
Abstract
Programmed cell death (PCD) is an ordered and tightly controlled set of changes in gene expression and protein activity that results in neuronal cell death during brain development. This article reviews the molecular pathways by which PCD is executed in mammalian cells and the potential relation of these pathways to pathologic neuronal cell death. Whereas the classical patterns of apoptotic morphologic change often do not appear in the brain after ischemia, there is emerging biochemical and pharmacologic evidence suggesting a role for PCD in ischemic brain injury. The most convincing evidence for the induction of PCD after ischemia includes the altered expression and activity in the ischemic brain of deduced key death-regulatory genes. Furthermore, studies have shown that alterations in the activity of these gene products by peptide inhibitors, viral vector-mediated gene transfer, antisense oligonucleotides, or transgenic mouse techniques determine, at least in part, whether ischemic neurons live or die after stroke. These studies provide strong support for the hypothesis that PCD contributes to neuronal cell death caused by ischemic injury. However, many questions remain regarding the precise pathways that initiate, sense, and transmit cell death signals in ischemic neurons and the molecular mechanisms by which neuronal cell death is executed at different stages of ischemic injury. Elucidation of these pathways and mechanisms may lead to the development of novel therapeutic strategies for brain injury after stroke and related neurologic disorders.
Keywords
GENETIC CONTROL OF PROGRAMMED CELL DEATH IN NEURONS
Programmed cell death (PCD) is the process by which neurons die during normal development. Much of our fundamental understanding of the genetic control of this process comes from the studies of Horvitz and colleagues in the roundworm C. elegans (reviewed in Horvitz et al., 1994). Their approach led to the identification of several genes that control PCD, the C. elegans death genes (ced). A large number of ceds have been identified, but three proved to control the key committed step in neuronal cell death: ced-3, ced-4, and ced-9 (Fig. 1). ced-3 and ced-4 activity commits the cell to PCD (Yuan and Horvitz, 1990), whereas ced-9 inhibits this process (Hengartner et al., 1992). A number of other ced genes are involved in the execution of PCD but do not prevent eventual cell death. Mutation in these genes affects the morphology of the dying neuron, their engulfment by surrounding neurons, or biochemical changes such as DNA fragmentation (Ellis et al., 1991). Other genes, known as C. elegans specification genes (ces), initiate PCD of neurons in C. elegans (Ellis and Horvitz, 1991).
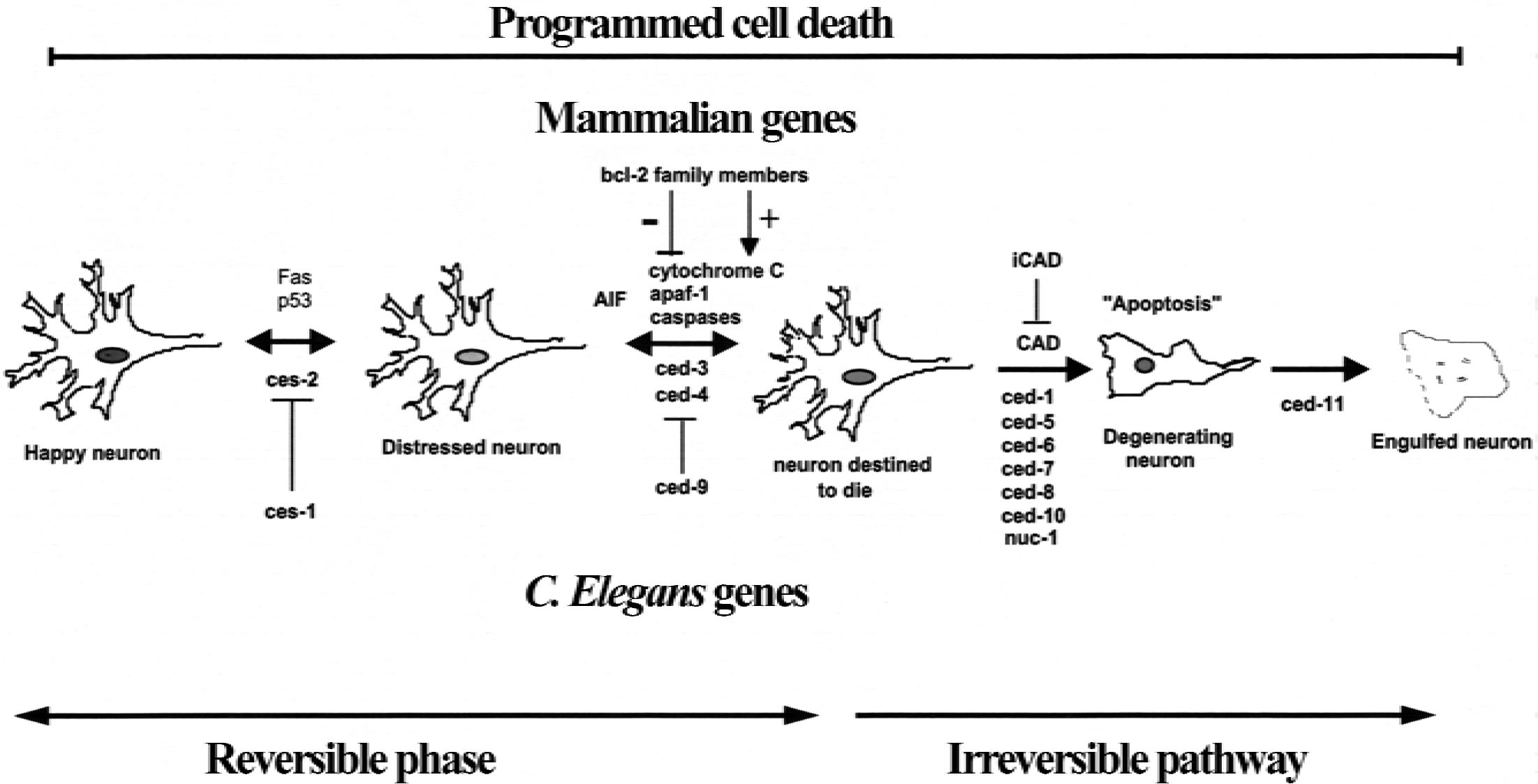
Schematic diagram illustrating the cascade of genes involved in programmed cell death and their counterparts in C. elegans.
