
Abstract
Cell death–regulatory genes like caspases and bcl-2 family genes are involved in delayed cell death in the CA1 sector of hippocampus after global cerebral ischemia, but little is known about the mechanisms that trigger their expression. The authors found that expression of Fas and Fas-ligand messenger ribonucleic acid and protein was induced in vulnerable CA1 neurons at 24 and 72 hours after global ischemia. Fas-associating protein with a novel death domain (FADD) also was upregulated and immunoprecipitated and co-localized with Fas. Caspase-10 was activated and interacted with FADD protein to an increasing extent as the duration of ischemia increased. Moreover, caspase-10 co-localized with both FADD and caspase-3. These findings suggest that Fas-mediated death signaling may play an important role in signaling hippocampal neuronal death in CA1 after global cerebral ischemia.
Delayed neuronal death in the hippocampal CA1 region after transient global cerebral ischemia is associated with altered expression of proteins that also have been implicated in programmed cell death. Expression and activity of cell death–regulatory genes, such as bcl-2 (Martinou et al., 1994; Linnik et al., 1995) and caspase (Chen et al., 1998; Li et al., 2000) family genes, may determine the fate of ischemic neurons, but the identity of the upstream signaling molecules and receptors that initiate delayed ischemic neuronal death is unclear.
The cell surface receptor Fas (also termed CD95 and APO-1) is a member of the tumor necrosis factor receptor (TNFR) superfamily and is central to some mammalian programmed cell death processes. The mature human Fas antigen consists of extracellular, membrane-spanning, and cytoplasmic domains. The cysteine-rich extracellular domain is required for interaction with Fas ligand (Fas-L) (Orlinick et al., 1997) and is toxic to Fas-expressing cells (Suda and Nagata, 1994), whereas the cytoplasmic domain appears to be required for death signal transduction (Itoh and Nagata, 1993) and, therefore, has been named the “death domain” (Tartaglia et al., 1993). Fas death domain may be a protein–protein interaction motif that signals cell death by interacting with a common intracellular adapter protein (Tartaglia et al., 1993), such as Fas-associating protein with a novel “death effector domain” (DED) (FADD) (Chinnaiyan et al., 1995; Boldin et al., 1996) or Fas receptor interacting protein (Stanger et al., 1995). Therefore, DED is thought to act as an adapter domain by linking Fas and other members of the TNFR superfamily to downstream signaling pathways.
Fas-mediated cell death requires the activation of a class of cysteine proteases. Caspase-8 possesses a sequence homologous to the DED of FADD within its pro–caspase-8. Caspase-8 also contains an IL-1beta-converting enzyme–like protease domain and is capable of cleaving caspase-3 substrates (Srinivasula et al., 1996). The cleavage of pro–caspase-8 protein was observed in Fas-induced apoptosis (Chinnaiyan et al., 1996). A death-inducing signaling complex, including Fas, FADD, and caspase-8, was found using a protein–protein interaction assay (Scaffidi et al., 1999), suggesting that caspase-8 is a downstream component of Fas-mediated signaling. Caspase-10 encodes two functional DEDs as well. One of these, like caspase-8, binds to the corresponding domain in the adapter molecule FADD. Caspase-10 is recruited to both the Fas and TNFR signaling complexes in a FADD-dependent manner because an active site mutant of caspase-10 inhibits Fas and TNFR-mediated apoptosis. Caspase-10, like caspase-8, is, therefore, a proximal component in Fas and TNFR signal transduction, which can be efficiently blocked by inhibitors of caspases, CrmA and p35 (Vincenz and Dixit, 1997). Both CrmA and caspase inhibitors can block Fas-induced apoptosis (Enari et al., 1995; Genestier et al., 1998). Activation of Fas rapidly stimulates the proteolytic activity of caspase-3 (Armstrong et al., 1996). Inhibition of caspase-3–like (Schlegel et al., 1996) activity by protease inhibitors, as well as by transient expression of CrmA, substantially suppresses Fas-triggered cell death. Therefore, activation of caspase family genes is a critical event in Fas-mediated cell death (Los et al., 1995; Tewari et al., 1995).
Although Fas plays a role in immune system function, involvement of Fas in ischemic neuronal cell death is suggested by the observation that downstream targets of Fas, such as caspase-3 (Chen et al., 1998; Li et al., 2000), caspase-2 (Schulz et al., 1998), and caspase-8 (Velier et al., 1999), are activated after cerebral ischemic injury. In addition, Fas messenger ribonucleic acid (mRNA) is induced in mouse and rat brain after cerebral ischemia (Matsuyama et al., 1994; Felderhoff-Mueser et al., 2000). However, little is known about whether Fas mediates delayed neuronal death in CA1 after global ischemia, or whether Fas-mediated death signaling in neuronal and nonneuronal systems involves the same pathway. In this study, we found that Fas and Fas-L were induced in CA1 neurons destined to die at 72 hours after global ischemia, and neuronal cell death was mediated through Fas death signaling.
MATERIALS AND METHODS
Cerebral ischemic model
Transient global cerebral ischemia (15 minutes) was induced using the four-vessel method (Pulsinelli et al., 1982) with modifications described previously (Simon et al., 1991). Male Sprague-Dawley rats (300 to 330 g) were ventilated with 1.5% isoflurane in a mixture of 25% O2 and 73.5% N2 O. The left femoral artery was cannulated for blood pressure monitoring and blood gas sampling. Rectal temperature was continuously monitored and kept at 37° to 37.5°C with a heating pad. Brain temperature was monitored with a 29-gauge thermocouple implanted in the left striatum. Animals were placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, U.S.A.). Both vertebral arteries were coagulated and transected at a level between the first and second cervical vertebrae. After the external carotid arteries were ligated, global ischemia was induced by common carotid artery occlusion with microvascular clips for 15 minutes under continuous monitoring of the electroencephalograph, which became isoelectric in all rats studied within 5 seconds of common carotid artery occlusion. At various times after the completion of ischemia, the animals were killed.
Northern blot analysis
Northern blots were performed as described previously (Jin et al., 1996). Total RNA isolated from hippocampus of sham-operated and ischemic brains (n = 4 per experimental condition) was separated by electrophoresis on a 1.2% agarose-formaldehyde gel (20 μg per per lane) and transferred onto a Hybond-N membrane (Amersham Pharmacia Biotech, Inc., Piscataway, NJ, U.S.A.) using the capillary method in 10 × SSC (1 × SSC is 0.15 mol/L NaCl and 15 mmol/L sodium citrate). The RNA was ultraviolet cross-linked to the filter using a Stratalinker device (Stratagene, La Jolla, CA, U.S.A.). The Fas and Fas-L probes were designed from corresponding rat sequences (Fas antisense: 5′-GCCCACTTGATATAACCCTTCTGAGCAGTTGTTGTCGG-3′, 211–174. GenBank accession number: D26112; Fas-L antisense: 5′-GCAGTAATTCAACTTCTTCTCCTCCATTAGCACCAGATCC-3′, 763–724. GenBank accession number: U03470) and were labeled with terminal deoxyribonucleotidyl transferase using 3′-end labeling kit (NEN Life Science Products, Inc., Boston, MA, U.S.A.). Prehybridization was performed in a mixture containing 50% formamide, 5 × SSC, 1 × Denhardt's solution (100 × Denhardt's solution is 2% polyvinylpyrrolidone, 2% bovine serum albumin, and 2% Ficoll 400), 0.1% sodium dodecyl sulfate (SDS), and 100 mg/mL of salmon sperm deoxyribonucleic acid (DNA) at 42°C for 2 hours. Hybridization was carried out for 16 hours at 42°C in the same buffer with32P-labeled oligonucleotide probe at 2 × 106cpm/mL. The membrane was washed for 15 minutes at room temperature in 1 × SSC/0.1% SDS, and three times for 15 minutes at 60°C in 0.1 × SSC/1% SDS. Autoradiographic study was performed overnight at −80°C with an intensifying screen.
