
Abstract
Apoptotic cell death plays an important role in the cascade of neuronal degeneration after traumatic brain injury (TBI), but the underlying mechanisms are not fully understood. However, increasing evidence suggests that expression of Fas and its ligand (FasL) could play a major role in mediating apoptotic cell death in acute and chronic neurologic disorders. To further investigate the temporal pattern of Fas and FasL expression after experimental TBI in the rat, male Sprague Dawley rats were subjected to unilateral cortical impact injury. The animals were killed and examined for Fas and FasL protein expression and for immunohistologic analysis at intervals from 15 minutes to 14 days after injury. Increased Fas and FasL immunoreactivity was seen in the cortex ipsilateral to the injury site from 15 minutes to 72 hours after the trauma, respectively. Immunohistologic investigation demonstrated a differential pattern of Fas and FasL expression in the cortex, respectively: increased Fas immunoreactivity was seen in cortical astrocytes and neurons from 15 minutes to 72 hours after the injury. In contrast, increased expression of FasL was seen in cortical neurons, astrocytes, and microglia from 15 minutes to 72 hours after impact injury. Concurrent double-labeling examinations using terminal deoxynucleotidyl tranferase-mediated deoxyuridine-biotin nick end labeling identified Fas- and FasL-immunopostive cells with high frequency in the cortex ipsilateral to the injury site. In contrast, there was no evidence of Fas- and FasL-immunopositive cells in the hippocampus ipsilateral to the injury site up to 14 days after the trauma. Further, Fas and FasL immunoreactivity was absent in the contralateral cortex and hippocampus at all time points investigated. These results reveal induction of Fas and FasL expression in the cortex after TBI in the rat. Further, these data implicate an involvement of Fas and FasL in the pathophysiologic mechanism of apoptotic neurodegeneration after TBI. Last, these data suggest that strategies aimed to repress posttraumatic Fas- and FasL-induced apoptosis may open new perspectives for the treatment of TBI.
The mechanisms underlying cellular death after traumatic brain injury (TBI) are poorly understood. However, evidence from experimental models of TBI suggest that traumatically induced cell death may be necrotic in nature, which is characterized by swelling of the nucleus and cytoplasmic organelles as well as by an early loss of plasma membrane integrity and cell lysis (Dietrich et al., 1994; Hicks et al., 1996). Studies also show that TBI may induce apoptosis (i.e., programmed cell death; for review see McIntosh et al., 1998). In contrast to necrosis, a cell undergoing apoptosis is characterized by uniform internucleosomal DNA fragmentation, nuclear shrinkage, chromatin compaction as well as by cytoplasmatic condensation, and disintegration (for reviews see Kerr et al., 1972; Bredesen, 1995).
Although apoptotic neuronal cell death has been suggested to play an important role for neurodegeneration after TBI, little is known about the intracellular cascade that leads to the induction of apoptosis after acute experimental brain injuries in vivo. However, evidence shows that the interaction between the Fas/APO (apoptosis)-1/CD95 molecule with its ligand (Fas ligand, FasL) may induce programmed cell death (for review see Becher et al., 1998).
Fas, a type I 48-kDa transmembrane receptor glycoprotein is a member of the nerve growth factor/tumor necrosis factor receptor superfamily of surface molecules whose members have a wide range of functions related to cell survival, differentiation, and immune responses. Ligation of Fas by its natural ligand, FasL, a type II membrane protein belonging to the tumor necrosis factor (TNF) family, or by agonistic anti-Fas monoclonal antibody, rapidly induces apoptosis in sensitive cells. In addition, it has been demonstrated that FasL-induced apoptosis involves the activation of death proteases of the caspase family (for review see Nagata, 1997).
Few data exist on the involvement of Fas/FasL in apoptotic cell death in acute and chronic CNS diseases, respectively. For example, Fas is overexpressed in neural-derived tumors (Tachibana et al., 1996) and is associated with apoptosis in neuroblastoma cells (Bernassola et al., 1999). In addition, reports identify Fas in adult postmortem brains of patients with neurodegenerative disorders including Alzheimer's disease (Nishimura et al., 1995) and multiple sclerosis (D'Souza et al., 1996; Bonetti and Raine, 1997). Further, intrathecal release of Fas has been observed in patients with bacterial meningitis (Fassbender et al., 1999) and in patients with acute ischemic stroke (Tarkowski et al., 1999). Moreover, Fas messenger RNA (mRNA) also is expressed in periventricular cells in postischemic adult mouse brains (Matsuyama et al., 1994). Last, a report provides evidence that overexpression of FasL may induce apoptosis after experimental brain ischemia in the rat (Martin-Villalba et al., 1999).
Despite increasing evidence that Fas and FasL may have a potential role for inducing apoptosis in cerebral ischemia and chronic CNS diseases, little is known about the expression of Fas and FasL after TBI in vivo. Only one report provides evidence that FasL is overexpressed in CSF of patients after severe head injury (Ertel et al., 1997). To further investigate potential changes of Fas and FasL expression after experimental TBI, rodents were subjected to a widely used model of experimental mechanical brain injury: lateral cortical impact injury (Dixon et al., 1991). The current study used Western blot analyses of cortical and hippocampal samples to determine the temporal profile of Fas and FasL protein changes from 15 minutes to 14 days after experimental TBI. Further, immunohistochemical examinations were performed to investigate the cellular distribution of Fas and FasL expression after TBI in vivo.