In more recent years, the identity of the mammalian counterparts of the ced genes has been determined. One of the first homologues identified was bcl-2, an oncogene that is overexpressed in B cell lymphoma, the mammalian homologue of ced-9 (Hengartner and Horvitz, 1994). The bcl-2 gene encodes a 26-kD membrane-associated protein that is localized in multiple subcellular sites including the mitochondria, nuclear membrane, and endoplasmic reticulum (Lithgow et al., 1994). Functionally, bcl-2 is a potent cell death suppressor, whose overexpression can prevent cell death in response to a variety of stimuli including serum and growth factor deprivation, glucocorticoids, chemotherapeutic agents, and baculoviral infection (Reed, 1994). These death suppressor effects explain why bcl-2 is an oncogene in lymphoma and other types of cancer. Subsequently, over 20 additional genes that comprise the bcl-2 family have been identified. Many of these proteins, like Bcl-2, inhibit PCD, but others, such as Bax (Oltvai et al., 1993), promote cell death.
Caspases are a family of proteases that are homologous to ced-3. Interleukin-1 β-converting enzyme (now called caspase-1) was the first caspase discovered and was found to share extensive homology with a critical cell death gene, ced-3, in C. elegans (Yuan et al., 1993). Since this discovery, more than 14 new caspases have been identified (Thornberry and Lazebnik, 1998; Wellington and Hayden, 2000). Based on their relative role in apoptosis, caspases can be categorized into three groups. Caspases 1, 4, 5, 11, 12, 13, and 14 (Group I) mainly function as cytokine processors. Caspases 2, 8, 9, and 10 (Group III) are initiator caspases that are usually the first to be activated upon receiving death signals and serve to couple cellular signaling pathways to the activation of downstream effector caspases. Effector caspases (Group II), including caspases 3, 6, and 7, are cleaved and activated by initiator caspases and in turn cleave crucial cellular substrates.
The molecular mechanisms by which these proteins interact to control PCD have recently been determined (Fig. 2). bcl-2 family genes play a key role in regulating mitochondrial permeability. Increased mitochondrial permeability occurs in response to down-regulation of Bcl-2 or other antiapoptotic bcl-2 family members, or in response to overexpression and translocation of proapoptotic Bcl-2 family members, such as Bax, to the mitochondrial membrane. Increased mitochondrial permeability results in egress of cytochrome c from the mitochondria to the cytosol (Jurgensmeier et al., 1998; Narita et al., 1998). Cytochrome c can then form a complex with Apaf-1 (the mammalian homologue of ced-4), dADP, and procaspase-9 in the cytosol to form the aposome (Budihardjo et al., 1999). As a result, caspase-9 is cleaved and activated and then can cleave other caspase family members including caspase-3 (Li et al., 1997a). Activation of caspase-3 appears to be a key event in initiating PCD. Numerous cellular substrates for caspase-3 have been identified, among the most crucial are the following: poly(ADP-ribose)polymerase (PARP), DNA protein kinase (DNA-PK), lamin B, and inhibitor of caspase-dependent DNase (ICAD, also called DNA fragmentation factor, DFF). The proteolytic cleavage of these important proteins is directly responsible for the characteristic changes associated with apoptosis, including the cessation of DNA repair, chromatine condensation, and internucleosomal DNA fragmentation (reviewed in Thornberry and Lazebnik, 1998).