In situ hybridization
Frozen sections from sham-operated and ischemic brains (n = 4 per experimental condition) were pretreated with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 minutes and 1 μg/mL of proteinase K for 10 minutes, and washed with PBS. Fas or Fas-L antisense probes (same as earlier) and sense probes were labeled using terminal deoxyribonucleotidyl transferase. In situ hybridization was performed as previously described (Jin et al., 1999a). Hybridization was performed for 18 hours at 42°C using hybridization solution consisting of 50% formamide, 5 × SSC, 2 × Denhardt's solution, 10% dextran sulfate, 100 mmol/L dithiothreitol, 10 mg/mL of salmon sperm DNA, 5% sarcosyl, and35S-labeled antisense or antisense probe at 1 × 107cpm/mL. After hybridization, the slides were washed twice for 10 minutes in 2 × SSC, treated with 20 μg/mL RNase A for 30 minutes at room temperature, then washed twice for 10 minutes in 1 × SSC, once for 2 hours at 55°C in 0.1 × SSC, and twice for 10 minutes in 0.5 × SSC at room temperature. The sections were dehydrated, dried, and autoradiographed on Kodak x-ray film. For cellular localization of signal, slides were dipped in NTB-2 emulsion, exposed for 5 weeks at 4°C, developed, and counterstained with cresyl violet.
Immunprecipitation and Western blot
Cell lysates were extracted from hippocampus of sham-operated or ischemic brains (n = 6 per experimental condition) using lysis buffer (0.1 mol/L NaCl, 0.01 mol/L Tris-HCl, pH 7.6, 1 mmol/L ethylenediaminetetraacetic acid, pH 8.0, 1 μg/mL of aprotinin, and 100 μg/mL of phenylmethanesulfonyl fluoride). Proteinconcentration was measured by Bio-Rad protein assay (Bio-Rad, Hercules, CA, U.S.A.). For immunoprecipitation, protein was incubated with primary monoclonal or polyclonal antibodies for 2 hours at 4°C and then with protein-A/G agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) for an additional 1 hour at 4°C. After washing three times with lysis buffer and dissolving in loading buffer, the immunocomplex or protein (50 μg per sample) was separated by 12% SDS–polyacrylamide gel electrophoresis under reducing conditions, and then transferred to polyvinylidene difluoride membranes (Bio-Rad). Membranes were incubated with the following primary antibodies: (1) affinity-purified rabbit polyclonal antibody against amino acids 308 to 327 mapping at the carboxy terminus of Fas of mouse origin; (2) affinity-purified rabbit polyclonal antibody against amino acids 225 to 244 mapping at the carboxy terminus of Fas-L of rat origin; (3) mouse monoclonal antibody against amino acids 354 to 373 mapping at the carboxy terminus of caspase-8 p20 of human origin and reacting with the p20-subunit and pro–caspase-8; (4) affinity-purified goat polyclonal antibody against amino acids 187 to 205 mapping at the carboxy terminus of FADD of mouse origin; (5) affinity-purified goat polyclonal antibody against amino acids 220 to 350 mapping within an internal region of caspase-10 of human region, and reacting caspase-10 p20 and pro–caspase-10 (all from Santa Cruz Biotechnology); or (6) affinity-purified rabbit polyclonal antibody produced by immunizing rabbits with a synthetic peptide corresponding to residues surrounding the cleavage site of human caspase-3, and detecting only the larger fragment of activated caspase-3 (BioLabs, Beverly, MA, U.S.A.). Secondary goat anti-mouse (for primary mouse monoclonal) or anti-rabbit (for primary rabbit polyclonal), or pig anti-goat (for primary goat polyclonal) antibodies conjugated with horseradish peroxidase (Jackson ImmunoResearch, West Grove, PA, U.S.A.) were used to visualize immunoreactive proteins using NEN Western blot chemiluminescence reagents (NEN Life Science Products, Inc., Boston, MA, U.S.A.). Controls for nonspecific binding included incubation in the absence of the primary antibody and preabsorption with peptide antigen. Differences in protein expression on Western blots were quantified by a GS-710 Calibrated Imaging densitometer and Quantity One software (Bio-Rad Laboratories).
Immunocytochemistry
Sham-operated or ischemic brains (n = 4 per experimental condition) were perfused with 0.9% saline followed by 4% paraformaldehyde in PBS (pH 7.4) and embedded in paraffin. The paraffin-embedded sections (6-μm thick) were deparaffinized with xylene and rehydrated with ethanol. Immunocytochemical study was performed as described previously (Jin et al., 1996). Briefly, after blocking endogenous peroxidase activity by incubation with 1% H2 O2 in PBS for 15 minutes, sections were incubated with blocking buffer (2% horse serum, 0.2% Triton X-100, and 0.1% bovine serum albumin in PBS) for 1 hour at room temperature. The sections were incubated overnight at 4°C with affinity-purified rabbit polyclonal antibody against Fas or Fas-L, diluted 1:50, followed by washing in PBS. The second antibody was a biotinylated goat anti-rabbit IgG (Vectastain Elite ABC, Vector Co., Burlingame, CA, U.S.A.) diluted 1:200. Sections were processed with a Vector ABC kit (Vector Co.) according to the manufacturer's instructions. The horseradish peroxidase reaction was detected by incubation with diaminobenzidine (0.05% in PBS) and H2 O2 (0.03%). Alternating sections were incubated without primary antibody as a control.
Triple-fluorescence labeling
Sections were deparaffinized with xylene, rehydrated with ethanol, and treated with unmasking solution (Vector Co., Burlingame, CA, U.S.A.). They then were incubated with blocking buffer for 1 hour at room temperature, and subsequently with a mixture of two primary antibodies raised in different species, at 4°C overnight, at a dilution of 1:50 to 100 in blocking buffer. After incubation with the primary antibodies as described earlier, sections were washed three times for 10 minutes each in PBS and then incubated with secondary antibodies. The secondary antibodies were as follows: fluorescein isothiocyanate–conjugated goat anti-rabbit or anti-mouse IgG diluted 1:200 (Vector Co.) or pig anti-goat IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, U.S.A.), and rhodamine (TRITC)–conjugated rat-absorbed donkey anti-mouse or anti-rabbit IgG diluted 1:200 (Jackson ImmunoResearch Laboratories, Inc.). DAPI (Vector Co.) was used to counterstain nuclei. The three fluorescence signals were detected with a Nikon E800 epifluorescence microscope (Nikon Inc, Melville, NY, U.S.A.) using excitation/emission wavelengths of 535/565 nm for rhodamine (red), 470/505 for fluorescein isothiocyanate (green), and 360/400 for DAPI (blue). Results were recorded with a Magnifire digital color camera (ChipCoolers Inc., Warwick, RI, U.S.A.).
