
Abstract
Subarachnoid hemorrhage (SAH) is a unique disorder commonly occurring when an aneurysm ruptures, leading to bleeding and clot formation, with a higher incidence in females. To evaluate the influence of 17-β estradiol (E2) in the outcome of subarachnoid hemorrhage, SAH was induced by endovascular puncture of the intracranial segment of internal carotid artery in 15 intact females (INT), 19 ovariectomized females (OVX), and 13 ovariectomized female rats with E2 replacement (OVX + E2). Cerebral blood flow was recorded before and after SAH. All animals were decapitated immediately after death or 24 hours after SAH for clot area analysis. Brains were sliced and stained with 2,3,5-triphenyltetrazolium chloride (TTC) for secondary ischemic lesion analysis. The cortical cerebral blood flow (CBF), which was measured by a laser–Doppler flowmeter, decreased to 29.6% ± 17.7%, 22.8% ± 8.3%, and 43.5% ± 22.9% on the ipsilateral side (P = 0.01), and decreased to 63.4% ± 14.1%, 57.4% ± 11.0%, and 66.6% ± 17.9% on the contralateral side (P = 0.26) in INT, OVX, and OVX + E2, respectively. The subcortical CBF, which were measured by the H2 clearance method, were 7.77 ± 12.03, 7.80 ± 8.65, and 20.58 ± 8.96 mL 100 g−1 min−1 on the ipsilateral side (P < 0.01), and 21.53 ± 2.94, 25.13 ± 3.01, and 25.30 ± 3.23 mL 100 g−1 min−1 on the contralateral side in INT, OVX, and OVX + E2, respectively. The mortality was 53.3%, 68.4%, and 15.4% in INT, OVX, and OVX + E2, respectively (P = 0.01), whereas no significant difference in clot area was noted among the groups. The secondary ischemic lesion volume was 9.3% ± 8.4%, 24.3% ± 16.3%, and 7.0% ± 6.4% in INT, OVX, and OVX + E2, respectively (P < 0.01). This study demonstrated that E2 can reduce the mortality and secondary ischemic damage in a SAH model without affecting the clot volume.
Stroke is the third most common cause of death in the adult population in the United States, after ischemic heart disease and all forms of cancer (Camarata et al., 1994). Subarachnoid hemorrhage (SAH) accounts for approximately 10% of all strokes (Selman et al., 1999). However, SAH affects a younger population and results in death in more than 50% of subjects, most of whom die within the first 24 hours. Subarachnoid hemorrhage accounts for more premature mortality than ischemic stroke (Broderick et al., 1994; Zhang et al., 1998). Subarachnoid hemorrhage can result in vascular changes such as acute vasospasm and intracranial hypertension, which lead to decrease of cerebral perfusion pressure and cerebral blood flow (CBF). All of these can contribute to secondary ischemic damage after SAH. Histologic studies of brains of patients who died shortly after SAH show extensive ischemic damage, and secondary ischemia has been reported to be one of the major causes of death shortly after SAH (Adams et al., 1981).
Unlike other kinds of strokes, aneurysmal SAH occurs more frequently in women than in men (Davis, 1994). Gender differences in the outcome of SAH are controversial, and the influence of the female sex hormone is unclear (Kongable et al., 1996; Simpson et al., 1991; Johnston et al., 1998). However, estrogens have been found to exert neuroprotective effects in models of ischemic stroke both in vitro and in vivo (Simpkins et al., 1997; Alkayed et al., 1998; Dubal et al., 1998; Zaulyanov et al., 1999; Yang et al., 2000). Whether estrogens exert similar protective effects in SAH as in ischemia is currently unknown. The purpose of this study was to determine whether 17β-estradiol (E2) influences the outcome of SAH and, if so, whether the influence relates to the ischemia associated with SAH.
MATERIALS AND METHODS
Preparation of animals
Female Charles River Sprague–Dawley rats (250 g, Wilmington, MA, U.S.A.) were maintained in laboratory acclimatization for 3 days before ovariectomy. Bilateral ovariectomy was performed two weeks before SAH under methoxyflurane inhalant anesthesia. All animal procedures were approved by the University of Florida Animal Care and Use Committee.
E2 administration and serum concentration
To obtain a sustained stable elevation in serum E2 concentration, implantation of a Silastic® pellet containing the steroid was used. To assess serum concentrations of E2 after this treatment regimen, a group of OVX animals (n = 5) was anesthetized with methoxyflurane inhalant and a control blood sample was taken through the jugular vein. Then a 30-mm-long Silastic® tube (1.57 mm inner diameter, 3.18 mm outer diameter) containing E2 (4 mg/mL in corn oil) was implanted subcutaneously in 5 OVX animals. Animals were returned to their cages and blood samples then were taken through the jugular vein at 24 and 48 hours after steroid administration, under methoxyflurane inhalant anesthesia. Serum was separated from blood cells by centrifugation and stored frozen (–20°C). Serum E2 concentrations were determined using duplicate serum aliquots in an ultrasensitive estradiol radioimmunoassay kit (Diagnostic Systems Lab, Webster, TX, U.S.A.).