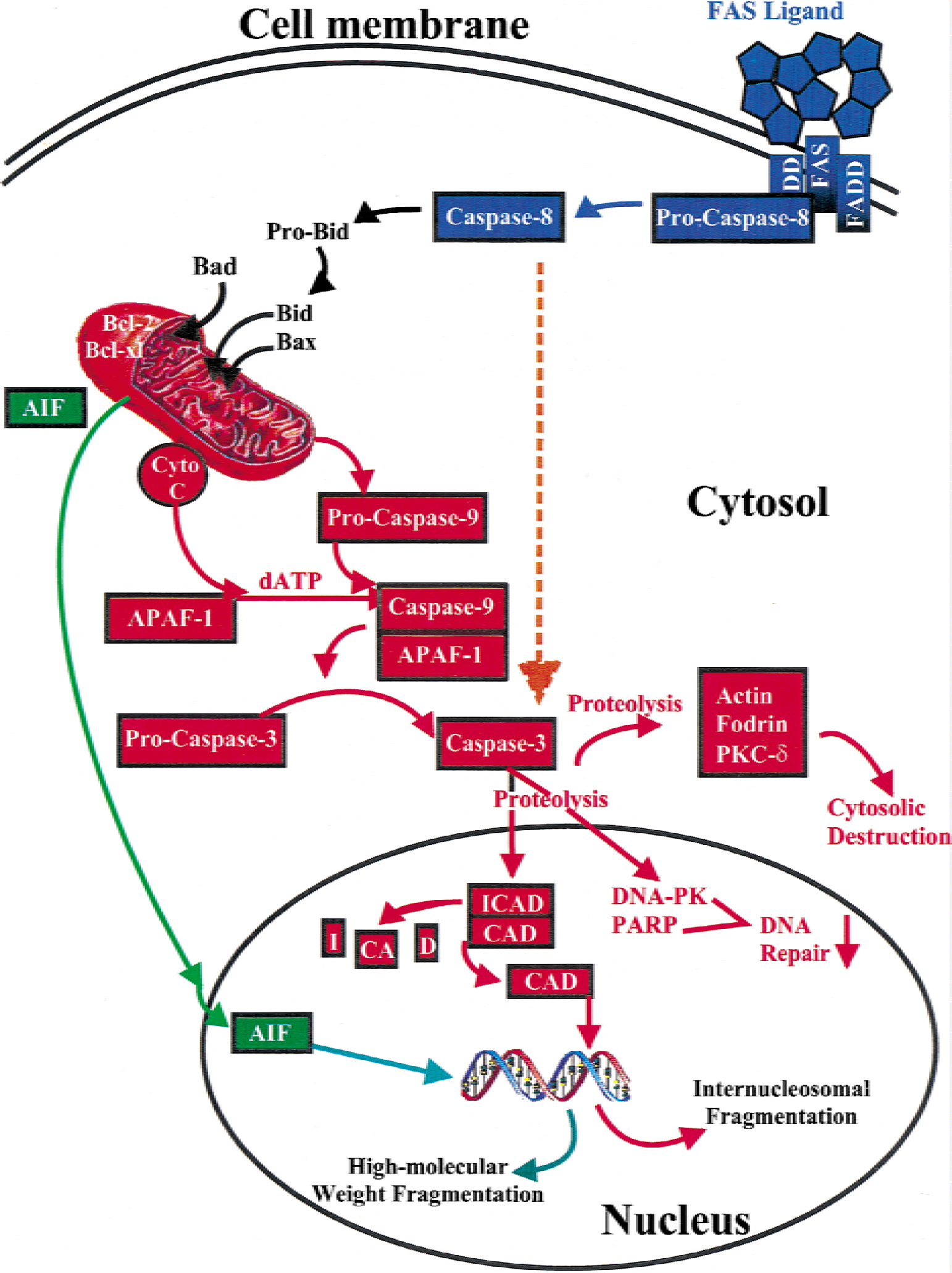
Schematic diagram illustrating key molecular events in programmed cell death in mammalian cells. Blue: Fas-system. Red: intrinsic pathway. Orange: extrinsic pathway. Green: apoptosis-induced factor pathway.
Activation of PCD is much more complex in mammalian systems than in C. elegans. There are at least three sites where PCD can be triggered—the mitochondria, cell membrane receptors, and chromosomal DNA. Mitochondrial injury produced by prolonged depolarization, oxidative stress, and opening of the mitochondrial permeability transition pore can all result in PCD (Reed et al., 1998). In response to these death stimuli, the mitochondria may then initiate PCD through release of cytochrome c and activation of the intrinsic caspase pathway, or the mitochondria may release apoptosis-inducing factor (AIF) and initiate apoptosis by caspase-independent mechanisms. Activation of cell surface receptors including Fas and tumor necrosis factor-α can also initiate PCD (reviewed in Wallach et al., 1999). These receptors activate caspase-8. Caspase-8 can activate the intrinsic pathway by cleaving the proapoptotic Bcl-2 family protein Bid (Gross et al., 1999; Wang et al., 1996), which translocates to the mitochondria, resulting in cytochrome c release. In liver, Fas-mediated apoptosis appears to require Bid and thus is executed primarily through the mitochondria (Yin et al., 1999). But caspase-8 can also directly cleave and activate caspase-3 (Stennicke and Salvesen, 2000). It is not known which pathway is active in neuronal apoptosis.
Finally, DNA damage can trigger PCD. DNA damage induced by a variety of methods including oxidative damage and ionizing radiation induces expression of the transcription factor p53 (Lowe et al., 1993). p53 expression can alter transcription of several genes that result in the initiation of PCD, including bax (Miyashita et al., 1994).
Once PCD is triggered, there are at least three major pathways by which it may be initiated—the intrinsic and extrinsic pathways that both lead to caspase-3 activation and the caspase-independent AIF pathway. The intrinsic caspase pathway is activated when cytosolic cytochrome c complexes with Apaf-1 and activates caspase-9. Caspase-9 then cleaves and activates caspase-3 (Li et al., 1997a). In the extrinsic pathway, caspase-8 is activated through the Fas or tumor necrosis factor-α membrane receptor systems (Scaffidi et al., 1998). Activated caspase-8 then cleaves and activates caspase-3. This pathway bypasses the mitochondria.
