
Abstract
Preconditioning with lipopolysaccharide (LPS), a toll-like receptor 4 (TLR4) ligand, provides neuroprotection against subsequent cerebral ischemic brain injury, through a tumor necrosis factor (TNF)α-dependent process. Here, we report the first evidence that another TLR, TLR9, can induce neuroprotection. We show that the TLR9 ligand CpG oligodeoxynucleotide (ODN) can serve as a potent preconditioning stimulus and provide protection against ischemic brain injury. Our studies show that systemic administration of CpG ODN 1826 in advance of brain ischemia (middle cerebral artery occlusion (MCAO)) reduces ischemic damage up to 60% in a dose- and time-dependent manner. We also offer evidence that CpG ODN preconditioning can provide direct protection to cells of the central nervous system, as we have found marked neuroprotection in modeled ischemia in vitro. Finally, we show that CpG preconditioning significantly increases serum TNFα levels before MCAO and that TNFα is required for subsequent reduction in damage, as mice lacking TNFα are not protected against ischemic injury by CpG preconditioning. Our studies show that preconditioning with a TLR9 ligand induces neuroprotection against ischemic injury through a mechanism that shares common elements with LPS preconditioning via TLR4.
Introduction
Toll-like receptors (TLRs) are a family of pattern recognition receptors involved in the identification of, and response to, foreign pathogens. Till date, at least 11 TLRs have been identified in mammals, and each recognizes different pathogen-associated molecular patterns. Although the stimuli are different, the resultant signaling cascades are all mediated through toll/IL (interleukin)1 receptor domain-containing adapters, the most prominent being MyD88, with subsequent activation of NF (nuclear factor)-κB. Toll-like receptors are broadly distributed on immune cells and thus play an important role in initiation of innate and adaptive immune responses.
Toll-like receptor 4 was the first TLR identified in mammals and is the most widely studied of all the TLRs. Endotoxin (lipopolysaccharide (LPS)), a cell surface component of Gram-negative bacteria, binds to TLR4 and at high levels can cause death through septic shock. An interesting counterpoint is that low concentrations of LPS actually induce a protective state against a subsequent lethal dose of LPS (reviewed in West and Heagy, 2002). This phenomenon, referred to as endotoxin tolerance, has been studied for more than 50 years, yet the molecular mechanisms are incompletely understood. It is known, however, that many proinflammatory cytokines and cytotoxic mediators that are normally elicited by LPS fail to be induced by a second exposure to LPS. Instead, new signaling proteins and antiinflammatory pathways are increased in the setting of endotoxin tolerance (reviewed in Fan and Cook, 2004; West and Heagy, 2002).
More recently, other TLRs have been shown to induce protection against a subsequent challenge with the same ligand, a state commonly referred to as self-tolerance. Priming of TLR2, TLR5, or TLR9 with their respective ligands induces a state of hyporesponsiveness to a subsequent challenge with their corresponding ligands (Dalpke et al, 2005; Mizel and Snipes, 2002; Sato et al, 2000; Yeo et al, 2003). This shared phenomenon is expected given the similarities in signaling pathways of the TLR family members. Interestingly, crosstolerance (or heterotolerance) between two differing receptors has also been reported, as ligands for TLR2 and TLR9 induce tolerance against a subsequent challenge with LPS (Dalpke et al, 2005; Lehner et al, 2001; Sato et al, 2000; Yeo et al, 2003).
However, heterotolerance is not induced with all combinations of TLRs, and differences in the ability to induce heterotolerance have been reported depending on the model (Dalpke et al, 2005; Dobrovolskaia et al, 2003). For example, Dalpke and colleagues, although able to show heterotolerance between TLR2, TLR4, and TLR9 in a macrophage cell line, and a similar heterotolerance with TLR2 and TLR4 in an in vivo paradigm, failed to induce heterotolerance with TLR9 in vivo. In fact, pretreatment with unmethylated CpG oligodeoxynucleotides (ODNs), the ligand for TLR9, actually enhanced tumor necrosis factor (TNF)α production in vivo in response to LPS and lipoteichoic acid, making the system hyperresponsive (Dalpke et al, 2005). These inconsistencies suggest a complicated interplay between the varying TLR signaling pathways, which is more complex than a simple feedback suppression of inflammatory signals within a cell.
In addition to the phenomenon of crosstolerance that exists among the different TLRs, tolerance against ischemic injury can be induced by LPS in various organs such as heart, brain, and kidney (Heemann et al, 2000; Rowland et al, 1997; Tasaki et al, 1997). Although the mechanism of protection in these models is even less well understood, the paradigm appears similar in that a small inflammatory response is initiated that mitigates the subsequent damaging inflammatory response associated with the secondary stimuli. In the case of brain ischemia, a systemic low dose of LPS delivered at least 1 day but not longer than 7 days before stroke reduces the ischemic injury (Bordet et al, 2000; Rosenzweig et al, 2004; Tasaki et al, 1997; Toyoda et al, 2000). A critical role for TNFα has been shown by us (Rosenzweig et al, 2007) and others (Tasaki et al, 1997) wherein LPS-induced tolerance to ischemia fails to occur in the absence of TNFα.