Statistical analysis
All values were expressed as the mean plus/minus standard error of the mean. Differences among means were analyzed using one-way analysis of variance (ANOVA). When ANOVA showed significant differences, pair-wise comparisons between means were tested by the Scheffe post-hoc test. A level of P < 0.05 was considered statistically significant.
RESULTS
Induction of Fas in CA1 neurons after ischemia
Because caspases are activated and expression of bcl-2 family genes is altered after cerebral ischemia with reperfusion, we examined whether Fas, which may initiate these changes in a variety of cell types, is induced after ischemic injury. Northern blotting was performed using an oligodeoxynucleotide probe (40-mer) designed from the rat Fas sequence. As shown in Fig. 1A, a specific hybridization band of the predicted size was detected in the ischemic sample, but little Fas mRNA was detected in sham-operated brain, suggesting that Fas mRNA is induced after ischemic injury. The cellular distribution of the Fas mRNA was further examined using in situ hybridization in sham-operated brain and in ischemic brain at 8, 24,and 72 hours of reperfusion. Consistent with the findings on Northern blots, expression of Fas mRNA was barely detectable in the sham-operated hippocampus (Fig. 1B, panel a). However, at 24 and 72 hours after ischemia, Fas mRNA was prominently induced in the hippocampal CA1 neurons, which are destined to die in this model (Fig. 1B, panel b). Only background signal was detected using a Fas sense probe (Fig. 1B, panel c).
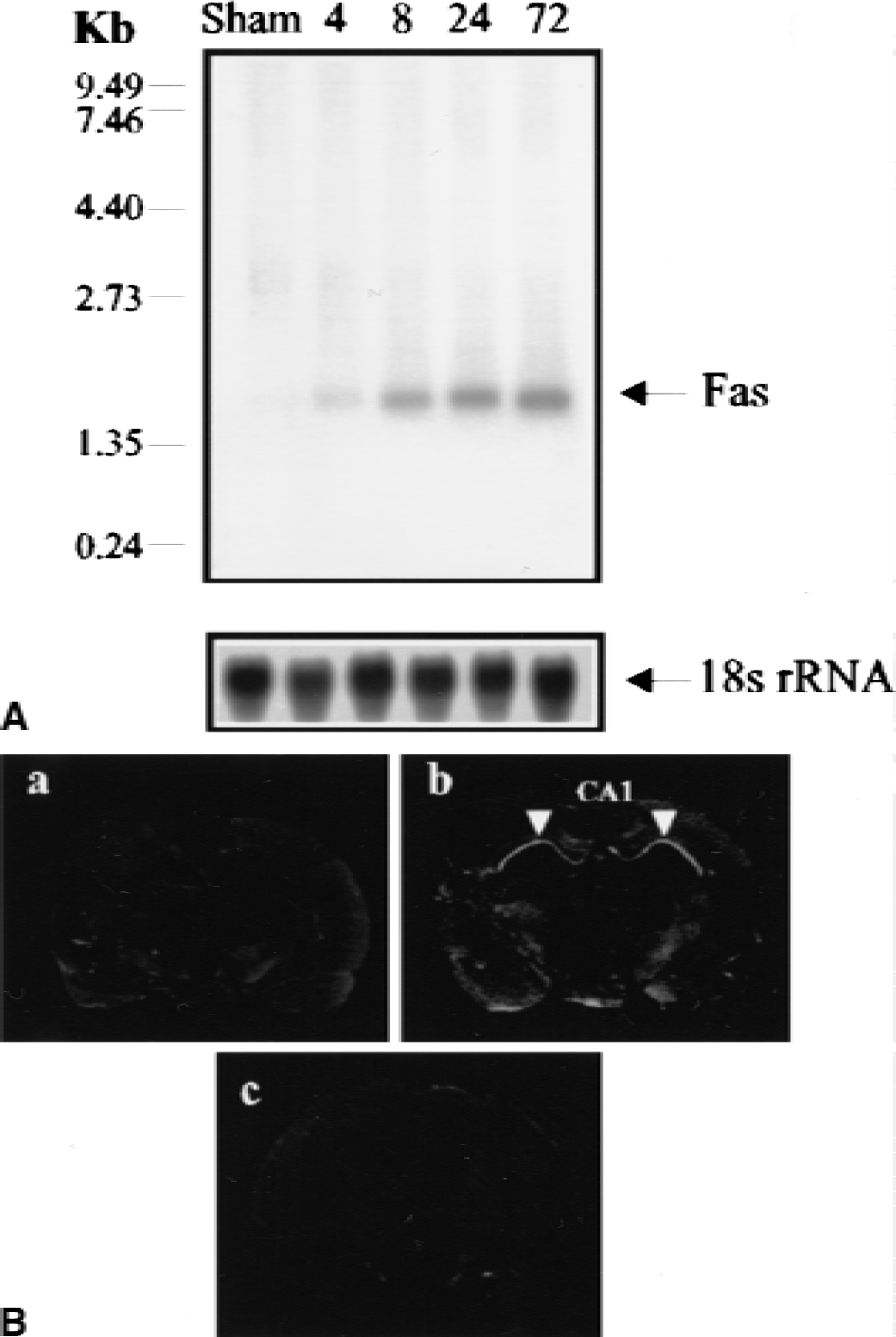
Expression and distribution of Fas messenger ribonucleic acid (mRNA) in the hippocampus after transient global ischemia.
Western blotting with an affinity-purified polyclonal antibody against Fas was performed next to examine whether Fas protein also was induced after ischemia. As shown in Figs. 2A and 2B, expression of Fas protein was increased in ischemic hippocampus, with highest levels of expression at 24 and 72 hours.