The third pathway by which PCD may occur is caspase-independent. A key factor in this pathway is AIF, a novel proapoptotic molecule that is involved in the final execution of apoptosis and that has been identified and partially characterized (Susin et al., 2000). The AIF cDNA codes for a protein of 612 and 613 amino acids in mouse and human, respectively (Susin et al., 1999). The AIF protein contains an amino-terminal mitochondrial localization sequence that confines AIF to residing exclusively in the mitochondria in healthy cells (Lorenzo et al., 1999). During apoptosis, AIF is released from mitochondria, loses its mitochondrial localization sequence domain (mature AIF), and expresses its apoptogenic effects (Susin et al., 1999). When microinjected into the cytoplasm of normal cells, recombinant AIF is sufficient to cause the following 4 apoptotic hallmarks: 1) the exposure of phosphatidylserine on the plasma membrane surface; 2) the condensation of nuclear chromatin (stage I); 3) large-scale DNA fragmentation; and 4) the dissipation of the mitochondrial transmembrane potential and release of cytochrome c (Ferri et al., 2000; Susin et al., 1999). These apoptogenic effects of ectopic (extramitochondrial) AIF are independent of the action of caspases and are not affected by overexpression of the antiapoptotic protein, Bcl-2 (Susin et al., 1999).
CHARACTERISTICS OF ISCHEMIC NEURONAL DEATH IN VIVO
Given these new insights into the mechanisms of PCD in mammalian cells, it is necessary to reexamine the evidence that PCD, or apoptosis, occurs after cerebral ischemia in vivo. In the following sections, the authors will review the presence and significance of apoptosis—that is, the morphologic changes associated with PCD and DNA fragmentation—and they will discuss the significance of these features. The authors will also review evidence that the expression and activity of genes that regulate PCD occur after ischemia. Finally, they will review evidence that alteration of expression or activity of these genes determines whether ischemic neurons die.
APOPTOSIS
Apoptosis is a term that is often used synonymously with PCD; however, as originally defined, apoptosis refers to distinct morphologic changes that occur after PCD. In developing neurons, these changes include condensation and cleavage of chromatin and the formation of cytoplasmic appendages called apoptotic bodies. These changes are distinct from the morphologic changes that characterize necrosis—swelling of cytoplasmic organelles and disruption of mitochondrial and plasma membranes.
Developing neurons that die through PCD have the typical morphologic changes known as apoptosis, whereas the pathologic hallmark of cerebral infarction is “coagulative necrosis.” These latter pathologic features characterize cell death in the core region of infarctions and after severe global ischemia. When ischemia is transient or less severe, some morphologic changes consistent with apoptosis have been reported. At the light microscopic level, some evidence of condensation and cleavage of the nucleus may be found. In global ischemia and in some cortical lesions after transient focal ischemia, relatively selective neuronal death occurs without the intense inflammatory response found in the core of infarctions. After global ischemia, at least, careful examination at the ultrastructural level suggests that ischemic neuronal death does not have the morphologic features of apoptosis. Neurons that at the light level appeared to be apoptotic with nuclear cleavage and cell fragmentation instead were found to be fragments of disintegrating cells. Furthermore, other features of apoptosis were not present at the ultrastructural level in more than 400 neurons examined by electron microscopy after global ischemia (Colbourne et al., 1999). These and other studies cast doubt as to whether the morphologic features of apoptosis occur at all in cerebral ischemia in adult brain.
The lack of morphologic changes that characterize apoptosis does not rule out the possibility that PCD contributes to death of the cell. The nature by which cell death is executed depends upon the expression and activity of several downstream genes that produce the morphologic changes of apoptosis. These genes may not be expressed in adult brain after ischemia for several reasons. First, cellular differentiation entails alteration of a cell's ability to express many genes. Thus, the adult postmitotic neuron may not be capable of expressing all of the genes that execute PCD in the developing neuron. For example, postmitotic neurons express some, but not all, of the cell cycle genes, several of which may also control PCD (Li et al., 1997b). Furthermore, translation of many gene products may be blocked because of energy failure after ischemic injury (Hossmann, 1993). Finally, expression and activity of Bcl-2 family genes has been shown to inhibit necrotic and apoptotic cell death (Kane et al., 1995). Thus, the presence of apoptosis strongly suggests that a cell died by PCD; however, the absence of apoptosis, often observed after cerebral ischemia, does not exclude PCD.
DNA FRAGMENTATION
Damage to nuclear DNA is a prominent feature of ischemic brain injury. It has been assumed frequently but erroneously that DNA fragmentation, often detected by in situ histochemical techniques such as TUNEL labeling, is synonymous with PCD. This view is overly simplistic. The significance of DNA fragmentation detected by TUNEL labeling must be carefully interpreted. TUNEL staining detects double-stranded DNA nicks bearing the 3′ OH group that can be produced by either endonuclease-dependent or -independent mechanisms. Oxidative stress, particularly prevalent in ischemia reperfusion models, can damage DNA and result in both single- and double-stranded DNA nicks (Sun, 1990), which are independent of any endonuclease activities. Significant oxidative DNA damage occurs shortly after reperfusion in rat temporary cerebral ischemia (Nagayama et al., 2000). This oxidative DNA damage is not limited to the boundaries between histones, but is randomly distributed, thus often producing a smear on DNA gels (Du et al., 1996); it is also readily detectible by TUNEL staining. Indeed, cells with either apoptotic or necrotic morphology have been stained TUNEL-positive under various pathologic conditions, including cerebral ischemia and traumatic brain injury (Chen et al., 1997; Clark et al., 1997; Liu et al., 1996; Portera-Cailliau et al., 1995; Rink et al., 1995). It is likely that ischemic neurons suffer a mixed type of nuclear DNA damage resulting from direct attacks by reactive oxygen species as well as endonuclease-mediated degradation. Thus, TUNEL labeling alone may not be sufficient to determine whether PCD is occurring.
