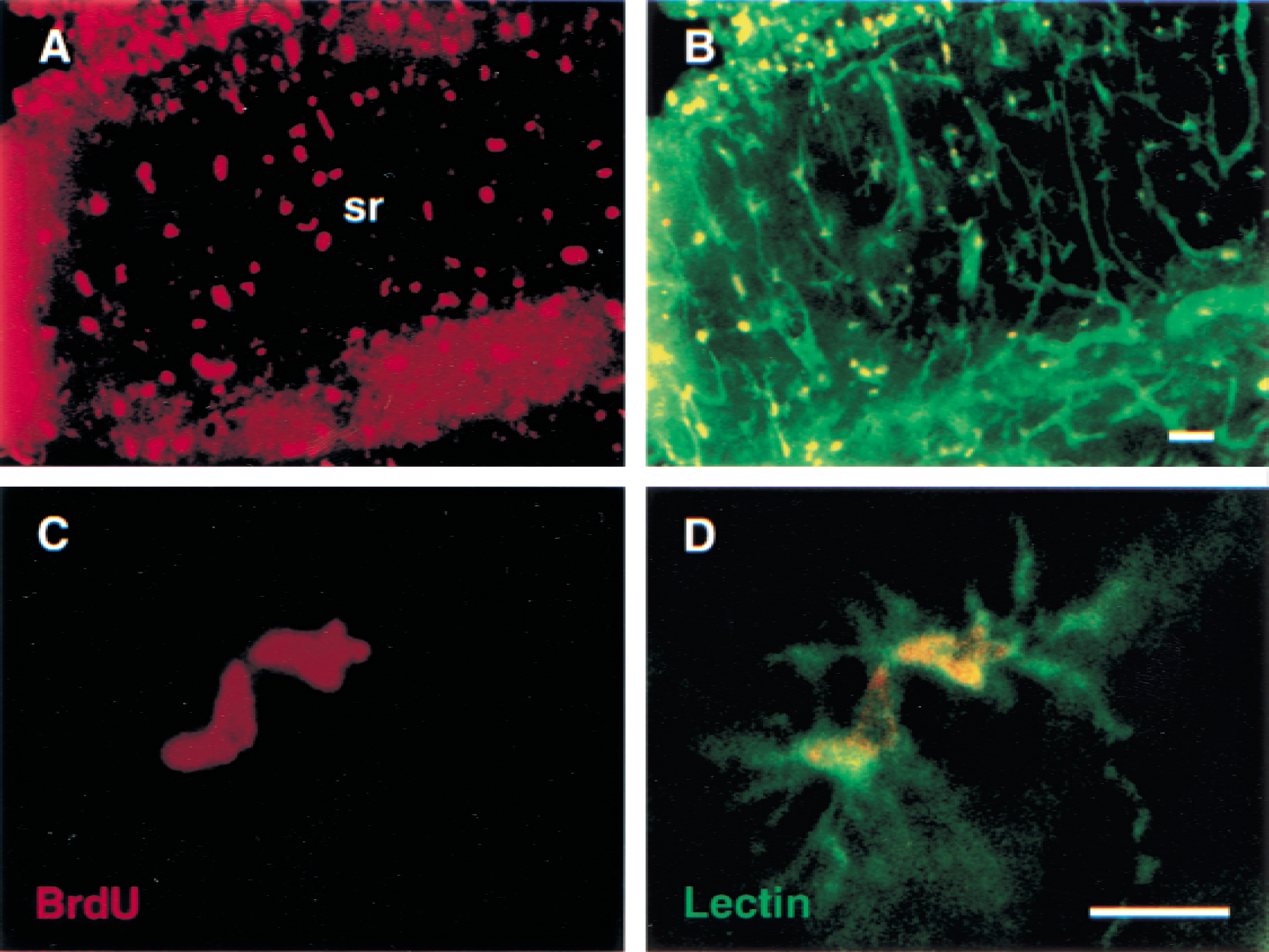
Most dividing cells at 4 days after global ischemia are microglia/macrophages. Double label fluorescent staining is shown for BrdU (
Myelin-associated glycoprotein (MAG) staining of myelin in striatum (
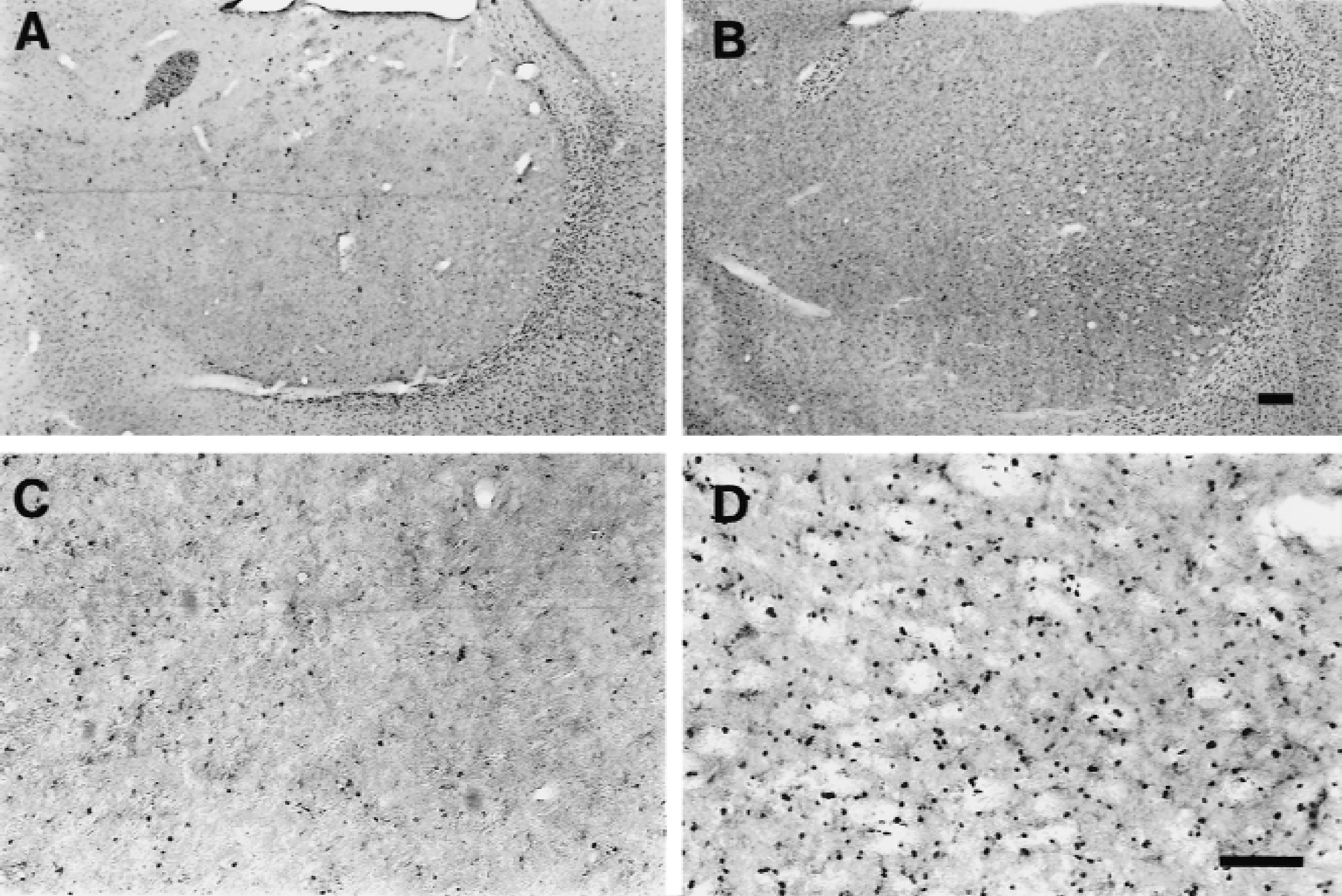
Adenomatous polyposis coli staining of oligodendrocytes in striatum (
Cell proliferation in striatum and cortex but not hippocampus after 2.5 minutes of ischemia
Animals subjected to 2.5 minutes of ischemia had a different distribution of BrdU-labeled cells. Four days after 2.5 minutes of global ischemia, most animals had marked increases of BrdU-labeled cells in the striatum (Fig. 1B) and neocortex (Fig. 1J), but not in the hippocampus (Fig. 1F). Of the 26 animals subjected to 2.5 minutes of global ischemia, 17 (65%) showed an increase in BrdU-labeled cells in the lateral striatum and neocortex, but not in the hippocampus (Table 1). In addition, 4 of 26 animals (15%) had an increase of BrdU-labeled cells only in the lateral striatum, not in neocortex or hippocampus (Table 1). Only 2 of 26 animals had an increased number of BrdU-stained cells in striatum, neocortex, and hippocampus, as was seen after 5 minutes of ischemia. Three animals had no increase in the number of BrdU-labeled cells (Table 1). Therefore, of the 23 animals that had an increase of BrdU-labeled cells after 2.5 minutes of global ischemia, 23 had increased numbers of BrdU-labeled cells in the striatum, 19 had increased numbers of BrdU-labeled cells in the neocortex, and only 2 had increased numbers of BrdU-labeled cells in hippocampus (Table 1).