MATERIALS AND METHODS
Rat model of traumatic brain injury
A controlled cortical impact device was used to induce a moderate level of TBI, as described previously (Dixon et al., 1991; Kampfl et al., 1996; Franz et al., 1999; Zhao et al., 1998). Briefly, adult male Sprague-Dawley rats (250 to 350 g) were intubated and anesthetized with 2% halothane in a 2:1 mixture of N2O/O2. Core body temperature was monitored continuously by a rectal thermistor probe and maintained at 37° to 38°C. Animals were mounted in a stereotaxic frame on the injury device in a prone position secured by ear and incisor bars. The head was held in a horizontal plane with respect to the interaural lines. A midline incision was made, the soft tissues reflected, and two 7-mm craniotomies were made between lambda and bregma and centered 5 mm laterally on either side of the central suture. The dura was kept intact over the cortex. Injury was induced by impacting the right cortex (ipsilateral cortex) with a 6-mm diameter tip at a rate of 4 m/s. The injury device was set to produce a tissue deformation of 2 mm. Velocity was measured directly by the linear velocity displacement transducer (Shaevitz Model 500 HR, Detroit, MI, U.S.A.), which produces an analog signal that was recorded by a PC-based data acquisition system for analysis of time/displacement parameters of the impactor. After cortical impact, animals were extubated and immediately assessed for recovery of reflexes (Dixon et al., 1991). Sham-injured animals underwent identical surgical procedures but did not receive impact injury. Naive animals were not exposed to any injury-related surgical procedures. Sixty-four animals were used in this study (naive, n = 4; sham-injured rats, n = 4; injured rats, n = 56). All animal studies carefully conformed to the guidelines outlined in the Guide for the Care and Use of Laboratory Animals from the Austrian Department of Health and Science and were approved by the University of Innsbruck Medical School Animal Welfare Committee.
Sample preparation, sodium dodecyl sulfate-polyacrylamide gel electrophoresis, immunoblotting, and quantification
Before killing, all animals (n = 32) were given a lethal dose of phenobarbital (20 mg/kg intraperitoneally; Tyrol Pharma, Kundl, Austria). The animals were killed by decapitation 15 minutes, 6 hours, 24 hours, 48 hours, 72 hours, 7 days, and 14 days after TBI (n = 4 for each time point after injury; n = 2 for sham and naive animals). Sham animals were killed at 15 minutes and 48 hours after sham surgery, respectively. Both cortices and hippocampi (ipsilateral and contralateral to the injury site) were removed. Excision of both cortices beneath the craniotomies extended approximately 4 mm laterally, approximately 7 mm rostrocaudally, and to a depth extending to the white matter. All samples were immediately frozen in liquid N2. The microdissected tissue was homogenized at 4°C in ice-cold homogenization buffer containing 20 mmol/L piperazine-N,N′-bis (2-ethanesulfonic acid) (pH 7.1), 2 mmol/L EGTA, 1 mmol/L ethylenediamine tetraacetic acid, 1 mmol/L dithiothreitol, 0.3 mmol/L phenylmethylsulfonylfluoride, and 0.1 mmol/L leupeptin. Protein concentrations were determined by bicinchoninic acid microprotein assays (Sigma, St. Louis, MO, U.S.A) with albumin standards. Protein-balanced samples were prepared for polyacrylamide gel electrophoresis in twofold loading buffer containing 0.25 mol/L Tris (pH 6.8), 0.2 mol/L dithiothreitol, 2% sodium dodecyl sulfate, 0.005% bromophenol blue, and 5% glycerol in distilled water. Samples were heated for 5 minutes at 95°C. Fifty micrograms of protein was routinely resolved in each lane. We used a vertical electrophoresis chamber with a 4% acrylamide stacking gel over a 13% acrylamide resolving gel. After separation, proteins were immediately transferred to nitrocellulose membranes using Western blotting. Lateral transfer was used with transfer buffer made up of 0.192 mol/L glycine and 0.025 mol/L Tris (pH 8.3) with 10% methanol at a constant voltage of 25 V for 90 minutes at room temperature (RT). Blots were immediately blocked for immunolabeling by overnight incubation using 5% nonfat milk in phosphate-buffered saline (PBS) and 0.05% Tween 20 (PBS-T) at 4°C. Coomassie blue (Biorad, Hercules, CA, U.S.A) and Ponceau red (Sigma, St. Louis, MO, U.S.A.) staining were routinely performed to confirm that equal amounts of protein were loaded in each lane. After incubation with a mouse monoclonal antibody against Fas (Transduction Laboratories, Lexington, KY, U.S.A.) diluted 1:2500 and anti-FasL mouse monoclonal antibody (Transduction Laboratories; dilution 1:2500) for 2 hours at RT, respectively, nitrocellulose membranes were incubated 45 minutes at RT with a secondary antibody linked to alkaline phosphatase (dilution 1:5000; Accurate Chemical and Scientific Corp., Westbury, NY, U.S.A). Visualization was performed with a p-nitroblue tetrazolium chloride (NBT; Boehringer-Mannheim, Mannheim, Germany) and 5-bromo-4-chloro-3-indoylphosphate (BCIP; Boehringer-Mannheim) alkaline phosphatase substrate.