Similarities among the known TLR signaling pathways and their shared ability to induce heterotolerance between certain members of the TLR family has led us to hypothesize that other TLR ligands may also provide neuroprotection against ischemic brain injury. Further, we postulated that TNFα may play a central role in conferring protection. To test our hypothesis, we examined the protective potential of CpG ODN 1826, a mouse-specific TLR9 ligand. We chose to examine TLR9 because, similar to TLR4, it is coupled to the signaling adapter, MyD88. In addition, activation of TLR9 by CpG ODNs increases serum TNFα levels in mice within 6 h of administration (Vasilakos et al, 2000; Vollmer et al, 2004). Toll-like receptor 4 and TLR9 display a similar cell-type distribution as both are expressed by multiple systemic immune cell types (Applequist et al, 2002; Zarember and Godowski, 2002), and on cells of the CNS (central nervous system) (McKimmie and Fazakerley, 2005; Olson and Miller, 2004; Tang et al, 2007). CpG ODNs are currently approved for human trials as vaccines and cancer therapies (reviewed in Krieg, 2006), which makes them particularly well suited for therapeutic development for use in stroke neuroprotection. Here, we report that ligand activation of TLR9 induces neuronal protection against brain ischemia. We show that neuroprotection is time-and dose-dependent. In addition, we report that TNFα plays an essential role in CpG ODN-induced ischemic tolerance, just as it does in LPS-induced tolerance to ischemic brain injury. These data are the first to indicate that TLR9 is a target for the induction of tolerance against ischemic injury in the brain.
Materials and methods
Mice
C57Bl/6 mice (male, 8 to 10 weeks) were obtained from Jackson Laboratories (West Sacramento, CA, USA). Tumor necrosis factor α knockout mice (B6.129S-Tnftm1Gkl/J) were also obtained from Jackson Laboratories. This strain is backcrossed at least five generations to C57Bl/6 at Jackson Laboratories. All mice were housed in a facility approved by the Association for Assessment and Accreditation of Laboratory Animal Care International. The animal protocols met National Institutes of Health guidelines with the approval of the Oregon Health and Science University Institutional Animal Care and Use Committee.
Reagents
ODN 1826 (tccatgacgttcctgacgtt), a mouse-specific phosphothioate CpG ODN ligand for TLR9, and ODN 2088 (tcctggcggggaagt), a mouse-specific TLR9 signaling inhibitor (Gursel et al, 2003; Stunz et al, 2002), were obtained from Invivogen (San Diego, CA, USA). Invivogen has confirmed the specificity of ODN 1826 for mouse TLR9 by testing against cells transfected with the other TLR family members (personal communication). In addition, endotoxin levels were determined to be negligible (<0.125 EU/mg). A control ODN (Invivogen; tccatgagcttcctgagctt) was used, which contains the same sequence as 1826 but the CpG dinucleotides are replaced by GpC dinucleotides (shown in bold). Therefore, it does not stimulate TLR9.
Oxygen-Glucose Deprivation In Vitro
Primary mouse mixed cortical cultures were prepared from E15 to E17 mouse fetuses. Cortices were dissected and dissociated with trypsin-EDTA (Gibco, Invitrogen, Carlsbad, CA, USA) and plated at a density of 1 × 106 cells/mL onto coverslips coated with poly-
Cell Death Evaluation In Vitro
Cell death in vitro was examined 24 h after OGD by means of fluorescent, cell-permeable, DNA-binding dyes: propidium iodide (PI), as an indicator of cell death, and 4′,6-diamidino-2-phenylindole (DAPI), as an indicator of the total number of cells. Coverslips were incubated with PI (1.5 μg/mL; Sigma-Aldrich) for 2 mins, washed with PBS, fixed for 30 mins in 10% formalin, and then mounted on slides with Vectashield mounting medium containing DAPI (Vector Labs). Stained cells were visualized with a fluorescent microscope (Leica GMBH, Bannockbum, IL, USA) and analyzed using Metmorph7 software (Molecular Devices Corp., Downington, PA, USA). The number of PI- and DAPI-stained cells was counted in two random fields of view on each coverslip, and percentage of cell death was calculated as ((mean of Pi-stained cells)/(mean of DAPI-stained cells)) × 100 per field of view. Each treatment was performed with triplicate coverslips within an experiment and the entire experiment was repeated three or more times.
Drug Treatments
A volume of 200 μL of CpG ODN 1826 and saline vehicle was administered by intraperitoneal injection. For dose-response studies, mice were injected 72 h before middle cerebral artery occlusion (MCAO). For determining the time window of protection, mice were treated from 1 to 14 days before MCAO.