Animals subjected to 2.5 minutes of global ischemia showed no evidence of neuronal loss in striatum on NeuN-stained sections (Fig. 2A) at 4 days after ischemia. As expected, there was no evidence of neuronal cell death in hippocampus at 4 days after 2.5 minutes of global ischemia (Fig. 2C). Therefore, neuronal cell death does not appear to be the stimulus for microglia/ macrophage cell proliferation in dorsal lateral striatum after 2.5 minutes of ischemia.
BrdU labeling of proliferating cells after ischemia-induced tolerance
The predominant pattern of BrdU labeling seen in the ischemia-induced tolerance group was similar to that observed in the animals subjected to 2.5 minutes of global ischemia. To produce ischemia-induced tolerance, animals were subjected to 2.5 minutes of ischemia, followed by 5 minutes of ischemia 3 days later. Four days after the 5-minute ischemia, the number of BrdU-labeled cells increased in the lateral striatum (Fig. 1D) and throughout all layers of the neocortex (Fig. 1L), but there was little change in the number of labeled cells in the hippocampus (Fig. 1H). Of the 16 gerbils in the tolerance group, 9 had an increase of BrdU-labeled cells in the striatum and neocortex, but not in the hippocampus, and an additional 3 had an increase of BrdU-labeled cells in the striatum, but not in neocortex or hippocampus (Table 1). Two of the 16 animals in the tolerance group had an increased number of BrdU-labeled cells in hippocampus, but not in striatum or neocortex, and 2 other animals had an increase in BrdU-labeled cells in striatum, neocortex, and hippocampus (Table 1). Therefore, the majority of the ischemia-tolerant gerbils (12 of 16) had an increase in the number of BrdU-labeled cells in striatum but not in hippocampus.
Animals subjected to the tolerance paradigm showed no evidence of neuronal loss on NeuN-stained sections of the striatum at 4 days after the 5-minute ischemia (Fig. 2B). As expected, there was no evidence of pyramidal cell death in CA1 or CA3 of hippocampus at 4 days after tolerance (Fig. 2D). Therefore, neuronal cell death does not appear to be the stimulus for microglia/macrophage cell proliferation in striatum after ischemia-induced tolerance.
BrdU and isolectin B4 double labeling of microglia/macrophages
To define what cell types were proliferating, double labeling experiments were performed at 4 days after 2.5 minutes of global ischemia. Double labeling for BrdU (red in Fig. 3A and 3C) and isolectin B4 (green in Fig. 3B and 3D) demonstrated that most of the BrdU-labeled cells were isolectin B4 positive microglia/macrophages (Fig. 3). Although the isolectin B4 stains microglia/ macrophages (Fig. 3B and 3D) and blood vessel endothelial cells (Fig. 3B) in the gerbil brain, careful inspection shows that the cells that have glial morphology in Fig. 3B usually have corresponding BrdU (red stained) positive nuclei in Fig. 3A. To confirm the colocalization, merging of the two channels shows green isolectin B4 processes in Fig. 3D, and the red BrdU-labeled microglia/macrophages in Fig. 3C are colocalized resulting in the two yellow cells in Fig. 3D.
Double label studies using BrdU and GFAP were limited. However, there were very few if any BrdU-labeled cells that colocalized with GFAP at 4 days after ischemia, suggesting very limited if any proliferation of astrocytes at 4 days after 2.5 minutes of ischemia. Further studies are needed to determine whether any astrocyte proliferation occurs after 2.5 minutes of ischemia or after ischemia-induced tolerance.
Myelin-associated glycoprotein and adenomatous polyposis coli staining of striatum
Because microglia/macrophage proliferation occurred without apparent neuronal cell death, the authors determined whether 2.5 minutes of ischemia produced damage to myelin using MAG staining, and whether there was any change in the number of oligodendrocytes using APC staining. Myelin-associated glycoprotein is expressed throughout central myelin sheets in a distribution identical to that for myelin basic protein (Favilla et al., 1984). The APC (adenomatous polyposis coli) tumor suppressor gene is localized to the cell body of oligodendrocytes (Bhat et al., 1996).