Endovascular subarachnoid hemorrhage model
Animals were anesthetized by intraperitoneal injection of ketamine (60 mg/kg) and xylazine (10 mg/kg). Rectal temperature was monitored and maintained between 36.5°C and 37.5°C during the procedure. With the aid of an operating microscope, the left common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were exposed through a midline cervical skin incision. A 3–0 monofilament suture with a blunt tip was introduced into the ICA through the ECA lumen, and advanced until resistance was encountered. The distance between the CCA bifurcation and the resistive point was approximately 1.8 cm. The suture was advanced another 5 mm and then withdrawn immediately. The CCA and ICA were coagulated and the skin incision was closed.
Measurement of regional cerebral blood flow
A laser–Doppler flowmeter (LDF) was used for cortical CBF measurements. The scalp was incised on the midline, and bilateral 2-mm burr holes were drilled 1.5 mm posterior and 4.0 mm lateral to the bregma. The dura was left intact to prevent cerebral spinal fluid leakage. Laser–Doppler flowmeter probes held in place by a micromanipulator were stereotaxically advanced to gently touch the intact dura mater. The lower stable readings were obtained and recorded for at least 10 minutes from both sides (baselines measurement) (Dubal et al., 1998; Cholet et al., 1997). For each animal, the lower CBF reading was recorded at the same sites within 30 minutes after SAH. The CBF values were calculated and expressed as a percentage of the baseline values. Cerebral blood flow values reported represent the mean ± SD for the average of the CBF recordings obtained.
Hydrogen clearance method was used for subcortical CBF measurement: two Teflon-coated platinum electrodes held in place by a micromanipulator were stereotaxically advanced to 3 mm posterior, 0.5 mm lateral to the bregma, and 4.0 mm deep into the subcortical region on both sides (He et al., 1995). For each animal, CBF were recorded 30 minutes after SAH.
Measurement of clot and lesion volume
Each group of animals was decapitated immediately after death or 24 hours after SAH, the brain was removed, and the base of the brain was photographed by a digital camera (Sony MVC-FD5, Tokyo, Japan) for measurement of the clot area. Then the brain was placed in a metallic brain matrix (Harvard, Holliston, MA, U.S.A.) for tissue slicing. Two-millimeter sections were made beginning at 3, 5, 7, 9, 11, and 13 mm posterior to the olfactory bulb. Each slice was incubated for 30 minutes in a 2% solution of 2,3,5-triphenyltetrazolium chloride (TTC) in physiologic saline at 37°C and then fixed in 10% formalin. The stained slices were photographed and subsequently measured for the surface area of the slices and the ischemic lesion (Image-Pro Plus 3.0.1, Silver Springs, MD, U.S.A.). Clot area was calculated as the percentage of the base of the brain covered by clot to represent the clot size. Ischemic lesion volume was calculated as the sum of the areas of the ischemic lesion across the six slices divided by the total cross-sectional area of these six brain slices.
Experimental protocol
Forty-seven animals were placed into 3 groups: 15 intact females (INT), 19 ovariectomized females (OVX), and 13 ovariectomized females with estrogen replacement (OVX + E2), respectively. In OVX + E2 animals, a 30-mm-long Silastic® tube containing E2 (4 mg/mL oil) was implanted subcutaneously 24 hours before SAH under methoxyflurane inhalant anesthesia. Intact and OVX animals received a Silastic® tube containing oil as a control.
Twenty-four hours later, after a baseline CBF reading was obtained, SAH was induced in each animal. Cerebral blood flow was recorded bilaterally at the same sites within 30 minutes after SAH. Then the animals were returned to their home cages under careful observation. Each animal was decapitated for clot and ischemic volume analysis immediately after death or 24 hours after SAH.
Subcortical CBF was measured in 15 animals 30 minutes after SAH—5 each for INT, OVX, and OVX + E2—using the hydrogen clearance method. The right femoral artery was catheterized for blood pressure monitor in each animal. Mean arterial pressure was recorded before, immediately after, and 30 minutes after SAH.
Statistic study
Statistical analyses were performed using SAS Software (SAS Institute, Cary, NC, U.S.A.). Paired t-tests were performed to assay serum concentration of E2 treatment after implantation. The authors compared the clot areas, lesion volumes, and CBF from the three groups. For each comparison, P values from one-way analysis of variance were provided. Chi-squared test was used to compare the mortality rate among the three groups.
RESULTS
E2 administration and serum E2 concentration
Implantation of a 30-mm E2 (4 mg/mL corn oil) pellet maintained stable serum E2 concentrations for at least 48 hours after administration. E2 concentration was 24.9 ± 6.6 pg/mL and 31.3 ± 11.5 pg/mL at 24 and 48 hours after administration, respectively, compared with 3.5 ± 1.2 pg/mL in OVX animals (P < 0.01) (Fig. 1).