Surgery
Cerebral focal ischemia was induced by MCAO as published previously (Clark et al, 1997). Mice were briefly induced with 3% isoflurane and maintained with 1.5% to 2% isoflurane throughout the surgery. The middle cerebral artery was blocked by threading silicone-coated 8-0 monofilament nylon surgical suture through the external carotid to the internal carotid, and finally blocking its bifurcation into the middle cerebral artery and the anterior cerebral artery. The filament was maintained for 60 mins (unless otherwise noted) while the mice were maintained under anesthesia; then, the filament was removed and blood flow restored. Cerebral blood flow was monitored with laser Doppler flowmetry (Transonic System Inc., Ithaca, NY, USA). Temperature was maintained at 37°C±0.5°C with a rectal thermometer-controlled heating pad and lamp (Harvard Apparatus, Holliston, MA, USA). All surgical procedures were performed under an operating stereomicroscope. After surgery, mice were kept alive for 24 h on a heating pad with access to soft food and water and were then killed. We consistently have a survival rate for the MCAO procedure that exceeds 85%.
Infarct Measurement
Mice were deeply anesthetized with isoflurane, then perfused with ice-cold saline containing 2 U/mL heparin. Brains were removed rapidly, placed on a tissue slicer, and covered with agarose (1.5%). The olfactory bulbs were removed and the remainder of the brain was sectioned into 1 mm slices beginning from the rostral end into a total of seven slices. The area of infarction was visualized by incubating the sections in 1.5% TTC (2,3,5-triphenyltetrazolium chloride; Sigma Aldrich) in PBS for 15 mins at 37°C. The sections were then transferred to 10% formalin (Sigma Aldrich). Images of the sections were scanned, and the hemispheres and areas of infarct were measured using ImageJ software (Abramoff et al, 2004). The measurements were multiplied by the section thickness and summed over the entire brain to yield volume measurements. Data of ischemic damage were calculated using the indirect method to minimize error introduced from edema. % Infarct = (contralateral hemisphere volume—volume of noninfarcted tissue of the ipsilateral hemisphere)/(contralateral hemisphere volume) × 100 (Swanson et al, 1990).
Quantification of Serum Tumor Necrosis Factor α
Blood was taken from mice (cardiac puncture) and allowed to clot for 2 h at room temperature. The blood was centrifuged (2,000 rpm, 20 mins) and the clear serum was removed and stored at −80°C until analyzed. Serum TNFα was measured using an ELISA available commercially from R&D Systems (Minneapolis, MN, USA). The sensitivity of the assay is ˜5.1 pg/mL. All samples were run in duplicate.
Statistical Analysis
All statistical analyses were performed using Prism (Graphpad). Mean differences were analyzed using one-way ANOVA with Bonferroni's post hoc test. Data were represented as mean ± standard error of the mean (s.e.m.) and differences were considered statistically significant when P<0.05.