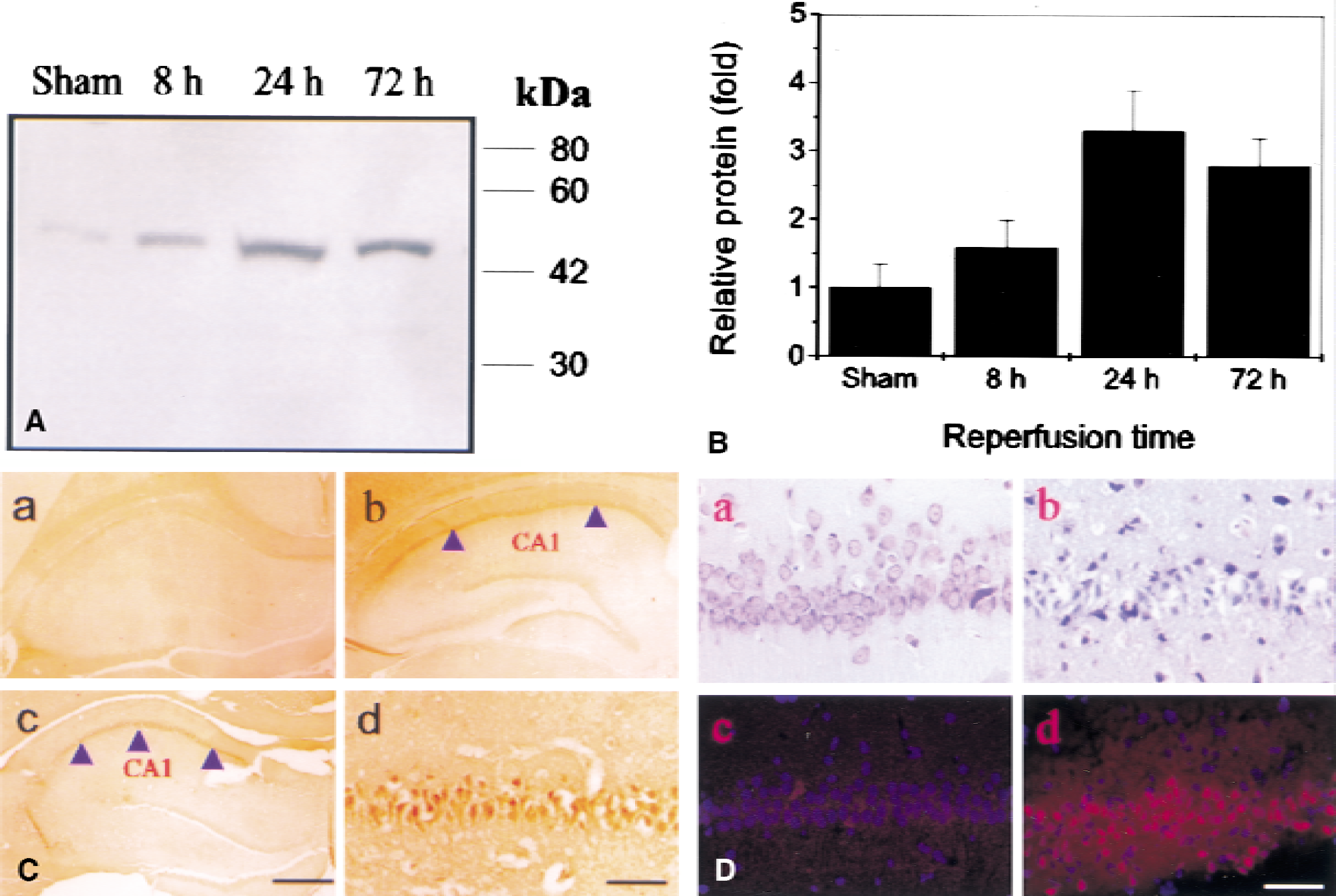
Expression and distribution of Fas protein in the hippocampus after transient global ischemia.
Immunohistochemical study was performed to examine the cellular distribution of Fas protein. Expression was barely detectable in neurons of sham-operated hippocampus (Fig. 2C, panel a). However, Fas immunoreactivity was significantly induced in CA1 at 24 and 72 hours after ischemia (Fig. 2C, panels b to d), in keeping with the results of in situ hybridization. The DNA damage staining (Klenow labeling assay) showed that damage only appeared in CA1 neurons at 72 hours, in keeping with our previous results (Jin et al., 1999b) and indicating that Fas protein expression increases in CA1 neurons before cell death (Fig. 2D). Some increase in the expression of Fas protein also is detectable in CA3 and dentate gyrus and in layers 4 and 5 of cerebral cortex at 24 hours of reperfusion compared with control.
Induction of Fas-ligand is induced in CA1 neurons after global ischemia
Receptor–ligand interactions are believed to be critical in regulating Fas-mediated cell death, suggesting that Fas activation in vivo may involve interaction with Fas-L. To determine whether Fas-L mRNA also was induced after global ischemia with reperfusion, Northern blots were performed using an oligodeoxynucleotide probe designed from the rat Fas-L sequence. A single band at about 1.6 kb was barely evident in control and more prominent in ischemic hippocampal samples (Fig. 3A). In situ hybridization using Fas-L antisense as a probe was performed for analysis of the cellular distribution of Fas-L mRNA. The Fas-L mRNA barely was detected in the normal hippocampus (Fig. 3B, panel a) but markedly induced in the CA1 region at 72 hours after global ischemia (Fig. 3B, panel b). Examination of emulsion-coated slides revealed that the silver grains occurred primarily over pyramidal neurons.
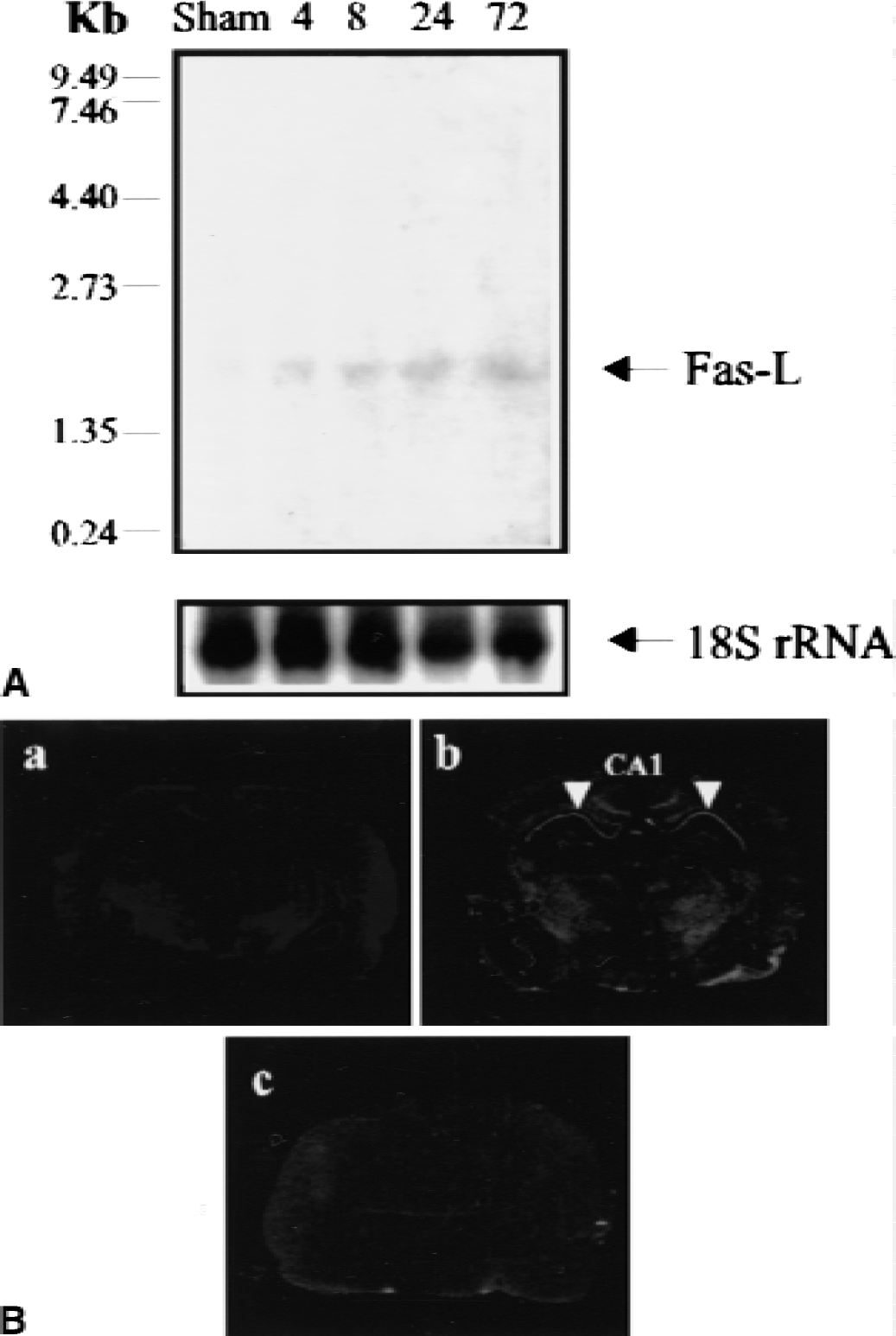
Expression and distribution of Fas-ligand (Fas-L) messenger ribonucleic acid (mRNA) in the hippocampus after transient global ischemia.