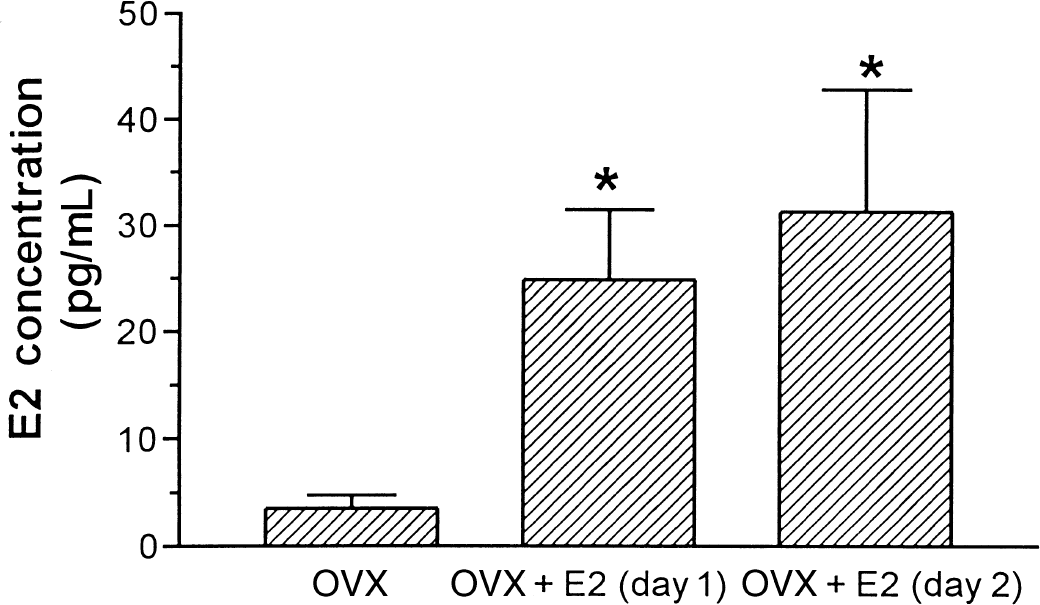
Serum E2 concentrations after treatment. E2 administration: subcutaneous implantation of a 30-mm-long Silastic® pellet filled with E2 oil (4 mg/mL). *OVX versus OVX + E2 (day 1), P = 0.0002; OVX versus OVX + E2 (day 2), P + 0.0028. Graph shows mean ± SD for 5 animals per group.
Effect of E2 on subarachnoid hemorrhage mortality
Most of the animals began to recover from the anesthetic at approximately 3 hours after SAH. Mean arterial blood pressure was 90.25 ± 9.00, 93.40 ± 11.41, and 97.17 ± 6.77 mm Hg in INT, OVX, and OVX + E2 before SAH, respectively. Blood pressure increased to 123.20 ± 6.61, 121.40 ± 9.34, and 114.70 ± 16.12 mm Hg immediately after SAH, then returned to 94.00 ± 17.72, 91.50 ± 17.23, and 81.80 ± 5.50 mm Hg 30 minutes after SAH in INT, OVX, and OVX + E2, respectively. No significant differences were noted among groups. The mortality was 53.3%, 68.4%, and 15.4% in INT, OVX, and OVX + E2 groups, respectively (P = 0.01). In the INT group, 3 animals died within 6 hours after SAH and 5 died between 6 and 24 hours after SAH. In the OVX group, 4 animals died within 6 hours and 9 died between 6 and 24 hours after SAH. In the OVX + E2 group, 1 each died within 6 hours and between 6 and 24 hours after SAH, respectively (Table 1).
Effects of ovarian steroids environment on mortality and time of death in rats subjected to SAH
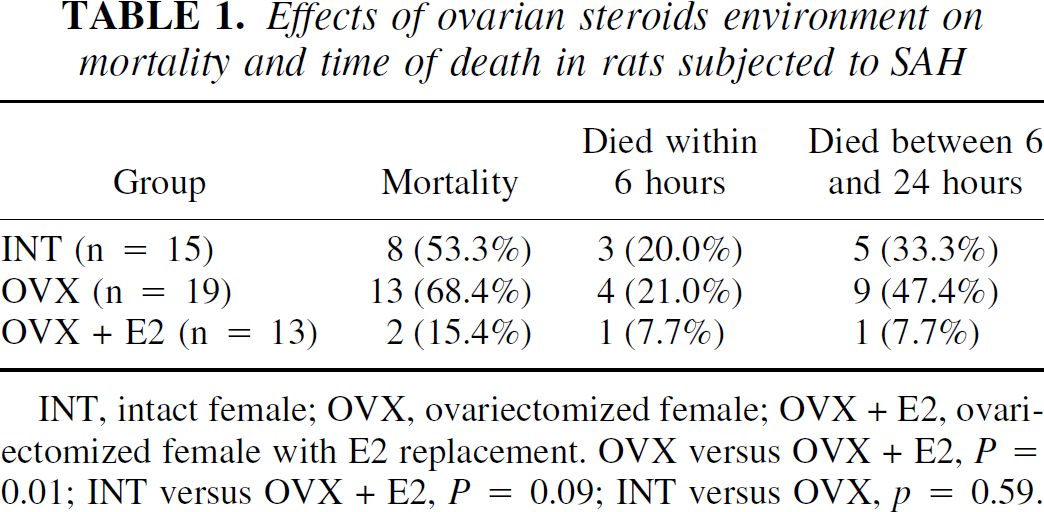
INT, intact female; OVX, ovariectomized female; OVX + E2, ovariectomized female with E2 replacement. OVX versus OVX + E2, P = 0.01; INT versus OVX + E2, P = 0.09; INT versus OVX, p = 0.59.