Results
CpG ODN Preconditions Against Ischemia-Induced Neuronal Cell Death In Vitro
We tested whether the TLR9 ligand, CpG ODN, would induce tolerance to ischemic cell death in an in vitro model of ischemia, OGD. Mouse mixed cortical cultures subjected to 3 h of OGD showed 56% cell death compared with untreated control cultures. Cultures preconditioned by exposure to CpG ODN 1826 (0.5 to 5.0 μg/mL) for 24h before OGD showed a dose-dependent reduction in cell death (Figure 1A). A dose of 1 μg/mL CpG ODN produced maximal protection, which resulted in a 60% reduction in cell death after exposure to OGD. No significant cell death was detected in cultures treated with CpG ODN alone (Figure 1A). Our findings show that pretreatment with CpG ODN provides significant protection from cell death induced by exposure to modeled ischemia (OGD) and suggest that TLR9 is a new target for preconditioning against ischemic neuronal injury.
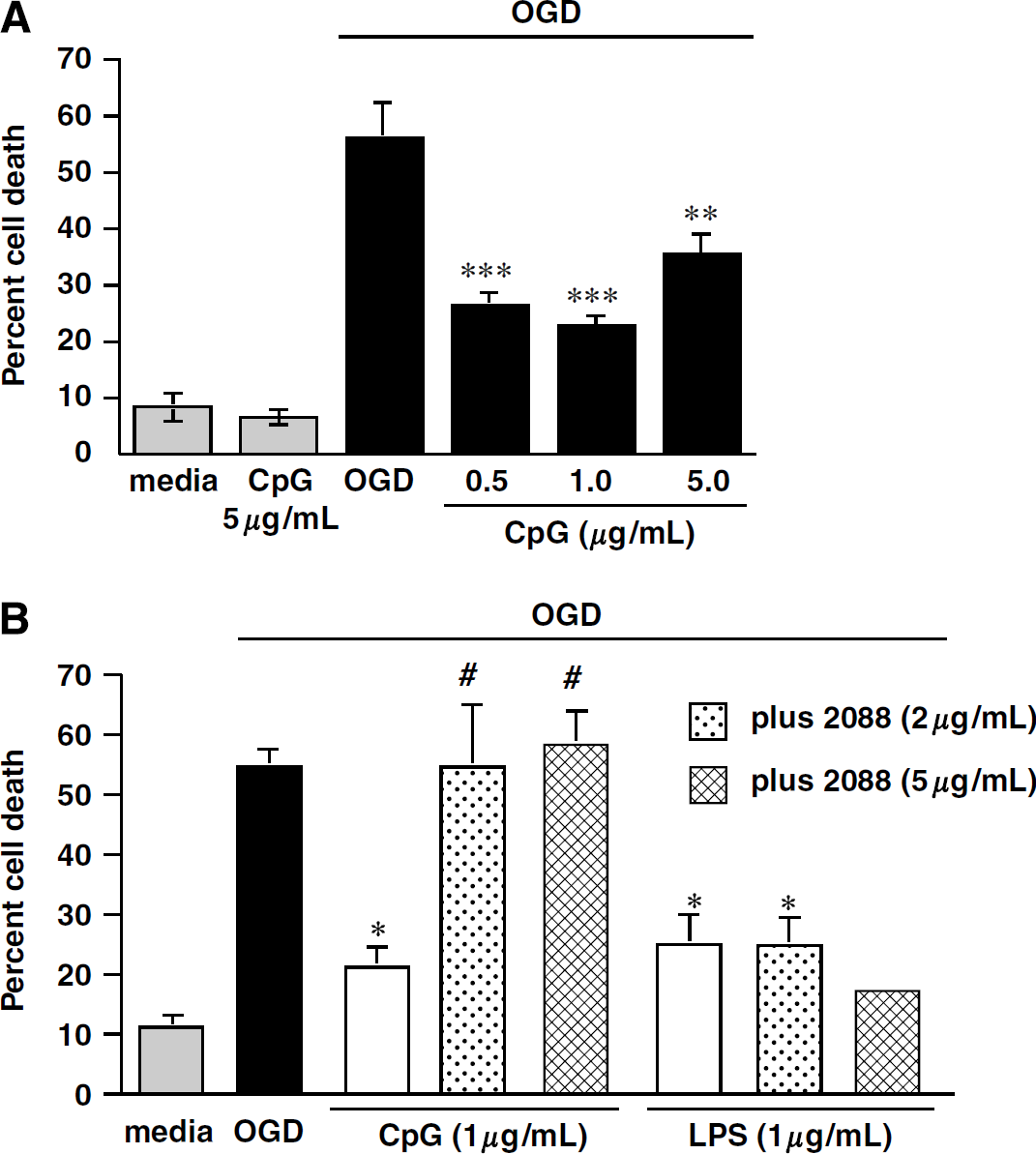
CpG protects primary mixed cortical cultures from OGD-induced cell death through TLR9. (
We used a TLR9-specific antagonist (ODN 2088) to confirm that protection was induced specifically via TLR9 signaling (Gursel et al, 2003; Stunz et al, 2002). Mixed cortical cultures were preconditioned with CpG ODN 1826 (1 μg/mL) in the presence or absence of the TLR9 antagonist ODN 2088 (2 or 5 μg/mL) for 24 h before OGD (3h). ODN 2088 abolished the protective effect of CpG 1826 at both doses, but failed to inhibit the protective effect of LPS (1 μg/mL; Sigma-Aldrich), a TLR4 agonist, which has been shown to induce tolerance to ischemia in vitro (Rosenzweig et al, 2007) (Figure 1B). These data suggest that CpG 1826 signals through TLR9 to induce protection against OGD.
Preconditioning with CpG Reduces Ischemic Damage in an In Vivo Model of Stroke
We next examined the protective potential of CpG ODN treatment in a mouse model of stroke. Mice were preconditioned with varying doses of CpG ODN 1826 (5 to 40 μg, intraperitoneally) 72 h before MCAO. Twenty-four hours after MCAO, mice were killed and infarct damage was determined. Pretreatment with CpG ODN at doses of 20 and 40 μg reduced the infarct size significantly (56.5% and 57.5% reduction, respectively; Figure 2). Thus, CpG ODN delivered systemically to mice preconditions the brain in a dose-dependent manner leading to marked tolerance to ischemic brain injury. To confirm that the protection was specific to CpG ODN, we tested a control ODN that contained the same sequence as 1826 but the CpG dinucleotides were replaced by GpC dinucleotides. No significant protection was observed in mice preconditioned with 20 μg of the control ODN (data not shown). The reduction in infarct size reported here at 24 h after MCAO remained evident in mice killed 72 h after MCAO (unpublished data).