Western blotting was performed using an affinity-purified antibody against Fas-L. As shown in Figs. 4A and 4B, Fas-L protein was expressed in both sham-operated and ischemic brains, but its expression was increased in the hippocampus after ischemia. Immunocytochemical staining using the same antibody showed that Fas-L immunoreactivity was significantly induced in hippocampal CA1 neurons at 72 hours after ischemia (Fig. 4C, panels c and d). This is similar to the expression pattern of Fas mRNA and protein, implying that Fas and Fas-L are co-localized and may, therefore, interact to promote delayed neuronal death in the CA1 sector of hippocampus after transient global ischemia.
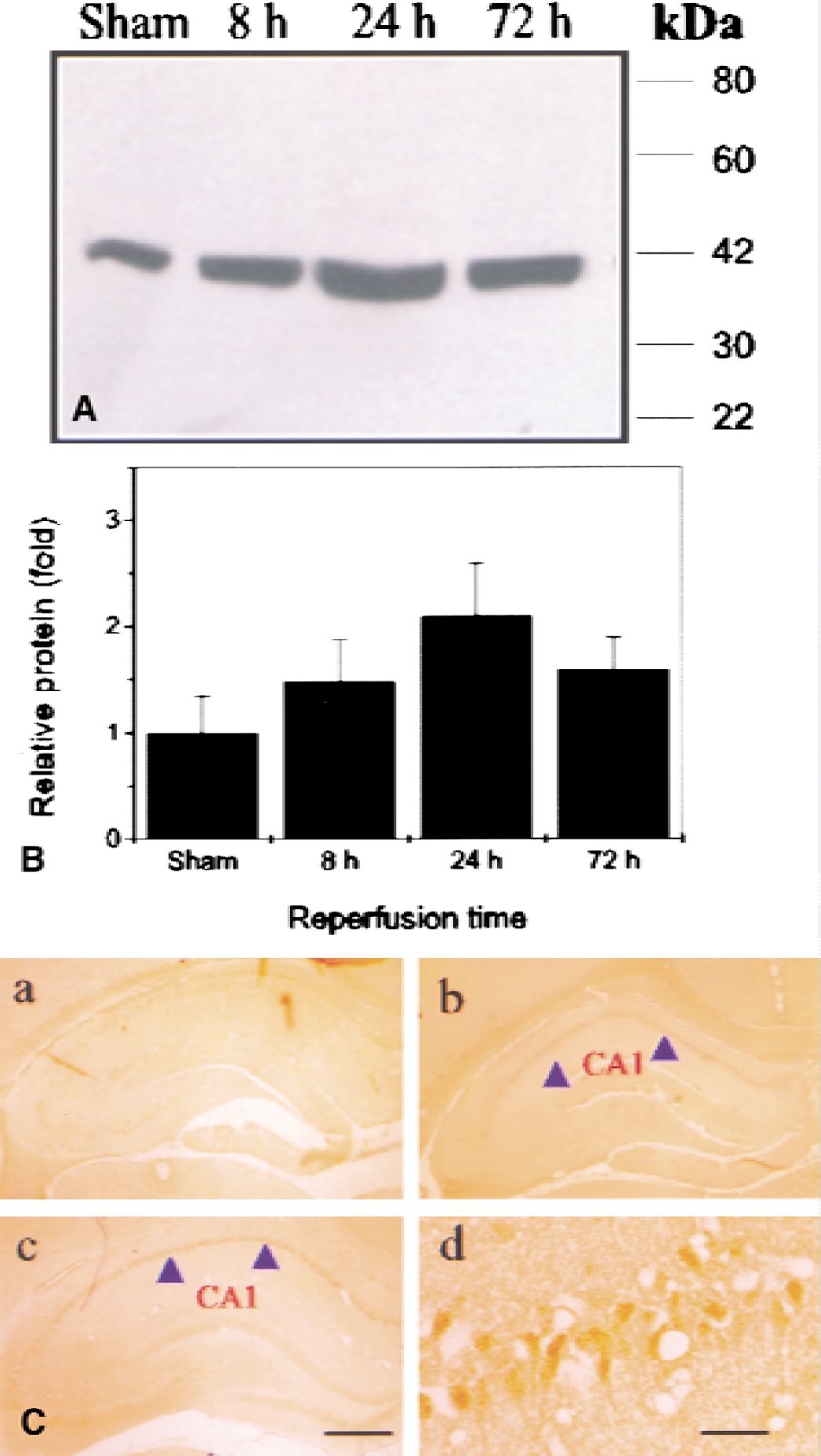
Expression and distribution of Fas-ligand (Fas-L) protein in the hippocampus after transient global ischemia.
Expression of FADD after global ischemia
The FADD molecule is regarded as a linker, or signal adapter, between Fas and the downstream components of Fas-mediated death signaling. To test whether FADD also was involved in Fas-mediated neuronal death after ischemic cerebral injury, Western blots were performed using an antibody against FADD. As shown in Figs. 5A and 5C, FADD protein was detectable in sham-operated hippocampus, and expression was increased at 8, 24, and 72 hours of reperfusion. Moreover, FADD co-immunoprecipitated with Fas (Figs. 5B and 5C), suggesting that the FADD protein interacts with Fas. Triple-fluorescence labeling showed that Fas was co-localized with FADD in cells in the CA1 sector at 72 hours (Fig. 5D), suggesting that FADD is a direct downstream target of Fas signaling.