Myelin-associated glycoprotein immunostaining showed the presence of multiple white matter tracts in medial and lateral striatum of sham-operated animals (Fig. 4A and 4C). These stained darkly and were well circumscribed in control animals (Fig. 4C). There was decreased staining of the white matter tracks in the lateral striatum after 2.5 minutes of global ischemia (Fig. 4B and 4D). At high power there was altered MAG staining in white matter tracks that was less circumscribed and less organized after ischemia (Fig. 4D) compared with sham controls (Fig. 4C). This was apparent in only 3 of the 7 animals examined at 7 days after ischemia.
Adenomatous polyposis coli staining showed stained cell bodies of oligodendrocytes throughout the striatum of sham-operated controls (Fig. 5A and 5C). After 2.5 minutes of ischemia, however, the APC-stained oligodendrocytes were more darkly stained and appeared to be more numerous (Fig. 5B and 5D) compared with sham-operated animals (Fig. 5C). However, this was only observed in 3 of the 7 animals that showed the altered MAG staining at 7 days after ischemia.
DISCUSSION
The finding that microglia/macrophages proliferate in striatum and neocortex but not in hippocampus after ischemia-induced tolerance was surprising because it occurred in the absence of any detectable neuronal cell death. Because it is likely that the microglia/macrophages proliferate in response to tissue injury, the data suggest that injury occurs to striatum and cortex after short durations of global ischemia that produce ischemia-induced tolerance. Although the injury may involve myelin or oligodendrocyte damage, or both, in a few animals, the nature of the injury that stimulates microglial/ macrophage proliferation in all animals is not known.
Injury-induced microglia/macrophages proliferation
Microglia proliferate in the hippocampus and specifically in CA1 after global ischemia (Bergstedt et al., 1993; Frank et al., 1993; Kato et al., 1994). Microglia/macrophages, as shown by electron microscopy, engulf and phagocytose the dying CA1 pyramidal neurons (Gehrmann et al., 1992b; Nitatori et al., 1995). Microglia proliferate after cortical stab wounds (Amat et al., 1996), MPTP-induced death of dopaminergic neurons (Czlonkowska et al., 1996), and phencyclidine- and MK–801–induced death of retrosplenial neocortex neurons (Rajdev et al., 1998). Kainic acid–induced excitotoxic death of neurons produces marked proliferation of microglia (Marty et al., 1991; Mitchell et al., 1993; Niquet et al., 1994).
It is less clear whether microglia ever proliferate when there is no cellular injury. For example, there is proliferation of microglia in the normal developing and normal adult primate brain (Eckenhoff and Rakic, 1988), although this could be a response to ongoing normal apoptosis (Ivacko et al., 1996). Microglia proliferate when microorganisms enter the brain (Kreutzberg, 1996) and when microglia are treated with lipopolysaccharide in vitro (Zielasek and Hartung, 1996). Thus it is possible, but unlikely, that the microglial/macrophage proliferation observed in striatum and neocortex after brief ischemia is related to chemical signals that may not be associated with detectable structural injury.
Microglia proliferate after injury to various cellular elements
Proliferating microglia not only engulf dying neuronal cell bodies, they also engulf the processes of dying neurons, including the dendrites of CA1 neurons in stratum radiatum of the hippocampus (Gehrmann et al., 1992b). After sciatic nerve section, microglia proliferate and engulf degenerating axons and synapses in the dorsal horn of the spinal cord (Gehrmann and Banati, 1995). Lesion of entorhinal neocortex stimulates microglial proliferation in dentate gyrus in the terminal zones of the perforant pathway inputs to the hippocampus (Hailer et al., 1999).
After focal cerebral ischemia, microglia proliferate in many regions remote from the infarct including ipsilateral substantia nigra, the contralateral neocortex and hippocampus, as well as corticospinal tracts in brainstem and spinal cord (Morioka et al., 1993). Microglia proliferate in the substantia nigra (Nishino et al., 1994), most likely in response to the death of striatal neurons and their axonal projections to substantia nigra.