Using combined in situ DNA end labeling and DNA gel electrophoresis methods, numerous studies have been able to detect internucleosomal DNA fragmentation in various ischemia models (Charriaut-Marlangue et al., 1996; Chen et al., 1997; Li et al., 1995; Linnik et al., 1993; MacManus et al., 1993). DNA fragmentation occurs at late stages of cell death after ischemia and is often accompanied by some morphologic characteristics of apoptosis (Charriaut-Marlangue et al., 1996; Chen et al., 1997b; Li et al., 1995), thus likely constituting one of the final steps in the execution of nuclear apoptosis in ischemic neurons. This provides strong evidence that the PCD execution pathway is activated in the ischemic brain. Activation of specific endonucleases is one important mechanism through which PCD is executed. Caspase-activated DNase (CAD), the endonuclease specifically activated by caspase-3, cleaves DNA between histosomes, resulting in fragments of 180 to 200 base-pair multiples (Enari et al., 1998). DNA cleaved in this fashion produces a pattern known as “laddering” on DNA gels. Despite the confounding effects of other types of DNA damage, such as oxidative damage in the evaluation of nuclear DNA, detection of the “laddering” pattern of internucleosomal DNA fragmentation in the ischemic brain nevertheless provides reliable evidence that PCD occurs in cerebral ischemia.
The mechanisms of DNA fragmentation after cerebral ischemia have been studied in some detail. MacManus et al. (1999) reported that formation of DNA fragmentation after ischemia is distinct from that found in classical apoptosis, in which most fragmented DNA contains 3′ ends with a recess of 8 to 10 nucleotides. Furthermore, induction of high molecular weight DNA fragmentation, demonstrated by the generation of 50 and 10 kbp fragments, often precedes that of internucleosomal DNA fragmentation in the brain after ischemia (MacManus and Linnik, 1997). These observations suggest that a complex and possibly unique process involving more than one endonuclease may mediate ischemia-induced neuronal DNA fragmentation. Several recent studies have focused on identifying the molecular basis of DNA fragmentation and the associated nuclear alteration in neuronal apoptosis and cerebral ischemia. The CAD/ICAD (inhibitor of caspase-activated DNase) genes have now been cloned from rat brain (Cao et al., 1999; Chen et al., 2000a), and the induction of gene expression and enzymatic activation of CAD in ischemic neurons strongly suggests that the caspase-3–activated CAD mediates internucleosomal DNA fragmentation in the brain after transient ischemia (Luo et al., 1999).
However, CAD may not be responsible for all types of apoptotic nuclear changes in neurons after ischemia. As determined in the cell-free apoptosis assays, the increased CAD activity appears to be responsible for ischemia-induced nuclear and chromatin fragmentation, but is not related to chromatin condensation and shrinkage of the nucleus (Fig. 3). Because shrinkage of nucleus and chromatin condensation is a common feature of ischemic injury associated with DNA fragmentation, it will be tempting in future studies to elucidate the molecular mechanism leading to these changes. The newly cloned genes Acinus and AIF are promising candidates for such studies. Acinus is another caspase-3–activated endonuclease that has been found to mediate chromatin condensation in certain nonneuronal cells during apoptosis (Sahara et al., 1999). Apoptosis-inducing factor is a molecule activated during apoptosis that, upon release from the mitochondria, induces chromatin condensation and nuclear shrinkage in a caspase-independent manner (Daugas et al., 2000; Susin et al., 1999). In addition, AIF is the endonuclease responsible for high molecular weight DNA fragmentation in apoptosis (Susin et al., 1999). Further work is warranted to determine whether these factors function synergistically with CAD and lead to nuclear degradation in ischemic neurons.