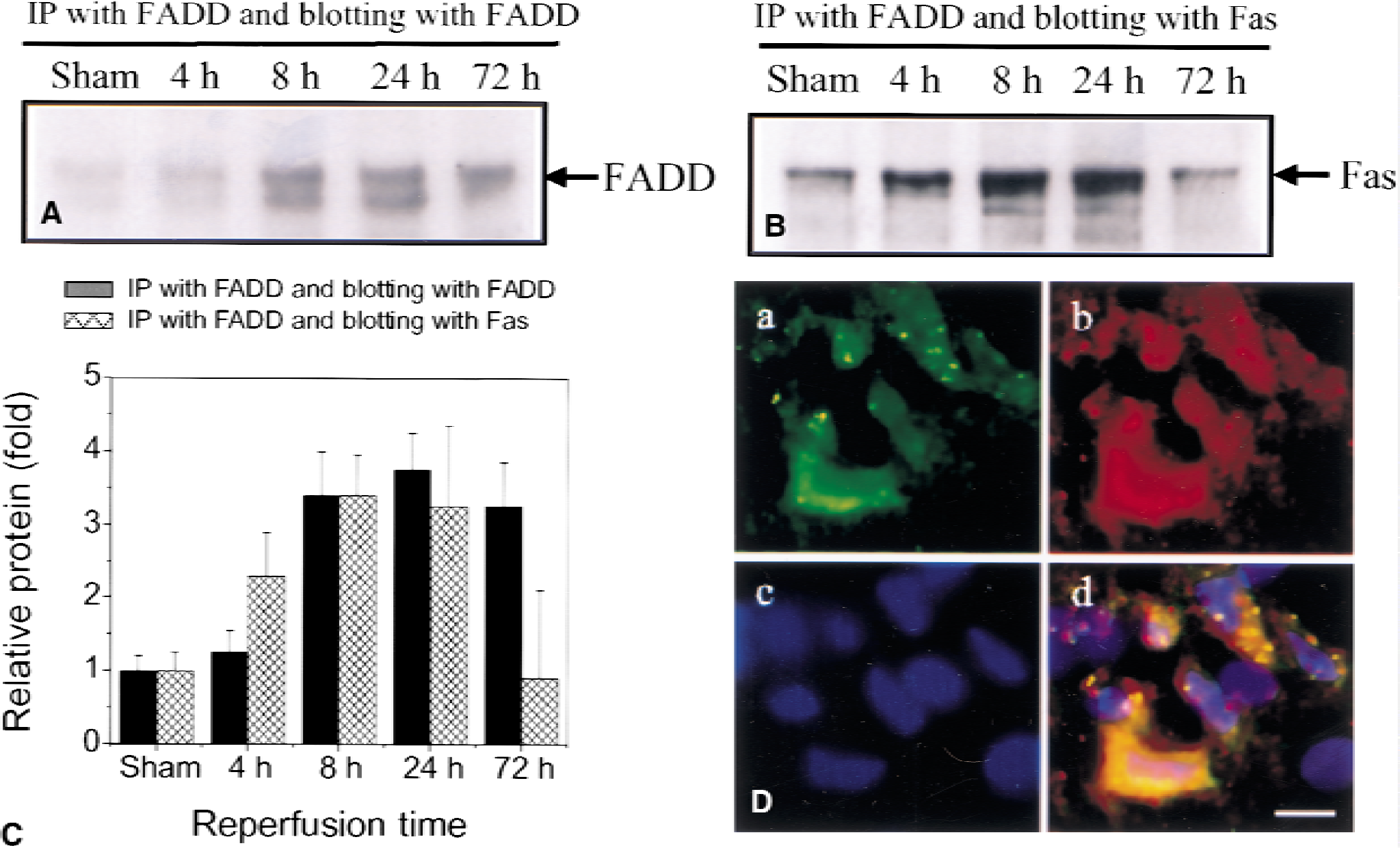
Expression of Fas-associating protein with a novel death domain (FADD) and its interaction with Fas.
Caspase-8 and caspase-10 activation after ischemia
Caspase-8 appears to be a proximal downstream target of Fas in many cell-death programs. Therefore, caspase-8 protein expression was analyzed by Western blotting using a monoclonal antibody against amino acids 354 to 373, mapping at the C-terminus of caspase-8 p20 of human origin. Accordingly, the antibody reacted with the p20-subunit and pro–caspase-8 and not with caspase-10 (Fig. 6A). As shown in Fig. 6B, pro–caspase-8 protein was detectable in normal hippocampus, but alteration of its expression was not detectable after ischemic injury. In addition, the cleavage form of caspase-8 was barely detectable after ischemia. Caspase-8 interacted slightly with FADD by co-immunoprecipitation analysis, and the protein–protein interaction was decreased with increasing reperfusion time (Figs. 6D and 6E).
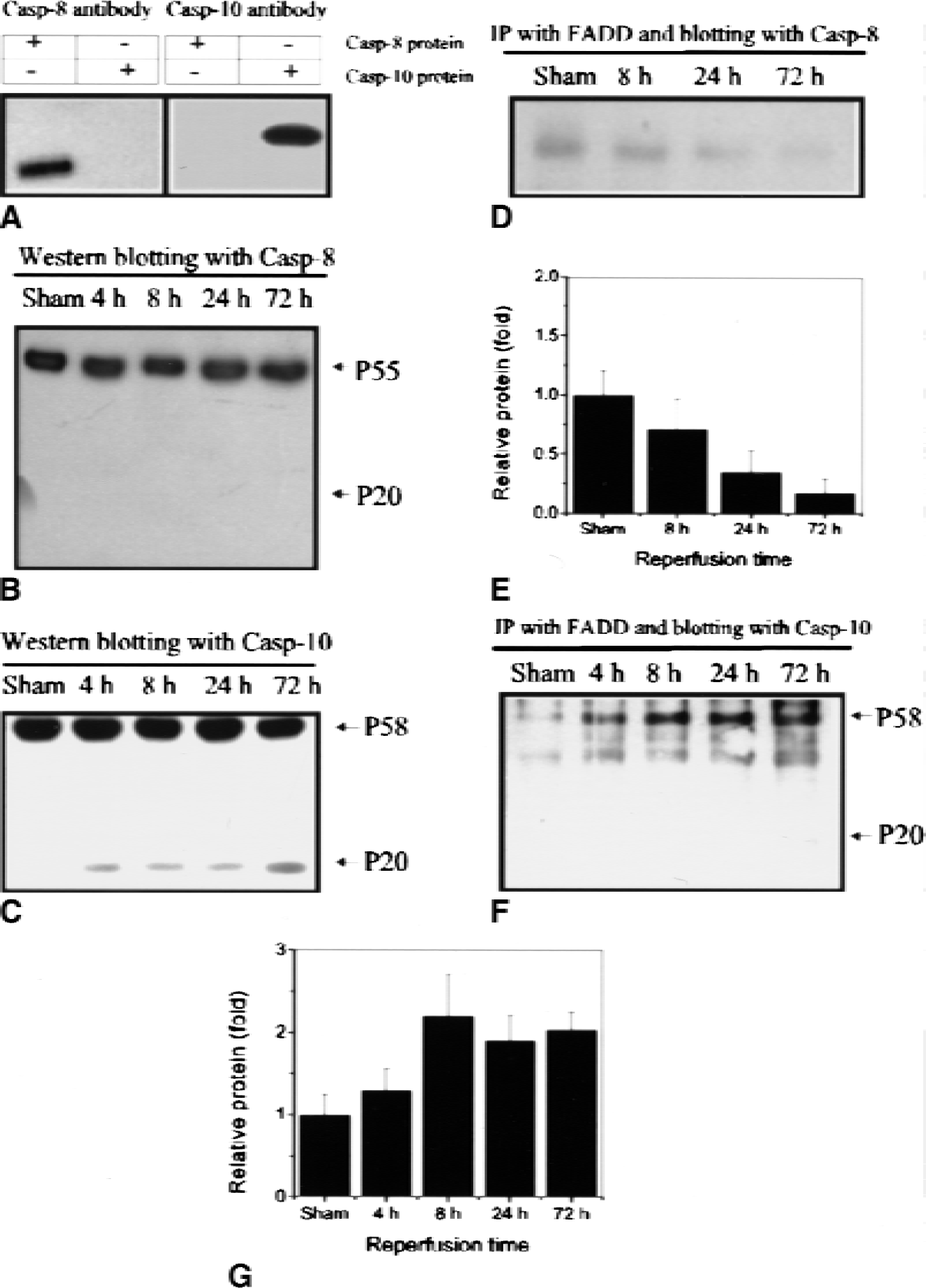
Expression and activation of caspase-8 and caspase-10 after ischemia.