Effect of E2 on clot area and secondary ischemic lesion volume
Clotted and unclotted blood was found around the circle of Willis distributed to both sides of the brain with the majority on the ipsilateral side. Blood was also found as a thin layer overlying both sides of the cortex. No clotted blood was found intracerebrally (Fig. 2). No significant differences of clot area were noted among INT, OVX, and OVX + E2 groups, which were 6.6% ± 5.0%, 5.9% ± 4.5%, and 7.6% ± 3.2%, respectively, of the brain base surface (P = 0.56) (Fig. 3).
Figure depicts sham
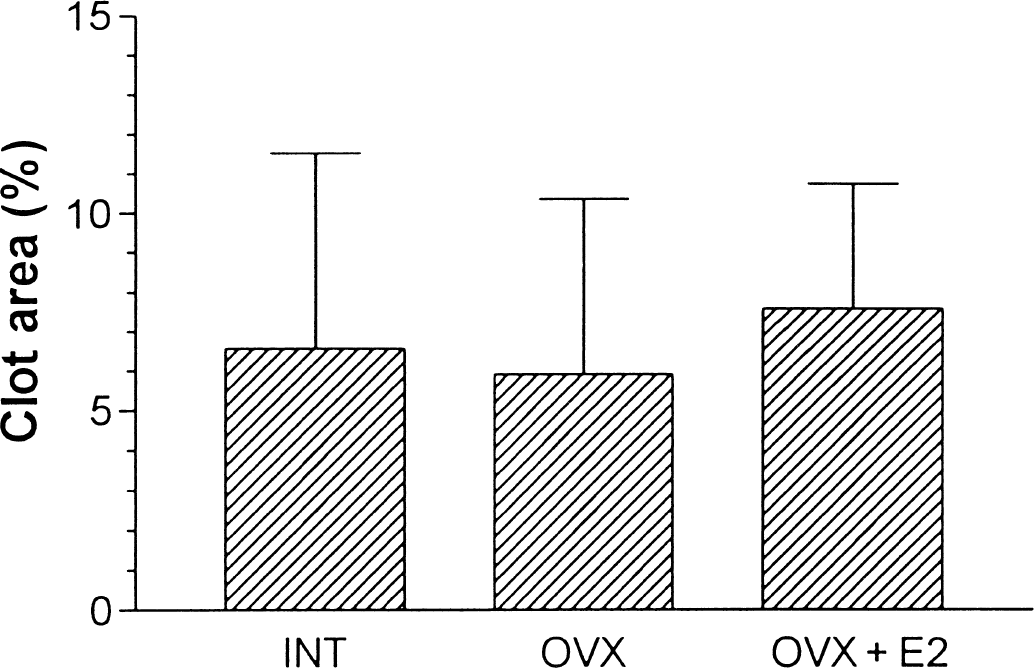
Clot volumes in INT (n = 15), OVX (n = 19), and OVX + E2 (n = 13). No significant differences were noted among the groups (P = 0.5617). Graph shows mean ± SD.
The secondary ischemic lesion was confined to the ipsilateral somatosensory cortex and basal ganglia in most of the animals. Contralateral somatosensory cortex was also involved in 8 of the 19 OVX (42.1%), 2 of the 15 INT (13.3%), and 3 of the 13 OVX + E2 animals (23.1%). The secondary ischemic lesion volume was 9.3% ± 8.4%, 24.3% ± 16.3%, and 7.0% ± 6.4% in INT, OVX, and OVX + E2 groups, respectively (P < 0.01) (Fig. 4).
