
Abstract
Direct injury of the brain is followed by inflammatory responses regulated by cytokines and chemoattractants secreted from resident glia and invading cells of the peripheral immune system. In contrast, after remote lesion of the central nervous system, exemplified here by peripheral transection or crush of the facial and hypoglossal nerve, the locally observed inflammatory activation is most likely triggered by the damaged cells themselves, that is, the injured neurons. The authors investigated the expression of the chemoattractants monocyte chemoattractant protein MCP-1, regulation on activation normal T-cell expressed and secreted (RANTES), and interferon-gamma inducible protein IP10 after peripheral nerve lesion of the facial and hypoglossal nuclei. In situ hybridization and immunohistochemistry revealed an induction of neuronal MCP-1 expression within 6 hours postoperation, reaching a peak at 3 days and remaining up-regulated for up to 6 weeks. MCP-1 expression was almost exclusively confined to neurons but was also present on a few scattered glial cells. The authors found no alterations in the level of expression and cellular distribution of RANTES or IP10, which were both confined to neurons. Protein expression of the MCP-1 receptor CCR2 did not change. MCP-1, expressed by astrocytes and activated microglia, has been shown to be crucial for monocytic, or T-cell chemoattraction, or both. Accordingly, expression of MCP-1 by neurons and its corresponding receptor in microglia suggests that this chemokine is involved in neuron and microglia interaction.
Direct as well as remote injury of the brain leads to substantial changes in the interaction of injured neurons and glial cells. In the former lesion type, regularly immune cells from the periphery are involved. Recent studies examined possible molecular mechanisms that might lead to the activation of glial cells during the inflammatory response (Feuerstein et al., 1998; Ghirnkar et al., 1998). Thus, there has been a focus on cytokines and chemokines, which are known to play a crucial role in regulation of migratory behavior and activation of immune cells (Liblau et al., 1995, Matsushima et al., 1989; Yoshimura et al., 1989; Allavena et al., 1994; Carr et al., 1994; Maghazachi et al., 1994; Karpus et al., 1998). Most of these studies investigated direct lesions of the brain and spinal cord tissue, which lead to a disruption of the blood–brain barrier. In these models, a plethora of chemokines are known to be produced by resident glial cells and infiltrating immune cells (Feuerstein et al., 1998; Ghirnikar et al., 1998). Astrocytes and microglia/macrophages have been considered to represent the main source for the observed chemokine release (Kim et al., 1995; Glabinski et al., 1996; Gourmala et al., 1997; Peterson et al., 1997; Grzybicki et al., 1998; Simpson et al., 1998). The participation of neuronal cells in immune-regulatory processes during central nervous system (CNS) pathology remains unclear. Whereas most studies ascribe neurons a passive role in inflammatory processes of the brain, there has been a recent report regarding neuronal IP-10 production after brain ischemia (Wang et al., 1998). Furthermore, under certain conditions neurons have been shown to express MHC molecules and to interact with T lymphocytes (Neumann et al., 1995; Flügel et al., 2000).
After peripheral injury of the facial or hypoglossal nerve, the blood–brain barrier remains intact and, at least in mice, a few blood-derived leukocytes traffic into the nuclei of the lesioned nerves (Raivich et al., 1998). Remote CNS injury triggers a cascade of morphologic and metabolic processes involving all cell types within the affected brain stem nucleus. The response of microglial cells toward the injury is one of the best-characterized processes (Kreutzberg, 1996; Blinzinger and Kreutzberg, 1968).
Although the microglial activation process is well characterized, the cellular origin of the external trigger is not known. In the case of a remote CNS lesion, neurons are damaged outside the CNS and hence, are the only injured cells. Initial signals for the activation of glial cells surrounding the injured neuronal cell bodies can therefore be assumed to originate from neurons. Yet only a few studies describe signaling molecules, secreted by the neurons, which may be involved in the direct interaction of microglia with neurons or in microglial attraction to the neuronal cell bodies (Harrison et al., 1998; Nishiyori et al., 1998). The main candidates are chemokines, which are released in the injured brain area. Indeed, different chemokines, including RANTES, MCP-1, and IP10, have been described to be involved in the recruitment of peripheral immune cells into the CNS in various injury models or experimental acute encephalitis (Karpus and Ransohoff, 1998; Kennedy et al., 1998).