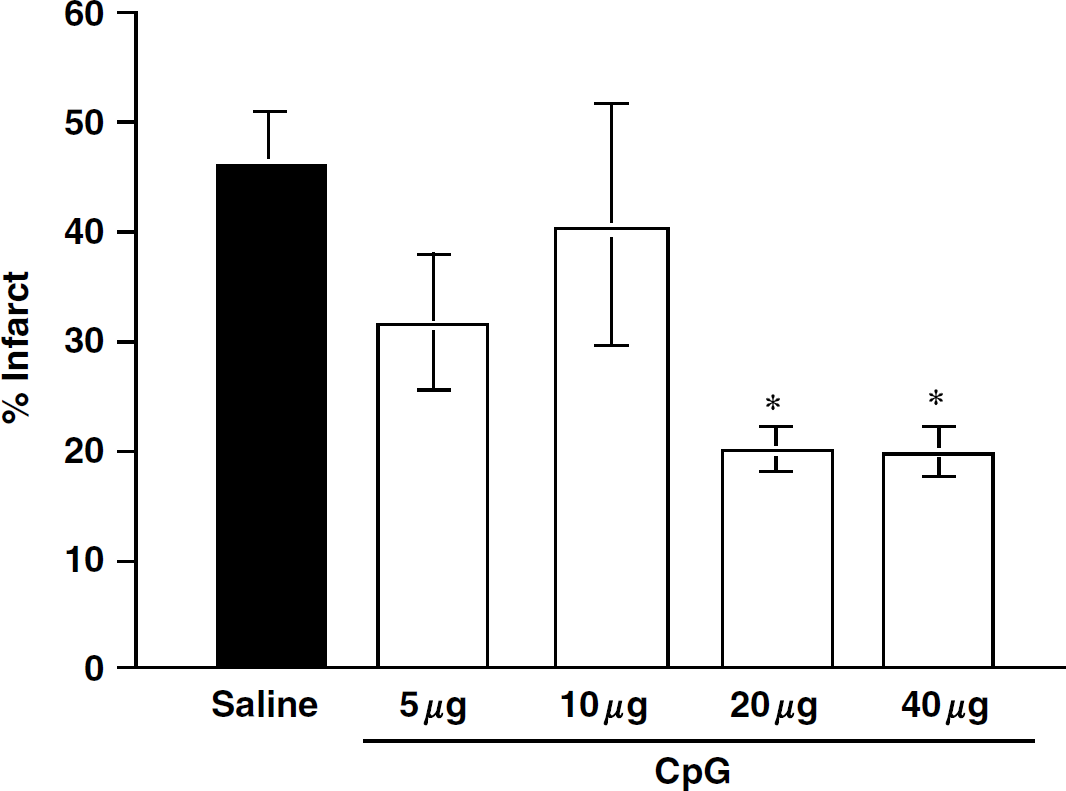
Preconditioning with CpG reduces infarct size in a mouse model of focal ischemia. C57BL/6 (males, 6 to 10/dose) received various doses of CpG 1826 (5 to 40 μg, intraperitoneally) 72 h before ischemic challenge (60 min MCAO). Infarct volume was determined 24 h after MCAO by TTC staining. Values denote group means ± s.e.m.; *P<0.05 versus saline controls by one-way ANOVA followed by Bonferroni's multiple comparison test.
Preconditioning Time Window of CpG-Induced Neuroprotection
Lipopolysaccharide preconditioning induces ischemic neuroprotection in the brain by 1 day, an effect that lasts for at least 7 days but is lost by 14 days after treatment (Rosenzweig et al, 2007). To determine the time window of neuroprotection induced by CpG ODN 1826, we administered CpG ODNs (20 μg, intraperitoneally) 1, 3, 7, and 14 days before MCAO. We found that pretreatment with CpG ODNs induced significant neuroprotection within 1 day (46% reduction in infarct volume), a neuroprotective effect that remained evident for 3 days (61% reduction) but diminished by 7 days. By 14 days postadministration, the protective effect was completely abolished in the CpG-treated mice (Figure 3). Thus, CpG preconditioning provides a time window of neuroprotection that lasts approximately 1 week and is lost by 2 weeks after administration. This time window of protection mirrors that seen with systemic administration of LPS.
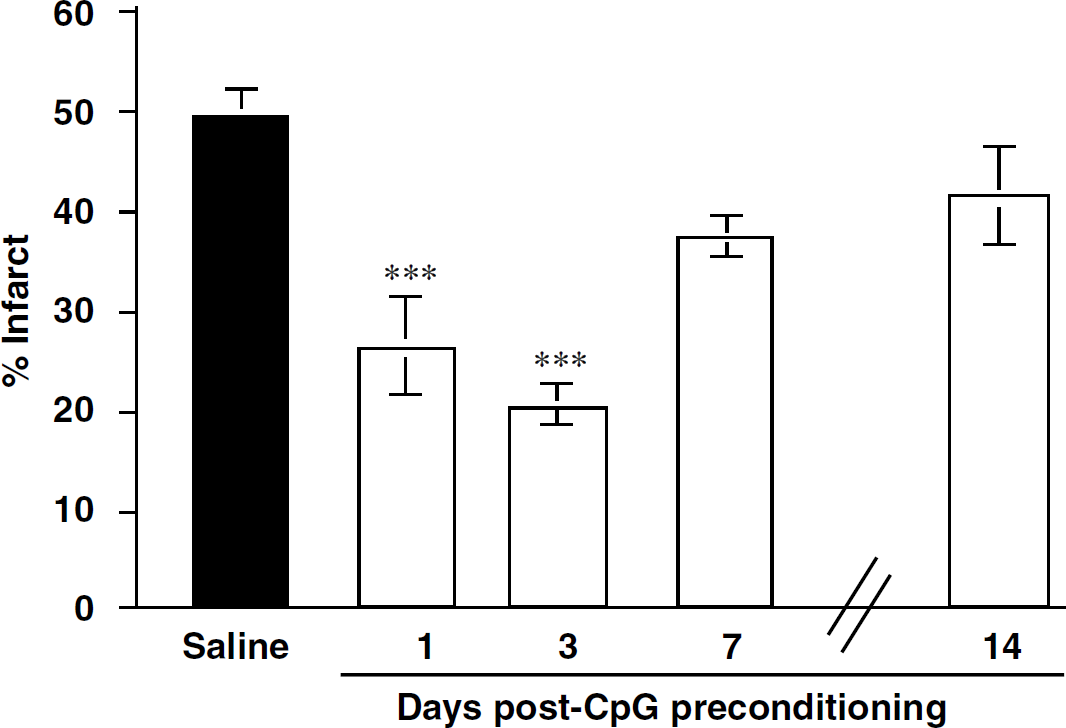
Time window of CpG preconditioning. C57BI/6 mice received an injection of saline (4 mice/time point) or CpG (20 μg, intraperitoneally) 1, 3, 7, or 14 days (6 mice/time point) before a 60 min MCAO. Infarct volume was determined 24 h after MCAO by TTC staining. No statistical difference was observed between the saline groups; thus, they were combined for analysis. Values denote group means ± s.e.m.; *** P<0.001 versus saline controls by one-way ANOVA followed by Bonferroni's multiple comparison test.