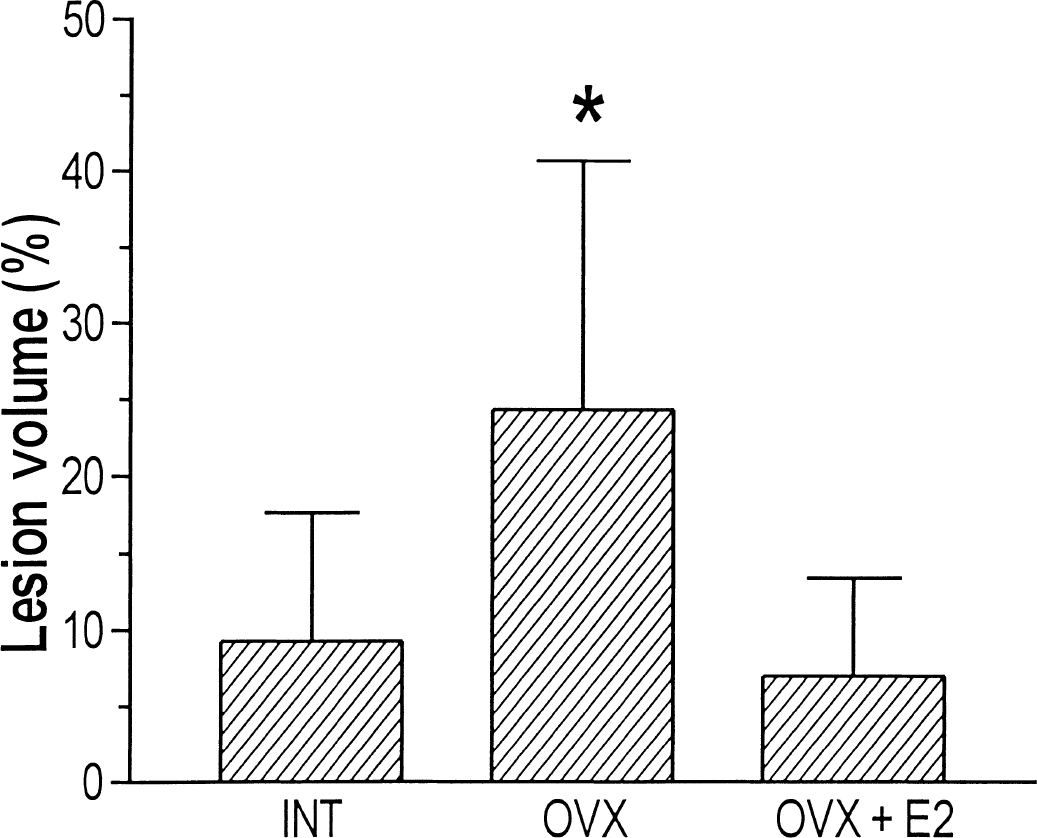
Secondary ischemic lesion volumes in INT (n = 15), OVX (n = 19), and OVX + E2 (n = 13). Lesion volume of OVX was significantly greater than that of INT and OVX + E2 (P = 0.0003). Graph shows mean ± SD.
Effect of E2 on cerebral blood flow after subarachnoid hemorrhage
The ipsilateral cortical CBF was reduced to 29.6% ± 17.7%, 22.8% ± 8.3%, and 43.5% ± 22.9% of the baseline in INT, OVX, and OVX + E2, respectively (P = 0.01) (Fig. 5). The contralateral cortex CBF was reduced to 63.4% ± 14.1%, 57.4% ± 11.0%, and 66.6% ± 17.9% of baseline in INT, OVX, and OVX + E2, respectively (P = 0.26) (Fig. 5).
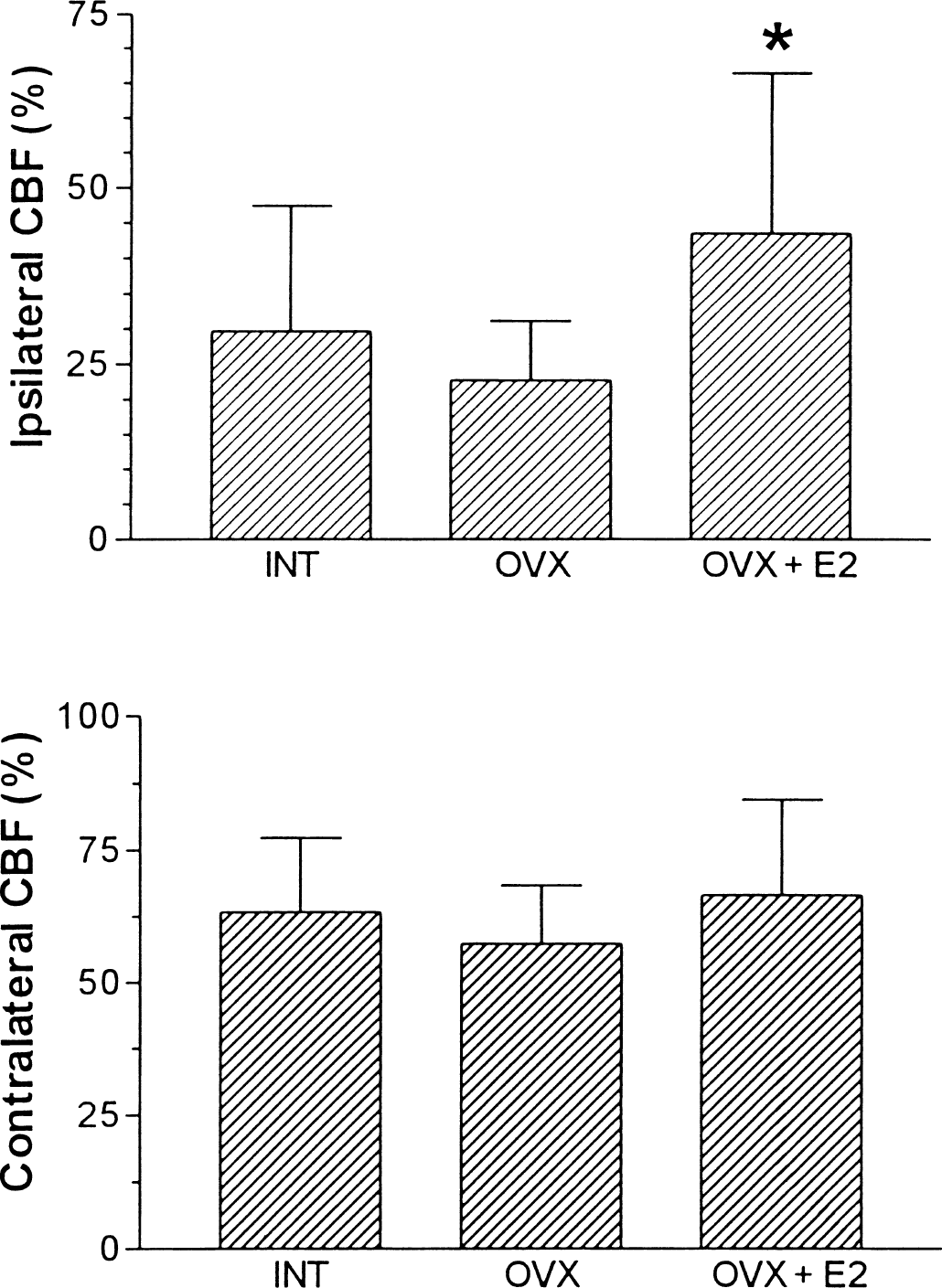
Cortical cerebral blood flow (CBF) in both sides of INT, OVX, and OVX + E2. In the left side (ipsilateral), cortical CBF of OVX + E2 was