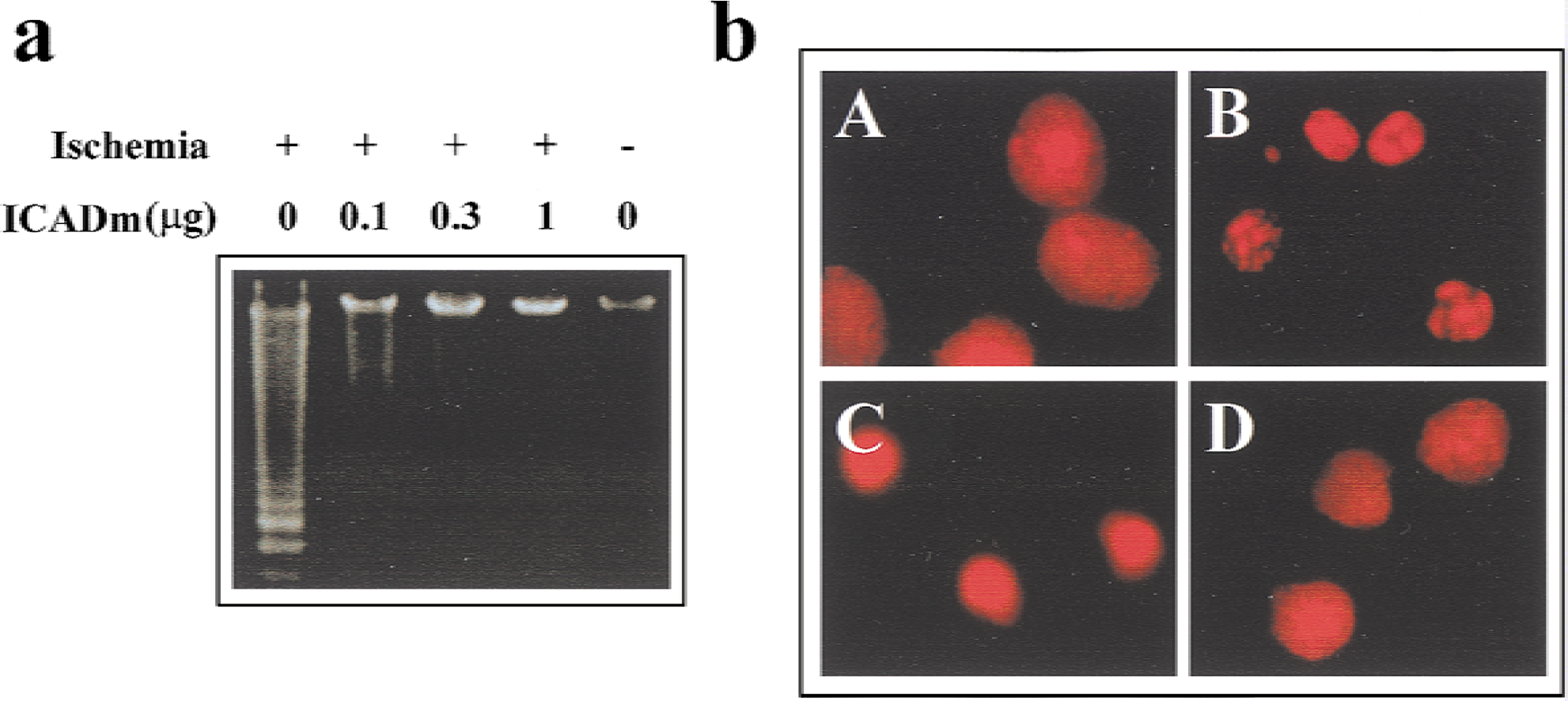
Detection of caspase-activated DNase (CAD) activity in the brain after transient focal cerebral ischemia using the cell-free apoptosis assay.
Internucleosomal DNA fragmentation is undoubtedly an important feature of PCD. But is such DNA degradation essential for the cell to die? In C. elegans, DNA fragmentation because of PCD is produced by nuc1 and, probably, other DNases. When nuc1 is mutated, DNA fragmentation does not occur, but neurons still die during development (Wu et al., 2000). Likewise, increased CAD activity is necessary to produce DNA fragmentation in mammalian neurons in response to apoptosis inducers, but neurons still die by other caspase-dependent mechanisms when CAD is inhibited (Chen et al., 2000a). Thus, DNA fragmentation characteristic of CAD activity, when present, strongly suggests that neurons are dying by PCD, but PCD can occur without CAD-dependent DNA fragmentation.
EXPRESSION OF GENES THAT REGULATE PROGRAMMED CELL DEATH
More definitive support for the hypothesis that PCD occurs after cerebral ischemia comes from studying the expression and activity of the genes that regulate PCD after ischemia. If expression and activity of these death regulatory genes is important in determining the fate of ischemic neurons, one would expect expression of genes, such as Bcl-2, that inhibit PCD to be found in neurons that are injured yet survive. However, death-promoting genes, such as caspases, should be expressed in neurons that die after ischemia. Because the cellular distribution of cell death is so clearly delineated in global ischemia models, the expression of death regulatory genes in this model provides a clear application of this thesis. In global ischemia, CA-1 is the most vulnerable region of hippocampus, whereas other regions of hippocampus, such as the dentate gyrus and CA-3, are relatively resistant to ischemia. The expression of death-promoting and death-inhibitory Bcl-2 family genes mirrors this pattern of selective vulnerability. bcl-2 and bcl-x-long, bcl-2 family members that inhibit PCD, are primarily expressed in dentate gyrus and CA-3. bax, a death promoting membrane family, is expressed primarily in CA-1. These findings support the hypothesis that bcl-2 family genes may play a role in regulating PCD.
Caspase-3, the caspase that has the highest homology to ced-3, the key step in committing a cell to PCD, is also expressed in CA-1 after cerebral ischemia (Chen et al., 1998; Gillardon et al., 1997). Caspase-3 is also expressed in focal ischemia models in regions where there is neuronal death. Furthermore, the cleaved active form of the enzyme can be detected with Western blot analysis (Chen et al., 1998). Caspase activity is also increased in cortex after more prolonged ischemia, consistent with its death promoting role (Fink et al., 1998; Krupinski et al., 2000; Namura et al., 1998). Thus, there is evidence that caspase-3 is expressed, cleaved, and activated after cerebral ischemia.