Tumor Necrosis Factor α is Required for CpG-Induced Neuroprotection
Previous work in the model of LPS preconditioning against brain ischemia has shown that the presence of TNFα plays an essential role in conferring neuroprotection (Rosenzweig et al, 2007; Tasaki et al, 1997). We postulated that CpG ODN preconditioning via TLR9 may have a similar requirement for the presence of TNFα. We first tested whether CpG ODN administration increased serum levels of TNFα. We found significant increases in TNFα levels in the serum as early as 1 h after injection (400 pg/mL compared with vehicle-treated mice, which were below the level of detection; Figure 4). The timing of the increase in TNFα was similar to what we have previously reported with LPS preconditioning (Rosenzweig et al, 2007); however, the magnitude is significantly less than that observed for preconditioning levels of LPS (3,092 pg/mL). As with LPS, the response diminishes quickly and returns to baseline by 72 h.
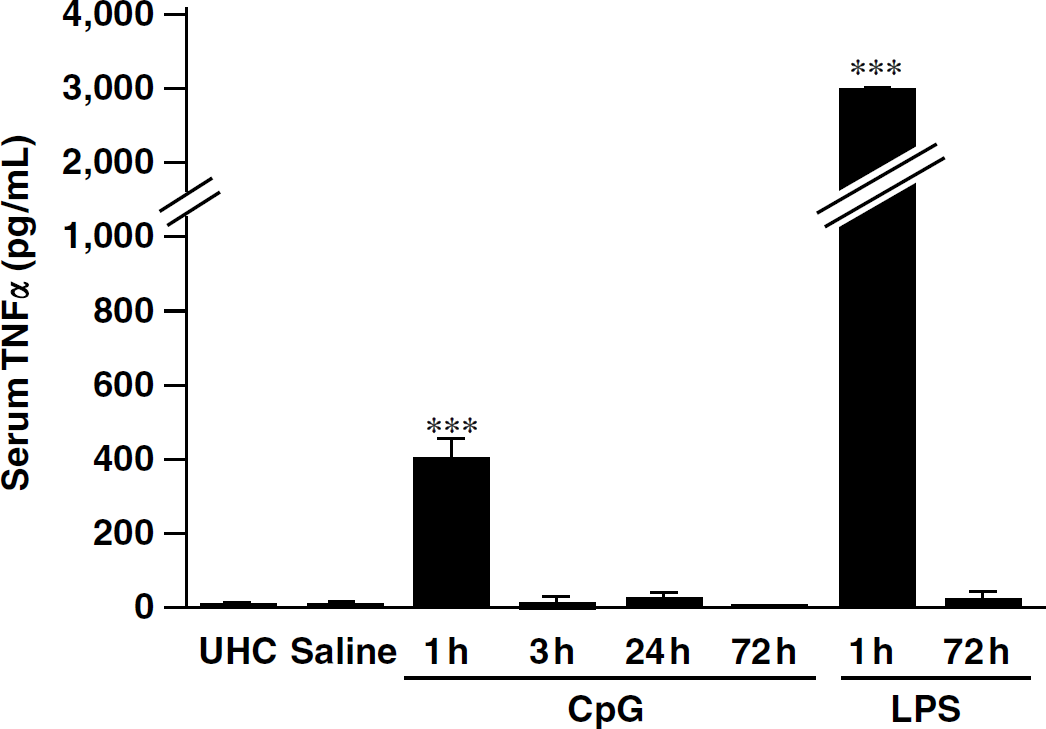
Serum TNFα levels significantly increased 1 h after CpG treatment. Mice (n = 4/time point) were administered CpG 1826 (20 μg, intraperitoneally) and blood was collected at 1, 3, 24, or 72 h after injection. Blood was allowed to clot for 2 h at room temperature and the serum was collected. Tumor necrosis factor α levels (pg/mL blood) were measured with a TNFα ELISA (R&D Systems). Values denote group means ± s.e.m.; *** P<0.001 by two-way ANOVA followed by Bonferroni's multiple comparison test. LPS (5 μg, intraperitoneally)-treated mice were included in the same experiment for comparison.