significantly greater than that of OVX and INT (P = 0.0119). In the right side (contralateral), no significant differences were noted among the groups. Graph shows mean ± SD.
The subcortical CBF, which were measured by H2 clearance, were 7.77 ± 12.03, 7.80 ± 8.65, and 20.58 ± 8.96 mL 100 g−1 min−1 on the ipsilateral side (P < 0.01), and 21.53 ± 2.94, 25.13 ± 3.23, and 25.30 ± 3.01 mL 100 g−1 min−1 on the contralateral side, in INT, OVX, and OVX + E2, respectively (Fig. 6).
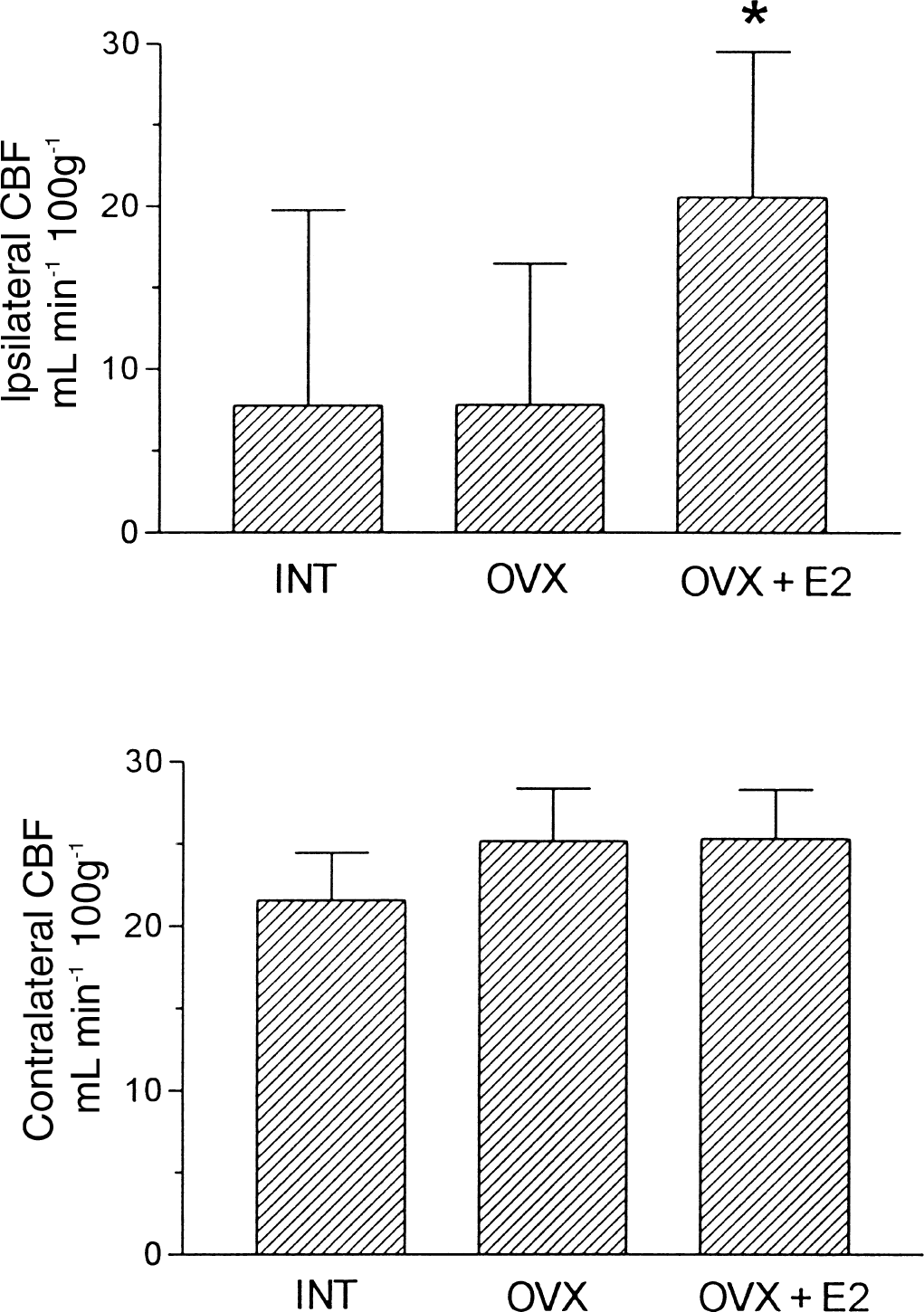
Subcortical cerebral blood flow (CBF) in both sides of INT, OVX, and OVX + E2. In the left side (ipsilateral), subcortical CBF of OVX + E2 was significant greater than that of OVX and INT (P < 0.01). In the right side (contralateral), no significant differences were noted among the groups. Graph shows mean ± SD.