Expression of bcl-2 family genes is also altered after cerebral ischemia. Expression of bcl-2 and other bcl-2 family members with death-suppressing activity is induced in neurons that are ischemic yet survive. Bcl-2 expression is induced primarily in ischemic neurons that do not die (Chen et al., 1995, 1997; Gillardon et al., 1996), including CA-3 and dentate neurons after transient forebrain ischemia and penumbral regions that do not infarct after temporary focal ischemia. Similar patterns of expression have been reported for Bcl-x-long, another Bcl-2 family member that inhibits PCD. Bcl-x-l is expressed in CA3 and dentate gyrus neurons in global ischemia, neurons that are ischemic yet survive (Chen et al., 1997). Two recent reports show that Bcl-w is induced after cerebral ischemia and is expressed in neurons that survive (Minami et al., 2000; Yan et al., 2000). Proapoptotic bcl-2 family members are expressed in neurons destined to die. For example, Bax is expressed in CA1 neurons that TUNEL-label after global ischemia (Chen et al., 1996; Krajewski et al., 1995). These results also are consistent with the proposed death regulatory roles of these gene products.
EFFECTS OF ALTERATION OF EXPRESSION AND ACTIVITY OF GENES THAT REGULATE PROGRAMMED CELL DEATH
Alteration of expression of the genes that regulate PCD is consistent with a possible role in determining the fate of ischemic neurons; however, the observation that their expression is altered does not exclude the possibility that their expression is simply an epiphenomenon in neurons that are already destined to live or die. The best evidence that these genes do indeed determine, at least in part, the fate of ischemic neurons comes from numerous studies where expression and activity of these genes is altered in ischemia models.
Expression of bcl-2 has been altered by transgenic methods, viral vectors, and antisense oligonucleotide treatment in ischemia models. Martinou et al. (1994) constructed a transgenic mouse that overexpressed Bcl-2 in neurons driven by the neuron-specific enolase promoter. The transgenic mice that overexpressed Bcl-2 in neurons had significantly smaller infarctions than wild-type control mice. Overexpression of Bcl-2 using herpes simplex vectors protects neurons against ischemia (Lawrence et al., 1997; Linnik et al., 1995). These studies show that Bcl-2 expressed before ischemia can protect the brain against ischemia, but do not determine whether Bcl-2 endogenously expressed in response to ischemia protects neurons. To address this issue, antisense oligonucleotide was used to block translation of endogenously expressed Bcl-2 after temporary focal ischemia. Animals treated with the antisense oligonucleotide to Bcl-2 had larger infarctions and decreased Bcl-2 protein expression compared with controls after temporary focal ischemia (Chen et al., 2000b). These results suggest that endogenous expression of Bcl-2 in response to ischemia has a neuroprotective effect and is not simply an epiphenomenon in neurons already destined to live.
Alteration of expression in activity of caspases by transgenic and pharmacologic means increases survival of ischemic neurons. Several studies have found that the infusion of peptide inhibitors of caspases decreased caspase activity in infarction volume after temporary focal ischemia (Cheng et al., 1998; Fink et al., 1998; Hara et al., 1997; Li et al., 2000; Schulz et al., 1998; Wiessner et al., 2000). Furthermore, overexpression of a dominant negative mutant of caspase-1 also ameliorated infarction volume in transgenic mice compared with wild-type controls (Friedlander et al., 1997). Caspase-1–deficient mice are resistant to neonatal hypoxia. Peptide inhibitors of caspase-3 improved neuronal survival after global ischemia in two studies (Chen et al., 1998; Gillardon et al., 1999), but had no effect in another study (Li et al., 2000). Taken together, these studies and other studies show that alteration of expression of caspases and Bcl-2 family proteins determine, at least in part, whether neurons live or die after an ischemic insult. These data provide compelling evidence that PCD mechanisms contribute to ischemic injury in animal models.
SUMMARY: A SPECTRUM OF CELL DEATH
Pharmacologic studies provide strong evidence that expression and activation of bcl-2 family genes and caspases determine the fate of the ischemic neuron. Cell death in ischemia is determined by multiple interdependent mechanisms including PCD. The relative contribution of PCD (versus necrosis) may depend upon the duration and severity of ischemia. These principles have been well documented in in vitro models of hypoxic ischemia and excitotoxicity. Excitotoxins can produce cell death that is necrotic in nature if neurons are exposed to high concentrations of glutamate for a prolonged period of time. However, a brief exposure to glutamate results in cell death that is inhibited by treatment with cyclohexamide and also results in DNA fragmentation characteristic of PCD (Bonfoco et al., 1995). Furthermore, combined hypoxia and hypoglycemia produced neuronal death with necrotic features in vitro unless excitatory amino acid receptors were blocked (Gwag et al., 1995). Thus, there is a continuum between PCD and necrosis in vitro depending on the duration and severity of the insult.