Caspase-10, a close structural homologue of caspase-8, also encodes two functional death-effector domains. One of these, like that of caspase-8, binds to the corresponding domain in the adapter molecule FADD, and in turn activates the Fas-mediated cell-death cascade (Boldin et al., 1996). Therefore, we investigated whether caspase-10, rather than caspase-8, might be activated after ischemia. Western blotting was performed using an affinity-purified polyclonal antibody against caspase-10, which does not cross-react with caspase-8 (Fig. 6A). Pro–caspase-10 was detected at about 58 kd in sham-operated and ischemic hippocampus, and a cleaved p20 subunit was detectable in ischemic hippocampus beginning at 4 hours and increasing over 72 hours of reperfusion; these findings indicate that caspase-10 was activated after cerebral ischemia (Fig. 6C). Moreover, caspase-10 co-immunoprecipitated with FADD; this association increased with increasing duration of reperfusion (Figs. 6F and 6G), and caspase-10 co-localized with FADD in CA1 neuronal cells at 24 and 72 hours after ischemia by immunohistochemical reaction (Fig. 7A). These findings suggest that caspase-10 may function as a direct downstream target of FADD in Fas-mediated ischemic neuronal cell death. Finally, caspase-10 was co-localized with caspase-3 in vulnerable CA1 hippocampal neurons after ischemia (Fig. 7B). By analysis of immunocytochemical findings, Fas protein is co-localized with the neuronal marker NeuN (Fig. 7C) but not the astrocyte marker GFAP, suggesting that Fas is expressed mainly in neuronal cells. The molecular interactions that may be involved in signaling of Fas-mediated neuronal cell death after cerebral ischemia are shown in Fig. 7E.
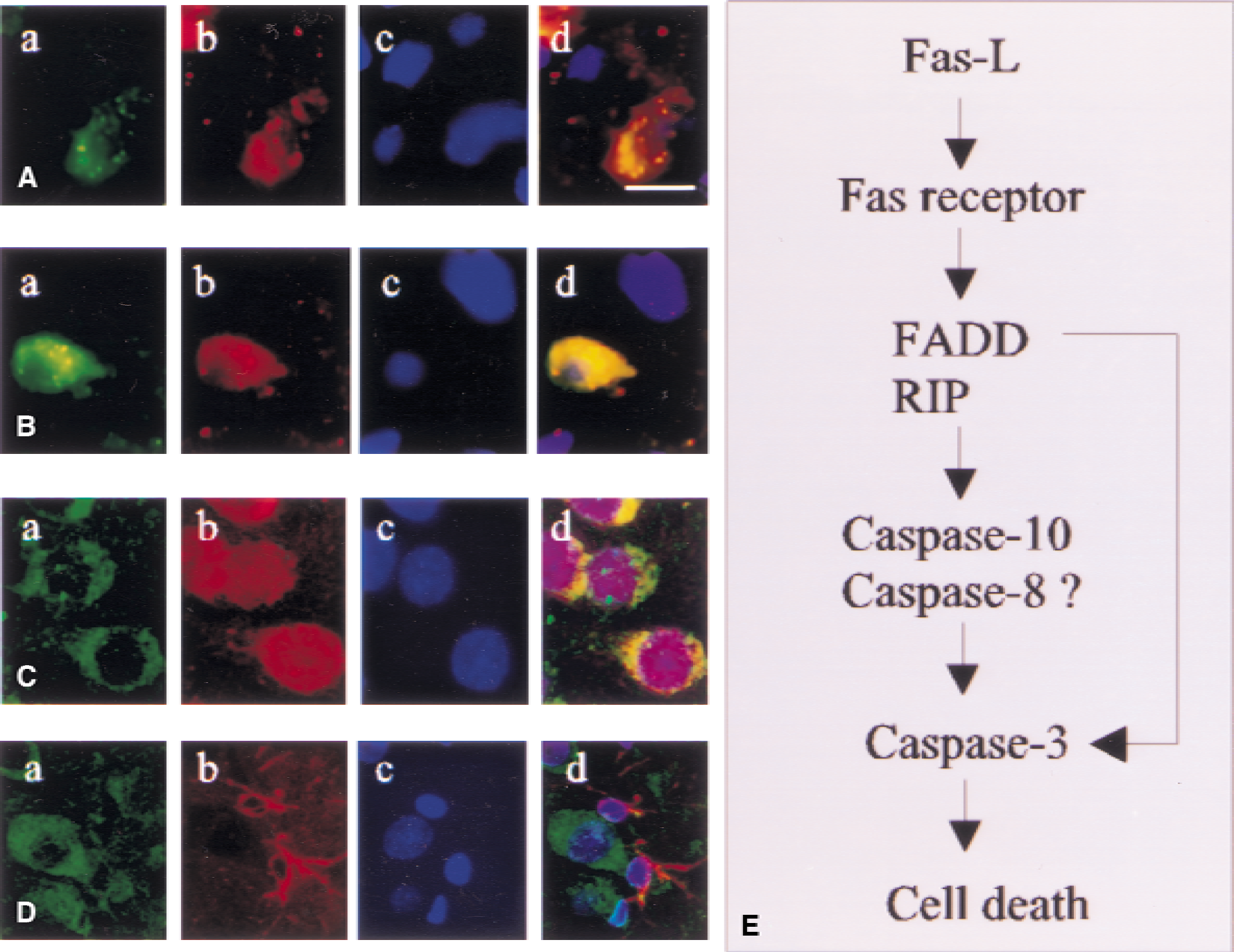
Co-localization of caspase-10 with Fas-associating protein with a novel death domain (FADD) and caspase-3 after ischemia.
DISCUSSION
In this study, we found ischemic induction of Fas and Fas-L mRNA and protein in vulnerable neurons of the hippocampal CA1 region, which are injured and eventually die by 72 hours after 15 minutes of global ischemia. The Fas adapter protein, FADD, also was upregulated after ischemia and co-localized with Fas protein by immunohistochemical reaction. In addition, immunoprecipitation experiments showed that FADD interacts with Fas, suggesting that it links Fas and downstream targets in Fas-mediated neuronal cell death signaling. Caspase-10, a proximal component in the Fas-mediated death pathway in many systems, was activated after ischemia, interacted with FADD by immunoprecipitation, and was localized to the same cells as FADD and caspase-3. These findings suggest that the Fas system may play an important role in the cascade of programmed cell death after global cerebral ischemia.