We investigated whether TNFα is required for CpG preconditioning by testing the neuroprotective effect of CpG ODN treatment in TNFα-knockout mice (B6.129S-Tnftm1Gkl/J; TNFα−/−). We administered CpG ODN 1826 to TNFα−/− and control mice (C57Bl/6; TNFα +/+) 72 h before a 40 min MCAO. Control mice pretreated with CpG showed a significant reduction in ischemic injury (46% reduction) as expected. In contrast, TNFα−/− mice pretreated with CpG did not show any reduction in infarct size (saline treated = 31.9% ± 7.7% versus CpG treated = 29.3% ± 6.8%; Figure 5). These data suggest that TNFα plays an essential role in mediating CpG-induced neuroprotection against ischemic injury.
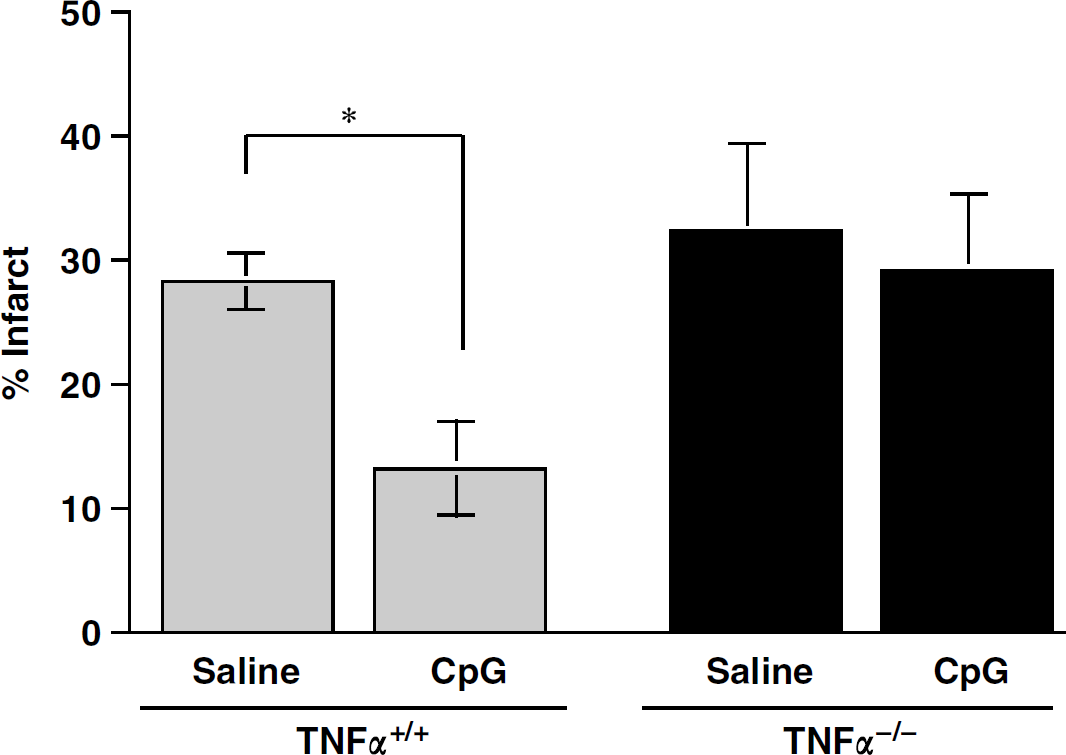
Tumor necrosis factor induction is required for CpG preconditioning. Control (C57BI/6; TNFα+/+) or TNFα-knock-out (B6.129S-Tnftm1Gkl/J; TNFα−/−) mice were treated with CpG 1826 (20 μ/g, intraperitoneally) at 72 h before a 40 min MCAO and infarcts were assessed 24 h after MCAO. Values denote mean ± s.e.m.; *P<0.05 versus saline treatment; n = 4 to 6 mice/group.
Discussion
We report the first evidence that a TLR9 ligand (CpG ODN) can serve as a potent preconditioning stimulus and provide protection against ischemic brain injury. This finding indicates that TLR9, in addition to TLR4, can induce preconditioning in the brain. Our studies show that systemic administration of CpG ODN 1826 in advance of brain ischemia reduces ischemic damage in a dose- and time-dependent manner. We offer evidence that CpG ODN preconditioning can provide direct protection to CNS cells, as we have found marked neuroprotection in modeled ischemia in vitro. In addition, using our in vitro model we show that CpG ODN specifically acts through TLR9 to induce neuroprotection. Finally, our studies support a critical role of TNFα in CpG-induced neuroprotection. This latter observation suggests that the mechanisms of neuroprotection by LPS and CpG preconditioning share common elements.
Previous studies have shown that LPS, acting through TLR4, can induce crosstolerance and provide neuroprotection against ischemic injury in the brain. We have posited that heterologous tolerance induction against brain ischemia extends beyond TLR4 to other TLRs. Here, we show that crosstolerance by TLR4 is not unique to this particular TLR, as we show that a TLR9 agonist, CpG ODN, also induces crosstolerance against an ischemic insult.