Immunohistochemistry
Animals from all treatment groups (n = 32) were given a lethal injection of phenobarbital (20 mg/kg intraperitoneally) before perfusion. Rats were transcardially perfused through the left ventricle (120 mL of 0.9% saline and 200 mL of 4% paraformaldehyde) at 15 minutes, 6 hours, 24 hours, 48 hours, 72 hours, 7 days, and 14 days after TBI (n = 4 for each time point after injury; n = 2 for sham and naive animals). Sham animals were killed at 15 minutes and 48 hours after sham surgery, respectively. The brains then were removed, grossly sectioned coronally at 2-mm intervals, and routinely embedded in paraffin. Sections were then cut at 3 to 4 μm on a rotary microtome, mounted on aminoalkylsilated glass slides, and processed for immunohistochemical staining as follows: Deparaffinized and rehydrated sections were microwaved for 15 minutes in 10 mmol/L sodium citrate buffer, pH 6.0, and allowed to cool to RT. Endogenous peroxidase was blocked by treatment with 0.3% H2O2 in methanol followed by incubation with 10% fetal calf serum (FCS) in PBS for 30 minutes. Rabbit polyclonal primary antibodies against Fas (M-20, Santa Cruz Technologies, Santa Cruz, CA, U.S.A.) or FasL (N-20, Santa Cruz) were diluted (1:1000) in 10% FCS in PBS and allowed to bind overnight at 4°C. Sections without primary antibodies were similarly processed to control for binding of the secondary antibody. Sections were washed in PBS three times for 10 minutes each and then incubated with a biotinylated anti-rabbit antibody (Vector Laboratories, Burlingame, CA, U.S.A.) at a dilution of 1:200 for 1 hour at RT followed by avidin-peroxidase (Sigma) for 1 hour at RT. The reaction was visualized by treatment with 0.05% 3,3-diaminobenzidine tetrahydrochloride solution in PBS containing 0.05% H2O2. The color reaction was stopped with several washes of PBS.
For double immunostaining using brightfield chromagens, sections were pretreated as described earlier. Rabbit polyclonal primary antibodies against Fas (M-20, Santa Cruz) or FasL (N-20, Santa Cruz) were diluted (1:1000) in 10% FCS in PBS and allowed to bind overnight at 4°C. Sections were washed in PBS three times for 10 minutes each and then incubated with a biotinylated anti-rabbit antibody (Vector Laboratories) at a dilution of 1:200 for 1 hour at RT followed by incubation with an alkaline phosphatase avidin-biotin substrate and then reaction with red (Vector Red, Vector Laboratories) or blue chromogen (Vector Blue; Vector Laboratories). Sections then were incubated with a mouse monoclonal anti-neuronal nuclei (NeuN) antibody (Chemicon, Temecula, CA, U.S.A.) (Wolf et al., 1996) for neuronal staining. For staining of astrocytes and microglia, a mouse monoclonal anti-glial fibrillary acidic protein (GFAP) antibody (Boehringer-Mannheim) (Debus et al., 1983) or an anti-ED1 monoclonal antibody (Serotec, Kidlington, U.K.) (Graeber et al., 1990) were used, respectively. All antibodies were diluted 1:500 in 10% FCS in PBS and allowed to bind overnight a 4°C. After being rinsed, sections were incubated with a biotinylated horse anti-mouse antibody (Vector Laboratories) followed by avidin-peroxidase (Sigma) for 1 hour at RT. The reaction was visualized by treatment with 0.05% 3,3-diaminobenzidine tetrahydrochloride solution in PBS containing 0.05% H2O2. The color reaction was stopped with several washes of PBS. Sections were dehydrated through graded ethanol, cleared in a xylene substitute (Histoclear, National Diagnostics, Atlanta, GA, U.S.A.), mounted in Permount (Fisher Scientific, Nepean, Ontario, Canada), and coverslipped. Control sections without primary antibody did not stain.
Histochemical detection of DNA fragmentation (terminal deoxynucleotidyl transferase-mediated deoxyuridine-biotin nick end labeling)
Terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine-biotin nick end labeling (TUNEL) was performed as described by Gavrieli et al. (1992) with minor modifications. Briefly, for double-label experiments, dewaxed and rehydrated sections of all animal groups from regions −1.5 to −3.4 mm bregma were pretreated with 5% proteinase K in 10 mmol/L Tris/HCl for 15 minutes at 37°C. After two washes in PBS, sections were incubated with blocking solution (0.3% H2O2 in methanol), rinsed twice, and permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2 minutes at 4°C. This was followed by incubation with labeling mix (1× TdT buffer containing 100 U/mL TdT and 20 nmol/L · mL biotin-conjugated 16 deoxyuridine) in a humidified chamber for 90 minutes at 37°C. After three washes in PBS, slides were incubated in Converter-POD for 10 minutes in a humidified chamber at 37°C. All reagents were purchased from Boehringer-Mannheim. The reaction was visualized by treatment for 3 minutes with 0.05% 3,3-diaminobenzidine tetrahydrochloride solution in PBS containing 0.05% H2O2. Sections then were processed for Fas and FasL immunohistochemical study as described earlier. Primary antibody, labeling mix, or secondary antibody were omitted in control sections. Sections were examined under the light microscope.