The Fas–Fas-L system has been implicated in the pathogenesis of immune disorders in humans, including lymphoproliferative syndrome, viral hepatitis, immunodeficiency syndrome, acute graft-versus-host disease, Hashimoto's thyroiditis, and autoimmune diabetes. Fas–Fas-L also has been shown to be involved in neurodegenerative disorders, such as Parkinson's disease (Mogi et al., 1996) and Alzheimer's disease (Nishimura et al., 1995; de la Monte et al., 1997). Involvement of Fas and Fas-L in cell death after focal ischemia is supported by the reduction of infarct volumes in lymphoproliferation mutant (lpr) mice expressing dysfunctional Fas-L compared with wild-type animals (Martin-Villalba et al., 1999). Our finding of Fas–Fas-L mRNA and protein induction in ischemic neurons suggests that the Fas–Fas-L system may mediate the processing of delayed cell death in CA1 sector of hippocampus after global ischemia and supports the hypothesis that delayed neuronal cell death requires de novo protein synthesis (Goto et al., 1990). The induction of Fas–Fas-L mRNA and protein was confirmed in primary neuronal cell culture after hypoxia with glucose deprivation (unpublished data). The mechanism of Fas–Fas-L induction in ischemic neuronal cells after ischemia is unknown. Reactive oxygen intermediates, which are increased after cerebral ischemia, may function as inducers of Fas–Fas-L. Oxidative stress induced by bleomycin or H2 O2 can trigger rapid induction of Fas and Fas-L mRNA and protein (Vogt et al., 1998), which can be blocked by the antioxidants thiol N-acetyl- l -cysteine (Roederer et al., 1990), deferoxamine (Hug et al., 1997), and intracellular glutathione (Um et al., 1996). Reactive oxygen intermediates also can enhance expression of Fas-L through the transcription factor NF-κB (Vogt et al., 1998), which has consensus elements in the Fas-L promoter (Harwood et al., 2000). Fas, in turn, induces NF-κB activation (Cheema et al., 1999) through FADD, caspase-8, and caspase-8-related protein (Casper) (Hu et al., 2000). Whether NF-κB ultimately exerts a pro-apoptotic or anti-apoptotic effect may be determined by the nature of the death stimulus rather than the cell type (Kuhnel et al., 2000). The observation that Fas expression was increased after ischemia not only in the valuable CA1 region of hippocampus, but also, to some extent, in CA3, dentate gyrus, and cerebral cortex, indicates that additional factors, besides the Fas–Fas-L system, contribute to determine the fate of ischemic neuronal cells.
Activated Fas associates with several signaling components including FADD, which is thought to act as an adapter protein by linking Fas to downstream signaling pathways in many systems (Chinnaiyan et al., 1995; Boldin et al., 1996). The FADD mutations confer complete resistance to Fas-induced cell death (Juo et al., 1999), which is consistent with our finding that FADD protein is upregulated in hippocampus after ischemia and interacts physically with Fas protein. Moreover, the extent of protein interaction was increased with increasing duration of reperfusion. In addition to FADD, several other proteins, such as Fas receptor interacting protein and Fas death domain-associate protein (Torii et al., 1999), also contain a DED, but it remains unclear whether these proteins function as key Fas adapters in cerebral ischemia (Stanger et al., 1995). In some cases, FADD and Fas receptor interacting protein might function as a combined signaling unit (Baker and Reddy, 1996).
Caspase-8 promotes Fas-mediated cell death in many apoptotic systems through protein–protein interactions and has protease activity toward most known caspases (Boldin et al., 1996; Muzio et al., 1996; Scaffidi et al., 1999). Our results indicate that alteration of caspase-8 expression is not detectable after cerebral ischemia, and interaction between caspase-8 with FADD decreased after cerebral ischemia. The observation that caspase-8 and caspase-3 are expressed in different populations of cortical neuronal cells undergoing delayed cell death after ischemia suggests that Fas-mediated neuronal cell death signaling after cerebral ischemia is independent of caspase-8 activation (Velier et al., 1999). However, it also is possible that caspase-8 activity might be sufficient to assist in the apoptotic death of some neurons in CA1 sector, despite caspase-8 levels being below the limits of detection. Therefore, whether caspase-8 plays a role in the Fas-mediated signaling of CA1 sector after ischemia remains to be explored. Caspase-10, a close structural homologue of caspase-8, also contains a DED. Caspase-10 is recruited to both the Fas and TNFR signaling complexes in a FADD-dependent manner because an active-site mutation of caspase-10 inhibits Fas and TNFR-mediated apoptosis. Our results indicate that caspase-10 protease is activated, caspase-10 protein interacts with FADD protein by immunoprecipitation analysis, and this interaction increases with increasing duration of reperfusion. Caspase-10 also is co-localized with FADD protein in CA1 neurons at 24 and 72 hours after ischemia, suggesting that caspase-10 may be a direct downstream target of FADD. However, it still is unclear whether binding of caspase-10 to FADD is sufficient for autoactivation because other factors can regulate Fas-induced programmed cell death.
In addition to activation of caspase-10 and co-expression of caspase-10 and FADD, our results also show that caspase-10 is co-localized with caspase-3 in CA1 neuronal cells destined to die after ischemia. Caspase-3 activation has been implicated as a critical event in Fas-mediated cell death (Los et al., 1995; Tewari et al., 1995). For example, Fas-mediated apoptosis in human carcinoma cell lines was inhibited by a caspase-3-like inhibitor (Hasegawa et al., 1996). We found previously that caspase-3–like activity was increased in CA1 after ischemia, and its role in delayed cell death was clarified further by inhibiting activation of caspase-3 in vivo, which significantly reduced neuronal death in CA1 after ischemia (Chen et al., 1998). Taken together, these findings suggest that caspase-3 may function as a distal downstream component of Fas-induced neuronal cell death signaling after cerebral ischemia.
Overall, our findings suggest that ischemic cell death in hippocampal CA1 sector may involve a cascade of Fas-mediated signaling in which ischemia induces Fas-L, Fas-L binds Fas receptor to induce its oligomerization, and the intracellular death domain of Fas protein interacts with FADD. In turn, FADD activates caspase-10 through the DED of FADD. Caspase-10 subsequently may activate executioner caspases such as caspase-3, either directly or indirectly, and eventually produce neuronal cell death. Our data do not prove that activation of the Fas–Fas-L pathway causes ischemic neuronal death, but they do show a temporal pattern of events that is consistent with such causation. Thus, Fas mRNA and protein, Fas-L mRNA and protein, FADD, and Fas–FADD interaction all increase beginning 4 to 8 hours after ischemia and reach maxima by 8 to 24 hours. By comparison, a more downstream signaling event—caspase-10 cleavage—continues to increase for at least 72 hours, at which time morphologic evidence of neuronal death and DNA damage first become apparent.