DISCUSSION
Subarachnoid hemorrhage is a unique disorder and a major clinical problem that commonly occurs when an aneurysm in a cerebral artery ruptures leading to bleeding, clot formation, early morbidity, and mortality. The incidence of SAH in females is greater than in males. But the influence of the female sex steroid on SAH outcome remains controversial. This study evaluated the influence of the female sex steroid on SAH outcome, and for the first time demonstrated that 17β-estradiol (E2), which is the most active natural estrogen, can reduce the mortality and secondary ischemic damage in a SAH model.
Several studies have demonstrated that E2 exerts neuroprotective effects in ischemic stroke (Simpkins et al., 1997; Shi et al, 1997; Dubal et al., 1998; Toung et al., 1998; Chen et al., 1998; Hawk et al., 1998; Wang et al., 1999; Rusa et al., 1999; Yang et al., 2000). But the effect of E2 on secondary ischemic damage of SAH, which has been demonstrated both in clinical and experimental studies and is one of the major reasons of death shortly after SAH, is unknown. The secondary ischemic damage can be caused by severe CBF reduction immediately after SAH, or processes involving vasospasm and edema leading to the reduction of CBF, or both (Warnell, 1996).
The endovascular SAH model, which uses focal puncture of the internal carotid artery, results in extensive distribution of blood throughout the subarachnoid space. This blood distribution is similar to that observed with SAH in human subjects and makes the studies of acute pathophysiologic changes of SAH, such as secondary ischemic damage, more clinically relevant (Veelken et al., 1995; Bederson et al., 1995; Matz et al., 1996). Diffusion magnetic resonance imaging study in this SAH model has shown that the acute secondary ischemic damage is confined primarily to the ipsilateral somatosensory cortex and basal ganglia and is involved in the contralateral somatosensory cortex in some of the cases, which was consistent with the authors' secondary ischemia result (Busch et al., 1998). Asymmetric ischemic damage could have resulted from the asymmetric clot distribution in this model, because there is a direct relation between the location of the thickest blood clots and the most severe vasospasm (Camarata et al., 1994).
Deprivation of ovarian steroids, which result from ovariectomy, increased secondary ischemic damage in the current study. In contrast, replacement of E2 in OVX animals decreased the secondary ischemic damage to the level below that of normal females. This study demonstrated that E2 exerted the similar neuroprotective effects in secondary ischemic damage of SAH as previously reported for ischemic stroke (Simpkins et al., 1997; Shi et al., 1997; Dubal et al., 1998; Toung et al., 1998; Chen et al., 1998; Hawk et al., 1998; Wang et al., 1999; Rusa et al., 1999; Yang et al., 2000). The neuroprotective effects of endogenous ovarian steroids or exogenous estradiol on secondary ischemic damage are of the same magnitude as is reported for primary ischemia. Thus, it appears that both endogenous and exogenous E2 exert neuroprotective effects in secondary ischemic damage of SAH in a manner that is not associated with changes of clot in SAH.