Statistical analysis
Western blotting band densities were quantified with computer-assisted two-dimensional densitometric scanning on a Macintosh computer using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/). Relative band densities on Western blots (n = 1 per blot) were expressed as arbitrary densitometric units for each time point. This procedure was repeated four times for a total of n = 4. Data acquired in arbitrary densitometric units were transformed to percentages of the densitometric levels observed on scans from sham animals on the same blot. Because no statistically significant differences for Fas and FasL immunoreactivity of sham samples were seen at 15 minutes and 48 hours after sham surgery, we used sham samples of 48 hours after sham surgery for our figure gels. Group differences were determined by analysis of variance and post hoc Tukey HSD test. Values given are mean ± SD. Differences were considered significant when P < 0.05.
RESULTS
Western blotting
Fas appeared as an immunoreactive band at approximately 45 kDa (Fig. 1A). Cortical impact injury resulted in an increase of Fas immunoreactivity in the ipsilateral cortex. Ipsilateral Fas immunoreactivity from injured animals increased by 275% relative to sham levels at 15 minutes after TBI (Fig. 1A). The increase in Fas immunoreactivity reached a maximum level by 24 hours after TBI (940% increase relative to sham animals) and declined to a 390% increase relative to sham at 72 hours after TBI. At 7 and 14 days after the injury, no statistical significant difference in Fas protein expression was observed between cortical samples ipsilateral to the injury site and cortical samples from sham injured animals.
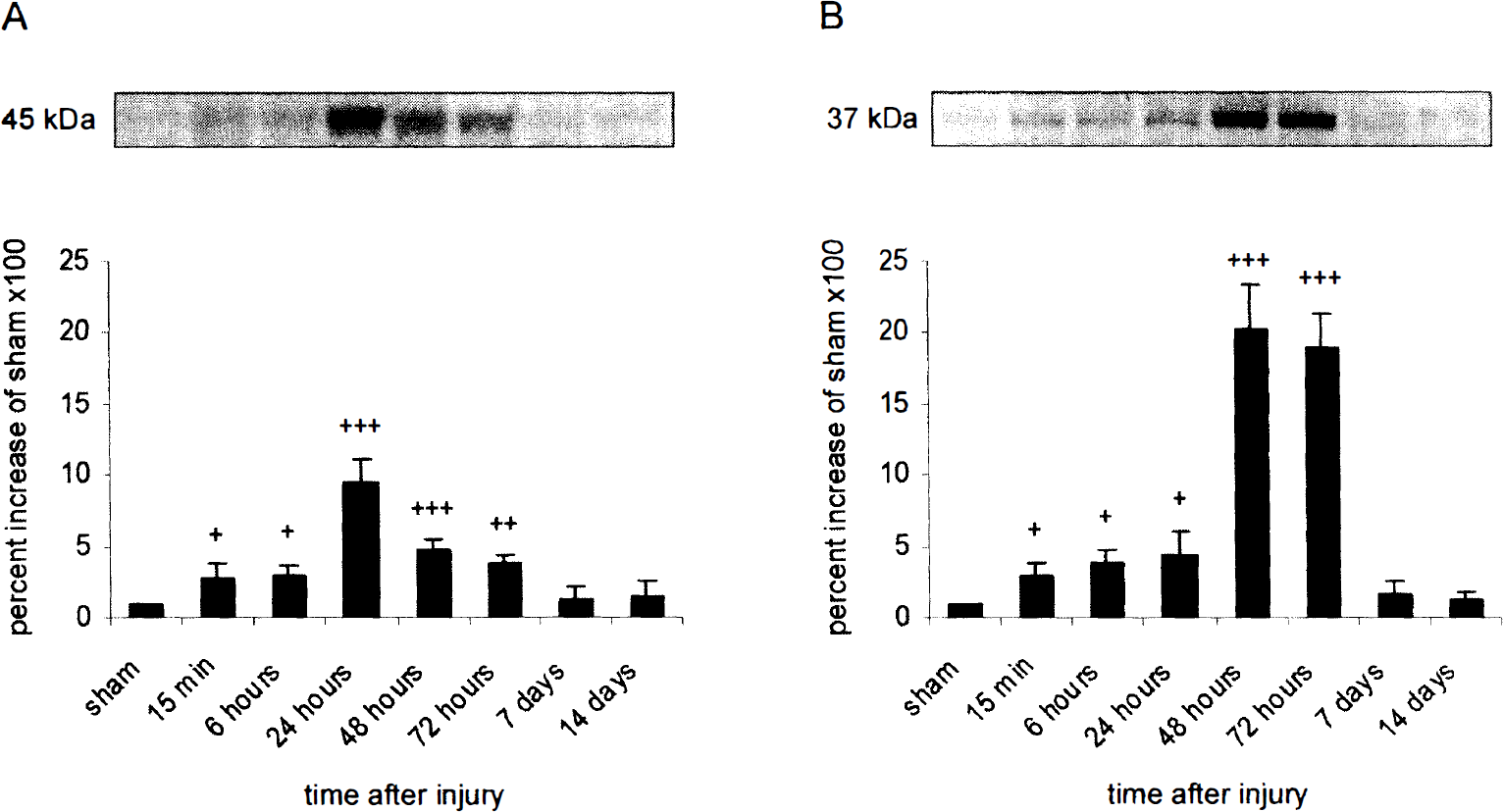
Time course of Fas
Western blot analysis for FasL indicated the presence of two immunoreactive bands at approximately 37 kDa (Fig. 1B), probably from different glycosylated isoforms (Suda et al., 1993; Runic et al., 1996). In the ipsilateral cortex, FasL levels were significantly higher from 15 minutes to 72 hours after injury compared with control (sham) animals (Fig. 1B). In the ipsilateral cortex, FasL immunoreactivity increased within 15 minutes after injury (290% increase relative to sham levels), peaked at 48 hours after TBI with an increase of 2020% relative to sham animals, and slowly decreased thereafter to a level of 1890% relative to controls at 72 hours after trauma.