Facial nerve axotomy that does not result in death of facial motor neurons stimulates proliferation of microglia (Kreutzberg, 1996; Streit and Graeber, 1993). These proliferating microglia presumably are involved in the synaptic stripping that occurs after axotomy, where the synapses on the axotomized motor neuron retract or degenerate (Svensson and Aldskogius, 1993; Streit and Graeber, 1993). Interestingly, microglia, but not astrocytes, proliferate after axotomy of the facial nerve (Graeber et al., 1988).
Microglia proliferate throughout the hippocampus
Although microglial/macrophage proliferation was not observed in hippocampus after 2.5 minutes of global ischemia, there was robust proliferation of these cells after the 5-minute ischemia that led to the death of most CA1 pyramidal neurons (Liu et al., 1998). The surprising feature to the microglial/macrophage proliferation in the hippocampus was that cell proliferation was observed in all regions including CA3 and the dentate granule cell neuron layer, regions that are resistant to global ischemia. Others also have noted proliferation of microglia throughout the entire hippocampus, including the CA3 and granule cell regions (Gehrmann et al., 1992a). It is likely that there is injury in these regions to which the proliferating microglia are responding. Although there may be damage to glia that is difficult to detect, there may also be neuronal injury in these regions. Some of the dentate hilar neurons die after 5 minutes of global ischemia (Johansen et al., 1987; Benveniste and Diemer, 1988; Gehrmann et al., 1992a). Microglial/macrophage proliferation in the dentate granule cell layer could be a response to hilar neurons that have dying processes in the granule cell layer (Hsu and Buzsaki, 1993). Moreover, there is death of some of the CA3 interneurons after global ischemia, which may also stimulate microglial macrophage proliferation in the CA3 layer (Hsu and Buzsaki, 1993).
White matter injury can stimulate microglial/macrophage proliferation
The most novel aspect of this study is questioning what occurs after 2.5 minutes of ischemia and ischemia-induced tolerance that stimulates microglial/macrophage proliferation in striatum and neocortex. One possibility is that there is neuronal death in the striatum or neocortex before that which has classically been shown to occur in CA1 pyramidal neurons of hippocampus. This is unlikely because previous studies have not detected neuronal cell death in striatum and neocortex before that in hippocampus after global ischemia in a wide variety of models using routine histology, terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) staining, and heat shock protein 70 (HSP70) immunocytochemistry (Ginsberg and Busto, 1989; Ito et al., 1975; Kirino, 1982; Lipton, 1999; Pulsinelli et al., 1982; Honkaniemi et al., 1996; Vass et al., 1988; Nowak, 1993; Gonzalez et al., 1991; Simon et al., 1991). Moreover, in the current study, there is no neuronal loss using NeuN neuronal staining after 2.5-minute ischemia. However, the possibility that nonlethal injury occurs to synapses, axons, or dendrites of neurons in striatum and neocortex that stimulate microglial/macrophage proliferation cannot be eliminated based on the results in the current study and needs to be addressed in future studies.
Some animals do have myelin and oligodendrocyte injury in striatum after 2.5 minutes of global ischemia. Oligodendrocytes are the most vulnerable glial cell, followed by microglia and then astrocytes, to combined glucose and oxygen deprivation in vitro (Lyons and Kettenmann, 1998; McDonald et al., 1998). Global and focal ischemia can damage white matter (Kurumatani et al., 1998; Pantoni et al., 1996; Valeriani et al., 2000; Yam et al., 1997). Death of oligodendrocytes has been documented at the margins of the infarct and some of the dying oligodendrocytes are TUNEL positive (Mandai et al., 1997). Most interestingly, the number of stained oligodendrocytes increased at the margins of the infarcts in white matter, suggesting proliferation of oligodendrocytes (Mandai et al., 1997). Similarly, the increased numbers of APC-stained oligodendrocytes in lateral striatum in the current study support the possibility that oligodendrocytes could divide from precursors and increase in number after global ischemia (Reynolds and Hardy, 1997).
Injury to oligodendrocytes and myelin is a sufficient stimulus for inducing both microglial/macrophage proliferation and oligodendrocyte proliferation. Rodent disease models in which injury is confined to the white matter lead to proliferation of both microglia and oligodendrocytes. In the twitcher mouse, a genetic murine model of dysmyelinating globoid cell leukodystrophy, there is proliferation of BrdU-labeled microglia and oligodendrocytes in the degenerating white matter (Taniike and Suzuki, 1995). The dysmyelinating mutant mouse jimpy (mp), which has a mutation in the proteolipid protein gene that leads to oligodendrocyte cell death and dysmyelination, also has an increased number of BrdU-labeled microglia and newborn oligodendrocytes (Wiessner et al., 1996). Therefore, degeneration of oligodendrocytes and white matter can elicit microglial/ macrophage and oligodendrocyte proliferation.