It is known that TLR4 couples to both MyD88- and TRIF-dependent pathways and that the MyD88 cascade culminates in NF-κB-mediated induction of proinflammatory cytokines (i.e., TNFα, IL6, and IL1β). Tumor necrosis factor α, which is required for TLR4-induced tolerance against ischemic injury (Rosenzweig et al, 2007; Tasaki et al, 1997), may be induced via the MyD88 cascade. We hypothesize that signaling through TLR9 would also induce tolerance via an MyD88-dependent mechanism, as TLR9 signals exclusively through MyD88 with no evidence of TRIF-dependent signaling (Horng et al, 2001; Schnare et al, 2000). Studies to explore this possibility are in progress in our laboratory and should provide important information broadly regarding the molecular mechanisms underlying tolerance to injury.
Thus far, the known ligands for TLR9 include bacterial DNA containing unmethylated CpG motifs, certain double-stranded DNA viruses, and synthetic CpG ODNs such as CpG ODN 1826 used in the studies described here (Bauer et al, 2001; Hemmi et al, 2000; Krug et al, 2004). Synthetic CpG ODNs have been shown to confer protection in mice against subsequent challenge from a variety of bacteria, viruses, parasites, and prions (Krieg, 2003). Protection against pathogen challenge typically occurred within 48 h and lasted for several weeks (Elkins et al, 1999; Krieg et al, 1998)—such a time window is similar to our findings with CpG-ODN-induced ischemic tolerance shown here and our recent report of LPS-induced ischemic tolerance to stroke injury (Rosenzweig et al, 2007).
The data presented here provide the first evidence that in addition to tolerance (protection) against foreign pathogens, CpG ODN administration protects against a stimulus that is unrelated to a foreign pathogen, namely ischemic injury. CpG-ODN-induced neuroprotection occurs after systemic administration in a mouse model of stroke. Tumor necrosis factor α appears to be critical to the induction of ischemic tolerance by CpG preconditioning, as we show here that CpG ODN administration fails to protect TNFα-deficient mice against ischemic brain injury. In wild-type mice, systemic administration of CpG increased TNFα in the plasma, which suggests that the actions of TNF may occur in the periphery. Further support for this lies in the observation made by Nawashiro et al (1997) that systemic administration of TNFα itself preconditions against stroke injury. In addition, LPS preconditioning against ischemic brain injury is abolished by systemic blockade of TNFα using TNF-binding protein (Tasaki et al, 1997). Thus, CpG ODN preconditioning in vivo very likely provides neuroprotection through a TNFα-dependent mechanism similar to that seen with LPS preconditioning.
We also report that CpG induces neuroprotection when directly applied to mixed cortical cultures in vitro, which are subsequently subjected to oxygen-glucose deprivation. The mechanism of this more direct route of CpG interaction in the CNS is still unclear. These cultures contain neurons, astrocytes, and microglia. Until very recently, astrocytes and microglia, but not neurons, were known to express TLRs, including TLR9 (Bowman et al, 2003; Olson and Miller, 2004). Neurons have also been reported to express TLRs, although the extent to which they are targets of cell signaling is not yet clear (Tang et al, 2007). Thus, CpG treatment may modulate the cytokine response to injury in glial cells, which in turn may have a protective effect on neurons either indirectly or through more direct modulation by activation of TLR9 on neurons, leading to altered neuronal signaling in the setting of injury. Whether TNFα has a critical role in CpG-induced tolerance in vitro is of interest and, as such, the subject of future studies in our laboratory.
We have recently reported genomic evidence that preconditioning via LPS activation of TLR4 produces a tolerant state in the brain via inflammatory mediators that are neuroprotective (e.g., Type I interferons) (Stenzel-Poore et al, 2007). In addition, in the setting of LPS-induced tolerance, there is a marked absence of deleterious inflammatory mediators (IL-6, macrophage inflammatory protein 1 alpha, TNF receptor-associated factor 6) generally found in ischemic brain injury. It is possible that these particular features of neuroprotection (suppressed proinflammatory mediators/increased neuroprotective cytokines) are common to preconditioning stimuli that act through TLRs.
The demonstration that ischemic tolerance in the brain occurs through TLR9, in addition to TLR4, raises the possibility that this is a conserved feature of all TLRs. The recognition that TLR9 is a new target for preconditioning broadens the range of potential antecedent therapies for brain ischemia, such as in the setting of coronary artery bypass grafting (˜300,000 procedures annually) where patients are at risk of cerebral morbidity (reviewed in Newman et al, 2006). Phase II clinical trials are already in progress with CpG ODNs for use in adjuvant and anticancer therapies (reviewed in Krieg, 2006). Thus, CpG ODNs may offer great translational promise as a prophylactic treatment against cerebral morbidity for ‘at-risk’ patients.
Conflict of interest
Dr Stenzel-Poore and Ms Stevens are the inventors of the technology used in this research. An exclusive licensing agreement exists between OHSU and Neuroprotect Inc. This potential conflict of interest has been reviewed and managed by OHSU and the Integrity Program Oversight Council.
Footnotes
Acknowledgements
We acknowledge Rebecca Hillary (PhD) for her outstanding technical support.