No significant differences were observed in Fas and FasL immunoreactivity between naive and sham-injured animals (data not shown). In addition, no significant changes in Fas and FasL protein expression were seen between sham and injured animals in cortical samples contralateral to the injury site and hippocampal samples ipsilateral and contralateral to the injury site at 15 minutes to 14 days after TBI (data not shown).
Immunohistochemical studies
Ipsilateral and contralateral cortical and hippocampal tissues were examined rostrocaudally from +0.2 to −3.8 mm bregma. No localized Fas and FasL immunoreactivity was present in the tissue from sham-injured (Figs. 2A and 2B; Fas and FasL, respectively, time point 48 hours after sham surgery) or naive control rats (data not shown). Fas immunoreactivity was observed in the ipsilateral cortex at the primary injury zone (−1.5 to −3.4 mm bregma) from 15 minutes to 72 hours after the trauma (Fig. 2C; time point 48 hours after trauma). Fas immunoreactivity was seen primarily in the soma of CNS cells (Fig. 2C) in cortical layers 1 to 5. To further investigate if Fas is expressed in glial or neuronal cells, we performed double-labeling experiments for Fas, the neuronal cell-specific marker NeuN, the astrocytic marker GFAP, and the microglial marker ED1. These immunohistochemical analyses of Fas-positive cells from 15 minutes to 72 hours after TBI (Figs. 2E and 2F; time point 48 hours after trauma) identified labeling with NeuN and GFAP and demonstrated the expression of Fas in cortical neurons (Fig. 2E) and astrocytes (Fig. 2F), respectively. In contrast to late time points after impact injury (i.e., 24 hours to 72 hours after TBI), only few astrocytes and neurons were immunoreactive for Fas at 15 minutes after the trauma (data not shown). No Fas immunoreactivity was detected in microglial cells (data not shown).
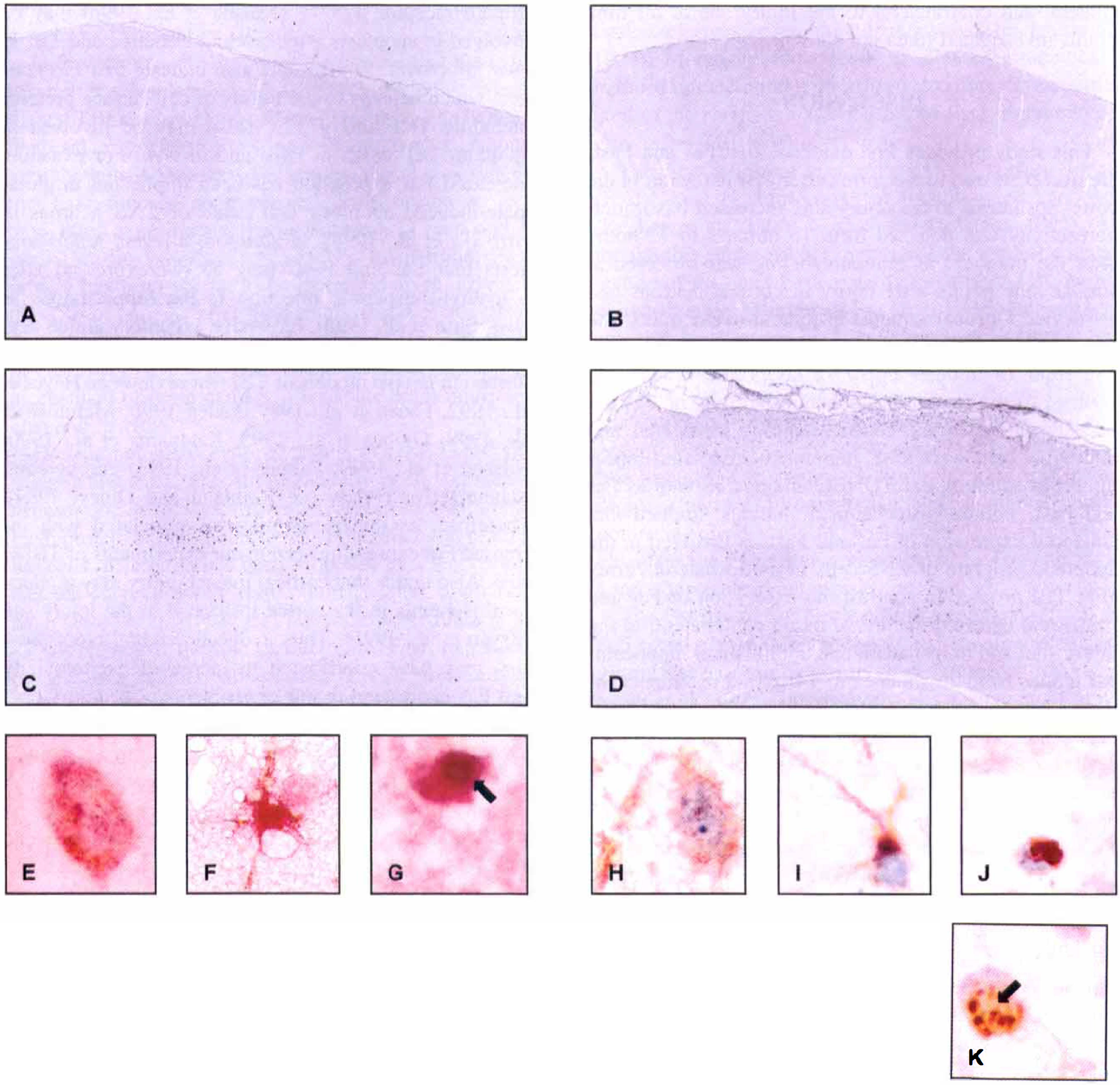
Immunohistochemical analysis of Fas and FasL expression in the ipsilateral cortex at 48 hours after TBI. Sham-injured brains showed no specific Fas
Cells positive for FasL were seen in the ipsilateral cortex from 15 minutes to 72 hours after the trauma. Similar to Fas, prominent FasL labeling was seen in the somata of CNS cells (Fig. 2D; time point 48 hours after impact injury) in the impact zone in cortical layers 1 to 5. In contrast to our Fas experiments, double-labeling investigations (Fig. 2H to Fig. 2J; time point 48 hours after TBI) provided evidence that FasL is expressed in neurons (Fig. 2H), astocytes (Fig. 2I), and microglial cells (Fig. 2J). At 15 minutes after the impact, only few astrocytes and neurons were immunoreactive for FasL. By 24 hours to 72 hours after TBI, the number of FasL-immunopositive neurons, astrocytes, and microlgial cells had increased unambiguously (data not shown).