There is a spectrum of severity in ischemic injury in vivo depending upon the model. Global ischemia is always transient in nature and thus would be expected to produce cell death with more characteristics of PCD. However, when global ischemia is prolonged, it also may produce cell death that is predominantly necrotic (Petito et al., 1997). Focal ischemia models show a gradient in the severity of hypoperfusion and in the rapidity of energy failure. In the core region of the infarction, blood flow may be close to zero (Tamura et al., 1981). Cell death in this region is often primarily necrotic in nature, unless the duration of ischemia is very brief. In the lateral cortex, the so-called penumbral region, hypoperfusion is less severe, allowing for the necessary energy production for expression of new proteins. Thus, these conditions are more favorable to PCD, especially in temporary focal models. In permanent focal ischemia models, however, conditions are favorable to necrotic mechanisms. In these models, glucose utilization is temporarily retained in the penumbral region; however, glucose utilization in the penumbral region approaches zero within 4 hours after middle cerebral artery occlusion (Shiraishi et al., 1989). Furthermore, there is rapid loss of protein synthesis in this region (Mies et al., 1991). Thus, variations in the duration and intensity of ischemia, energy failure, and protein synthesis inhibition may produce a continuum between the features of necrosis and PCD in ischemia models.
ANSWERS, QUESTIONS, AND FUTURE DIRECTIONS
In the last decade, the mammalian genes that control PCD and the mechanisms by which their proteins execute PCD in large part have been determined; however, it is less certain whether PCD occurs in the ischemic adult postmitotic neuron. The most convincing data supporting the hypothesis that PCD occurs after ischemia are the observations that expression and activity of death regulatory genes are altered by ischemia, and that treatments altering expression or activity of these gene products influence the outcome of ischemic injury. Treatments such as caspase inhibitors could provide practical stroke therapies because caspases are active many hours after the onset of ischemia. However, despite these advances, a number of important questions remain unanswered. The answers to these questions will provide important insights into the future therapeutic strategies aimed at limiting ischemic neuronal injury.
Could other approaches besides caspase inhibition be more effective?
Caspase activation is only one consequence of mitochondrial apoptosis. Mitochondrial dysfunction and opening of the mitochondrial permeability transition pore may result in activation of caspases through egress of cytochrome c into the cytoplasm; however, there are several other mechanisms by which mitochondrial dysfunction may contribute to ischemic neuronal death. Severely damaged mitochondria may be unable to maintain the electrochemical gradient necessary for respiration and glucose oxidation. Thus, mitochondrial dysfunction may exacerbate ischemic injury by exacerbating energy failure. In such circumstances, the mitochondria may be worse than dead—the dysfunctional mitochondria also produce oxygen free radicals that injure other cellular organelles and DNA. Hence, treatments that prevent mitochondrial dysfunction could be a more potent neuroprotective strategy than caspase inhibition.
How is mitochondrial apoptosis initiated and controlled in ischemic neurons by Bcl-2 family proteins?
bcl-2 family genes play a key role in maintaining and controlling mitochondrial integrity in response to injury. Development of practical conventional drugs that mimic or inhibit the effects of Bcl-2 and related proteins is difficult because Bcl-2 family proteins are integral membrane proteins and not enzymes. An alternative strategy for altering the function of these proteins, rather than mimicking or inhibiting their effects, may be to interfere with the initiation of Bcl-2 family protein expression or the mechanisms that control translocation of these gene products to the mitochondria. More than 20 bcl-2 family genes have been identified, and it is unclear which of these family members play key roles in controlling PCD in the ischemic neuron. Furthermore, it is not currently known how the expression and translocation of these proteins to the mitochondria is regulated in the ischemic neuron.
Is programmed cell death in neurons after ischemia initiated by cell surface receptors?
The Fas cell membrane receptor system is an important mechanism that triggers PCD in many mammalian cells. Because Fas is a proximal site in the cascade of events in PCD, treatments aimed at inhibiting Fas could conceivably block many facets of cell death execution. Several recent studies suggest that Fas-associated PCD can occur in brain and spinal cord as a result of ischemia and other insults (Felderhoff-Mueser et al., 2000; Martin-Villalba et al., 1999; Nogae et al., 1998); however, the relative contribution of Fas-induced PCD in ischemic brain is not yet defined. Furthermore, it is not known whether activation of Fas in ischemic neurons leads to the activation of the intrinsic pathway through caspase-8–mediated Bid cleavage or leads to the activation of the extrinsic pathway directly through caspase-8.
Are there caspase-independent mechanisms active in ischemia?
Caspase activation may not be the only mechanism through which PCD is executed in the ischemic brain. Apoptosis-inducing factor and other proteins could mediate neuronal cell death through caspase-independent mechanisms. It is not known whether AIF-induced PCD occurs in neurons or whether AIF is released from brain mitochondria and activated in response to ischemic injury. If AIF or other caspase-independent pathways are active in ischemic neurons, treatments aimed at blocking caspase-dependent and -independent pathways of PCD could be more effective than blocking either pathway alone.
The answers to the above questions will define the precise molecular mechanisms by which PCD is initiated and executed in ischemic neurons. To address these questions, precise molecular tools must be developed and used in ischemia models. These tools include the development of additional transgenic mice models where genes in these pathways are disrupted or overexpressed, the design of specific and highly membrane-permeable protease inhibitors, and the identification and overexpression of dominant negative inhibitors of key death effector proteins. The use of these tools in answering the above questions may lead to the development of novel therapeutic strategies for stroke and related neurologic disorders.
Footnotes
Acknowledgments:
The authors thank Carol Culver for editorial assistance and Anne Stetler and Chris O'Horo for assistance with illustrations.