Although myelin injury can stimulate proliferation of microglia and oligodendrocytes, and some of the animals appear to have myelin injury and oligodendrocyte proliferation after global ischemia in this study, the current results must be interpreted cautiously. Because only some animals demonstrated the decreased MAG and increased APC staining in striatum, damage to myelin may stimulate microglial/macrophage proliferation in some of these animals but may not stimulate microglial/ macrophage proliferation in most of the other animals. Alternatively, myelin or oligodendrocyte damage may have little to do with microglial/macrophage proliferation in any of the animals. Finally, it is possible that myelin damage is occurring in all of the animals after 2.5 minutes of ischemia and tolerance but that more sensitive techniques will be needed to reliably detect this damage.
Progenitors and mechanisms of microglial/macrophage proliferation
Precursor cells that give rise to BrdU-labeled microglia/macrophages appear to arise from the bone marrow, perivascular microglial/ macrophage cells that are enclosed within the basal lamina and undergo replacement with bone marrow-derived cells (Giordana et al., 1994; Gehrmann et al., 1995b), and possibly from juxtavascular microglia that reside on the parenchymal side of the central nervous system vascular basal lamina and undergo little replacement (Gehrmann et al., 1995b). Labeled microglia from blood enter areas of hippocampal cell injury after global ischemia (Imai et al., 1999). Wherever the precursor cells are derived, they probably divide when mitogens—including MCSF, GMCSF, and IL3 (Giulian et al., 1991; Lee et al., 1994; Liva et al., 1999)—are released by astrocytes, resident microglia, and perhaps other cells at the site of an injury (Gehrmann, 1995; Lee et al., 1994; Ganter et al., 1992). MCSF plays an important role because osteopetrotic mice that lack MCSF have no evidence of microglial/macrophage proliferation after axotomy of facial motor neurons (Raivich et al., 1994) and after ischemia (Fedoroff et al., 1997). However, many other factors must modulate the microglia proliferation. For example, neuronal release of transforming growth factor-beta2 stimulates microglial/ macrophage proliferation in vitro (Dobbertin et al., 1997), providing a possible mechanism by which injured neurons could release transforming growth factor and stimulate proliferation of microglia precursors.
Role of dividing microglia
Most studies suggest that activated or proliferating microglia, or both, harm the injured brain (Lees, 1993) and that suppressing the microglia may improve outcome from ischemic brain injury (Gehrmann et al., 1995a). This idea originates from the phagocytic (Banati and Graeber, 1994) and immune functions of microglia, and the expression of potentially injurious molecules like inducible nitric oxide synthase (Murphy et al., 1993; Nakashima et al., 1995). However, some data suggest that microglia might be beneficial in some circumstances. Microglia express some molecules that could be neuroprotective (Vannucci et al., 1997; Soriano et al., 1994). Mice with knockout of tumor necrosis factor receptors had greater damage and less microglia activation than wild-type controls after focal ischemia and seizures (Bruce et al., 1996).
A possible protective role for MCSF and microglial/ macrophage proliferation has been obtained in osteopetrotic op/op mice that lack functional MCSF. MCSF-deficient mice have larger infarcts and no evidence of microglial/macrophage proliferation after a stroke compared with their wild-type littermates. Delivery of MCSF-1 to op/op mice decreases infarct size and promotes microglial/macrophage proliferation and activation around infarcts (Fedoroff et al., 1997). However, because MCSF-1 administration to normal mice decreased infarct size without affecting the degree of microglial/macrophage proliferation (Fedoroff et al., 1997), it was suggested that MCSF effects on neuronal rather than microglial/macrophage MCSF receptors were responsible for protection against focal ischemia (Fedoroff et al., 1997). Nevertheless, it is possible that the dividing microglia are protective. Because microglial/macrophage proliferation occurs in the gerbil model of ischemic tolerance, the authors suggest that these proliferating cells may contribute to the phenomenon of ischemic tolerance and may be protective.
Footnotes
Acknowledgments:
The authors thank Dr. Myriam Bernaudin for her helpful suggestions and comments, and Dr. Holger Wille and Mr. Ed Caballero for assistance with the photography.