To verify further an apoptotic component of posttraumatic cell death, we used TUNEL staining technique on tissue sections from injured or control rat brains. The TUNEL-positive cells were detected in the ipsilateral cortex in layers 1 to 5 from 6 hours to 72 hours after injury (data not shown). No TUNEL-positive cells were observed in sham-operated animals, in the ipsilateral and contralateral hippocampus and in the contralateral cortex at all time points investigated (data not shown). To further support the possibility that Fas and FasL mediate trauma-induced apoptosis, we performed double-labeling experiments using TUNEL and Fas (Fig. 2G) and FasL (Fig. 2K) immunohistochemical studies. Double staining clearly demonstrated that TUNEL-positive cells with condensed and fragmented nuclei (Figs. 2J and 2K, arrows) also expressed Fas (Fig. 2G) and FasL (Fig. 2K) in the ipsilateral cortex at 48 hours after TBI.
Immunoreactivity for Fas and FasL was absent in contralateral cortical samples and hippocampal samples ipsilateral and contralateral to the injury site at all time points investigated (data not shown).
DISCUSSION
This study provides first evidence that Fas and FasL are overexpressed in the cortex after TBI in the rat. In the cortex ipsilateral to the injury site, increased Fas immunoreactivity was detected from 15 minutes to 72 hours after the trauma. Fas immunolabeling was observed at similar time points after injury in cortical neurons and astrocytes. Cortical samples ipsilateral to the injury site also revealed a significant overexpression of FasL protein from 15 minutes up to 72 hours after trauma. In contrast to the immunohistochemical results of Fas expression in the cortex, cortical neurons, astrocytes, and microglial cells were FasL immunopositive after impact injury. In addition, our TUNEL findings, as well as Fas and FasL immunohistochemical findings, implied that enhanced expression of Fas and FasL is involved in the apoptotic program of CNS cells in the ipsilateral cortex after TBI in vivo. In contrast, no expression of Fas and FasL were detected in cortical tissue contralateral to the injury site, and in ipsilateral and contralateral hippocampal tissues from 15 minutes to 14 days after impact injury.
Histologic and biochemical features of apoptotic cell death have been reported after experimental TBI. For example, reports provide evidence that posttraumatic apoptosis, as demonstrated by DNA fragmentation and TUNEL staining, is seen after experimental lateral fluid percussion (FP) injury (Rink et al., 1995; Yakovlev et al., 1997; Conti et al. 1998) and cortical impact injury (Colicos and Dash, 1996; Colicos et al., 1996; Newcomb et al., 1999). In addition, gene expression associated with apoptosis has been studied. For example, alterations in the Bcl-2 gene family, which appear to be an important factor in mediating trauma-induced cell death, have been observed after FP injury (Raghupathi et al., 1997) and controlled cortical impact (Clark et al., 1997). Further, current studies on caspase 3 expression, a cysteine protease that has been associated with the final steps in the apoptotic cascade, suggest a strong link between caspase activation and neuronal apoptosis after FP injury (Yakovlev et al., 1997; LaPlaca et al., 1999) and controlled cortical impact (Pike et al., 1998)in vivo.