The neuroprotective mechanisms of E2 are not yet elucidated, although both direct neuroprotective action on neurons and indirect effects on the cerebral vasculature are possible. Direct effects can include reduction in reactive oxygen species that accumulate during ischemia (Hall et al., 1991), blockade of excitatory amino acid toxicity (Singer et al., 1996; Weaver et al., 1997), modulation of calcium homeostasis (Collins et al., 1993, 1996; Mermelstein et al., 1996; Chen et al., 1998), induction of antiapoptotic protein (Singer et al., 1998; Pike, 1999), and/or enhancement of brain glucose uptake (Shi et al., 1997; Alvarez et al., 1997). Estrogens classically exert their effects by a nuclear estrogen receptor mechanism of action. It is still controversial whether the neuroprotective effect of estrogens is receptor-dependent or -independent. Several lines of evidence suggest that these neuroprotective effects are not a result of the classical estrogen receptor-mediated mechanism. First, tamoxifen, a mixed estrogen receptor agonist/antagonist, does not block the neuroprotective effect of 17-β E2 in serum-deprived SK-N-SH neuroblastoma cells (Green et al., 1997; Culmsee et al., 1999). Second, 17-α E2 has been demonstrated to have neuroprotective efficacy and potency similar to 17-β E2, although 17-α E2 binds only weakly to the estrogen receptors (Green et al., 1997). However, several studies demonstrated that estrogen's neuroprotective effects are receptor-dependent. ICI 182780, an estrogen receptor antagonist, can prevent E2 from protecting against cell death (Wilson et al., 2000). It seems that estrogens may act by multiple mechanisms. E2 could protect from the secondary ischemic damage through similar mechanisms as in ischemic stroke; however, other mechanisms could be operative. Estrogen has been reported to exert both neuroprotective and flow-preserving effects (Toung et al., 1998), but the ischemic protective effects of estrogens seem independent of flow-preserving effects (Dubal et al., 1998; Wang et al., 1999). The current results suggest that estrogen's protective effects on ischemia are flow-independent, because the secondary ischemia lesion volumes were also significantly reduced in the INT group even though both the cortical and subcortical residue CBF were at same level as that in OVX group.
In the E2 replaced animals, both cortical and subcortical residual CBF on the ipsilateral side of the SAH remained greater than that of the OVX and INT group after SAH, whereas no significant difference was noted on the contralateral side among groups. The reduction of CBF is closely related to the presence of blood and blood breakdown products within the perivascular spaces, acting either directly upon the cerebral vessel wall or perhaps more indirectly through perivascular nerves and central brain stem afferent connections to produce an acute vasospasm (Jackowski et al., 1990). Using the similar SAH model, Bederson et al. (1998) have shown that the internal circumference of internal carotid artery and anterior cerebral artery decreased to approximately 50% at 60 minutes after SAH. So the flow-preserving effect of estrogen could partly result from its interaction on vasospasm. Nitric oxide (NO) has been demonstrated to be related to the vasospasm after SAH (Sobey and Faraci, 1998; Sayama et al., 1998), and estrogen appears to alter mygenic tone by increasing cerebrovascular NO production, or action, or both (Geary et al., 1998). Estrogen can also cause the rapid dilation of blood vessels by stimulating endothelial nitric oxide synthase (Shaul, 1999). The current results suggest that the blood flow-preserving effect could only be induced by exogenous estrogen replacement. Because the exogenous estrogen level in the current study was 24 to 31 pg/mL, which is within the physiologic range (Butcher et al., 1974; Nequin et al., 1979), the lack of blood flow preservation in INT rats was not because of circulating estradiol levels. One explanation for the lack of this effect in endogenous steroids is the presence of progestin in intact female, which varied from 5 to 82 ng/mL, depending on the stage of the estrous cycle, in INT females and less than 5 ng/mL in OVX females (Butcher et al., 1974; Nequin et al., 1979; Murphy et al., 2000). Sarrel (1999) has shown that the vasodilation response to estrogen in women is blunted by progestin. However, the observation that secondary ischemia is similarly reduced in both INT and OVX + E2 rats suggests that progestin is not influencing the neuroprotective effects of estrogen and that the observed blood flow-preserving effects of exogenous estrogens are not the primary neuroprotective mechanism. In other words, in the SAH model, estrogen's neuroprotective effects are flow-independent.
Although both endogenous female steroids and exogenous E2 reduce secondary ischemic damage in SAH, endogenous female steroids (INT group) were not associated with a significant reduction of mortality, whereas E2 replacement markedly reduced mortality. The observation of effects of endogenous ovarian steroids on mortality is consistent with the clinical studies of the outcome of SAH. Kongable et al. (1996) showed that the SAH outcome of women and men is the same even though women were older and harbored more aneurysms. Simpson et al. (1991) found that men have a high risk of unfavorable outcome after SAH. In contrast to the above studies, Johnston et al. (1998) found the mortality of SAH was 62% greater in females than in males. Overall, sex differences in the clinical outcome of SAH are unresolved. The different effects of exogenous estrogen and endogenous female steroids on CBF could attribute to the different mortality between the INT and the OVX + E2 group. The vasodilation effect of estrogens can be reduced by progestin (Sarrel, 1999), suggesting that a progestin blockade of this important action of estrogens could account, in part, for the increased mortality of intact females. In addition, hydrocephalus, which occurs in 20% of patients with SAH and is associated with additional morbidity and mortality, is related to vasospasm after SAH (Black, 1986; Suarez-Rivera, 1998). Further studies of ovarian steroids and their interaction in acute vasospasm and vasospasm-related hydrocephalus are needed to explain the difference of endogenous female steroids and exogenous E2 replacement in mortality after SAH.
In summary, the current study demonstrated that E2 could reduce the secondary ischemic damage and mortality of SAH by exerting neuroprotective effects. These effects are not associated with the change of the clot volume in SAH.