Although the potential role for apoptosis in neuronal cell death after TBI has been suggested, the molecular and cellular mechanisms underlying the induction of apoptotic cell death after acute brain injuries remain to be further elucidated. However, evidence indicates that signaling through specific receptors may be responsible for apoptotic cell death in the CNS. For example, the expression of TNFα (Goodman et al., 1990; Liu et al., 1994; Shohami et al., 1996) and the low-affinity neurotrophin receptor p75NTR(Kokaia et al., 1998) may be involved in apoptosis after cerebral ischemia and TBI in vivo. Moreover, limited data also indicate that Fas antigen, which belongs to the family of cell surface proteins including TNF and p75NTR, also may be involved in apoptotic cell death in vitro and in vivo. For example, increased Fas expression has been implicated in glutamate-induced apoptotic cell death of CNS neurons in vitro(Li et al., 1998). In addition, a recent report suggests that Fas and FasL may be overexpressed after N-methyl-D-aspartate injection in the hippocampus in vivo(Shin et al., 1998). Excessive excitatory amino acid release with subsequent neurotoxicity also has been described in in vivo models of TBI (for review see Hayes et al., 1992, Faden et al., 1989; Faden 1996; McIntosh et al., 1998; Globus et al., 1995; Katayama et al., 1990; Nilsson et al., 1990; Palmer et al., 1993 and cerebral ischemia (for review see Rothman and Olney, 1989). Therefore, excitotoxicity may be associated with increased Fas expression seen in our experiments of TBI in vivo. Also notice that cortical impact injury may produce focal ischemia in the cortex ipsilateral to the injury site (Bryan et al., 1995). Thus, reduced cerebral blood flow also may have contributed to increased excitotoxicity and Fas expression in our experiments.
Our Western blotting data indicate that Fas protein is overexpressed in the ipsilateral cortex from 15 minutes to 72 hours after the injury. Interestingly, Fas mRNA expression after experimental cerebral ischemia in the rat also is increased in the acute phase after the ischemic insult (Matsuyama et al., 1995). Because our data and a recent report (Newcomb et al., 1999) suggest that apoptotic-like morphologic features in cortical cells after controlled cortical impact injury in the rat is most apparent up to 3 days after the trauma, it could be speculated that apoptotic cell death may indeed be mediated by Fas overexpression in the traumatically injured brain. Our results provide clear evidence of apoptotic-like morphologic features (indicated by TUNEL staining) in Fas-positive cortical cells after TBI in the rat.
Our immunohistochemical results suggest that Fas is expressed in astrocytes and neurons after TBI. A similar cellular distribution of Fas upregulation also has been described in different acute and chronic neurologic disorders. Using in situ hybridization, Matsuyama et al. (1994) report on Fas-immunoreactive neurons and glia from 6 to 24 hours after experimental cerebral ischemia in the rat. Further, increased expression of Fas also has been observed in glial cells after oxidative stress and hypoxia in vitro(Vogt et al., 1998), and in neurons and astrocytes of demyelinating lesions of multiple sclerosis patients (Dowling et al., 1996). These findings indicate that although Fas has been reported to be constitutively absent or weak in normal brain parenchyma (Park et al., 1998), brain is readily induced to express this receptor in a variety of pathologic stimuli.
In this report, we also provide evidence that the cytotoxic effector molecule FasL is upregulated after experimental TBI in vivo. Our Western blotting results indicate that FasL is overexpressed from 15 minutes to 72 hours after the injury. Similar to our results, recent data suggest that FasL-induced apoptotic cell death occurs up to 5 days after experimental brain ischemia in vivo and in vitro(Martin-Villalba et al., 1999). In addition, FasL protein has been shown to be increased up to 3 days in the CSF of patients after head injury (Ertel et al., 1997). Taken together, these data indicate that early expression of FasL may have a pivotal role in the pathophysiologic mechanism of brain damage after acute brain injuries in vivo and in vitro.
Our immunohistochemical results of FasL expression suggest that FasL is posttraumatically overexpressed in neurons and astrocytes. Moreover, and in contrast to Fas, FasL protein expression also was upregulated in microglial cells. Microglia has been proposed as a key cellular element in progressive tissue damage after brain ischemia (Stoll et al., 1998) and head trauma (Aihara et al., 1995). However, the role of microglial FasL expression in mediating cellular damage after brain injuries remains to be further elucidated. For example, current data indicate that FasL is not expressed in microglia after experimental cerebral ischemia in vivo(Martin-Villalba et al., 1999). In contrast, a report provides evidence that oxidative stress and hypoxia in vitro trigger FasL expression in microglial cells (Vogt et al., 1998), which may contribute to apoptotic cellular damage. It has been shown that activated microglia can cross the blood-brain barrier and accumulate in the cortex after TBI in vivo(Aihara et al., 1995). Therefore, it could be speculated that FasL-expressing microglia may migrate to Fas-expressing neurons and astrocytes, which, through receptor-ligand interaction, may result in subsequent apoptotic cell death.
Our data provide evidence that increased Fas and FasL immunoreactivity, as detected by immunoblotting, was seen at 15 minutes after impact injury. Our immunohistochemical results indicate that Fas and FasL are over-expressed in neurons and astrocytes at this early time point after TBI. Previous studies also suggest that increased expression of TNF-α, an antigen that belongs to the family of cell surface proteins including Fas, is overexpressed in cortical neurons within 1 hour after TBI (Knoblach et al., 1999; Shohami et al., 1994). However, we cannot exclude that other cells may have contributed to these early increases in Fas and FasL immunoreactivity in our experiments. For example, an immediate disruption of the blood-brain barrier, as seen after experimental TBI (Dietrich et al., 1994; Fukuda et al., 1995), may allow immune cells to enter the brain and may therefore contribute to the early increase of Fas and FasL immunoreactivity. In this regard, notice that Fas and FasL are expressed on blood leukocytes (Mincheff et al., 1998). Moreover, blood cells at sites of traumatically induced intracerebral hemorrhage also may have contributed to the early increase of Fas and FasL immunoreactivity after TBI in vivo.
The interaction of Fas with its cell surface ligand (FasL) may be important for induction of apoptosis (Becher et al., 1998; Nagata and Golstein, 1995). Although this study did not investigate the precise colocalization of Fas- and FasL-positive cells after TBI, immunohistochemical studies of Fas and FasL unambiguously showed that Fas and FasL are expressed in similar cortical regions after TBI. Further, our study clearly demonstrates the expression of Fas and FasL in TUNEL-positive cells in the same cortical regions after TBI, suggesting that the Fas/FasL system may play an important role in cortical apoptotic cell death after cortical impact injury. However, future studies using combined immunohistochemical study for Fas and FasL have to further elucidate the exact interactions between Fas and its ligand after acute brain injuries in vivo.
The Fas ligand can be cleaved proteolytically from the cell surface by membrane-associated metalloproteinases, giving rise to a soluble form (sFasL) of the protein (Kayagaki et al., 1995). Interestingly, sFasL may block apoptosis by inhibiting interaction between Fas and FasL on the cell surface (Cheng et al., 1994). However, sFasL also has been described to induce oligomerization of Fas on the target cell, resulting in activation of apoptosis (Matute-Bello et al., 1999; Becher et al., 1998). Although our study did not determine the precise role of soluble FasL in possible neurodegeneration or neuroprotection after TBI, our data suggest that the membrane form of FasL may be involved in apoptosis after cortical impact injury. The interaction between the membrane-bound form of FasL and Fas antigen also has been shown to play a crucial role in mediating cytotoxicity after CNS ischemia in vivo(Martin-Villalba et al., 1999) and in vitro(Vogt et al., 1998). Taken together, these results and our data suggest that Fas-carrying cells may indeed be a target of membrane-bound FasL attack after acute brain injuries in vivo. However, ongoing studies in our laboratory currently are investigating the precise role of sFasL on cellular damage or protection after TBI.
Our study failed to detect increased Fas and FasL expression in the hippocampus ipsilateral to the injury site from 15 minutes to 14 days after the trauma. This is in contrast to previous reports, where features of hippocampal apoptosis have been described after lateral FP injury (McIntosh et al., 1989)in vivo (Conti et al., 1998; Yakovlev et al., 1997). However, recent data suggest that there is no clear evidence of hippocampal cell death at early and late time points after unilateral cortical impact injury (Franz et al., 1999). Although higher magnitudes of cortical impact injury (Colicos et al., 1996; Colicos and Dash 1996) than used in our study have been reported to produce apoptotic hippocampal cell loss, the injury parameters in the current study did not result in overt apoptotic hippocampal damage (as indicated by TUNEL and Fas/FasL immunohistochemical study) at any of the time points examined. Moreover, another model of experimental TBI (i.e., central FP injury) may cause cortical neurodegeneration without evidence of hippocampal cell death (Taft et al., 1992; 1993). In addition, a report also indicates that cortical cells after cortical impact injury eventually die, whereas hippocampal cells, although probably functionally impaired as indicated by increased proteolysis, appear morphologically intact, even at later time points after lateral cortical impact injury in vivo(Newcomb et al., 1997). Also notice that the presence or absence of hippocampal apoptotic cells after cortical impact injury may be attributable to the use of different impact angles (Colicos et al., 1996).
The precise pathway for Fas/FasL signaling in the brain has not yet been determined. However, recent reports on Fas-mediated apoptosis indicate that Fas activation initially stimulates interleukin converting enzyme-like proteases and then caspase 3-like proteases (Saas et al., 1999; Cheema et al., 1999) downstream to induce the morphologic changes and DNA fragmentation typical of apoptosis. Evidence shows that activation of caspase 3-like proteases contribute to neuronal apoptosis after FP injury (Yakovlev et al., 1997) and controlled cortical impact injury in the rat (Pike et al., 1998).
In conclusion, our study demonstrates increased cortical expression of Fas and FasL after TBI in the rat. Further, our findings indicate a pivotal role of the Fas-FasL pathway in the pathologic mechanism of apoptotic neurodegeneration after cortical impact injury in vivo. Last, our results suggest that blockade of the Fas/FasL system may reduce posttraumatic apoptosis and therefore may represent a new molecular target for therapeutic intervention after TBI.
Footnotes
Abbreviations used
Acknowledgements
The authors thank Nadja Greier and Verena Pirchl for expert technical assistance.