In the current study, the authors investigated changes in the expression of chemokines that may be involved in neuron and glial interactions (Bolin et al., 1998; Wang et al., 1998). The authors report on the rapid up-regulation of neuronal MCP-1 after nerve injury. In contrast, the authors could not detect any regulation of IP10 and RANTES expression. Western blot analysis of the MCP-1 receptor, CCR2, revealed its constitutive expression in the facial nucleus, which did not change after axotomy.
MATERIALS AND METHODS
Animals and surgical procedures
The right facial or hypoglossal nerve of adult male Wistar rats (weighing approximately 250 g) was transected. Three rats were killed for each survival period (3 hours, 6 hours, 1, 3, 7, 14, 28, 42, and 84 days after surgery). Brains were removed and immediately frozen. Three normal (nonoperated) animals were used as controls.
In situ hybridization
Polymerase chain reaction (PCR) primers were constructed specific for rat MCP-1, IP10, and RANTES. DNA fragments were amplified by PCR and cloned into the pZErO vector (Invitrogen, Groningen, The Netherlands). For in vitro transcription, the cloned probes were amplified with PCR using primers specific for pZErO anchored with either the T7 or Sp6 RNA-polymerase recognition sequence (Schwaiger et al., 2000). One to two nanograms of plasmid was amplified with PCR for 25 cycles with denaturation at 94°C for 10 seconds, annealing at 64°C for 10 seconds, and elongation at 74°C for 20 seconds. The PCR product was purified from primers and salt using the QuiaQuick-kit (Quiagen, Hilden, Germany). Transcription using either T7 or Sp6 RNA-polymerase was performed according to the manufacturers instructions (Boehringer Mannheim, Mannheim, Germany) using 30μCi 35S-αSUTP (Amersham Pharmacia, Freiburg, Germany) and 250 ng of the respective PCR product as template. In situ hybridization (ISH) was performed with sense and anti-sense probe. The sense probes did not reveal a specific signal (data not shown). Dried cryostat sections (20 μm) were fixed for 20 minutes in 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (PBS) and washed 3 times for 5 minutes in PBS. In situ hybridization was performed as described previously (Schmitt et al., 1999). Sections were dehydrated, air-dried, covered with film emulsion (NBT-2, Kodak, Stuttgart, Germany), and exposed for 5 to 10 days.
The percentage of MCP-1 expressing facial motoneurons was determined in sections treated with ISH and Hemalum counterstaining by counting the number of ISH+ and ISH–neurons in an average of six complete facial nucleus sections including all levels of the facial nucleus of three independent animals for each time point.
Immunohistochemistry
Animals were perfused with 4% paraformaldehyde in PBS. After removal from the skull, brains were incubated in the same fixative at 4°C for 24 hours followed by cryoprotection in 15% sucrose solution in PBS overnight. For immunohistochemical staining, cryostat sections (20 μm) of the brain stem were fixed for 30 minutes in 4% paraformaldehyde/PBS. Sections were incubated with polyclonal rabbit-anti-MCP-1 antibody (Biozol, Germany) for 1 hour at room temperature (1:2000 dilution). Binding of primary antibodies was evaluated using a biotinylated goat anti-rabbit serum preabsorbed with heat-inactivated normal serum from Lewis rat strain 30 minutes before use. Avidin-biotin-peroxidase (Vectastain ABC-Kit; Vector Laboratories, Burlingame, CA, U.S.A.) served as the detection system with 3, 3'-diaminobenzidine as an enzymatic substrate. Preabsorption of the primary anti-MCP-1 antibody with recombinant MCP-1 (Biosource, Nivelles, Belgium) for 4 hours abolished specific MCP-1 stain. Alternatively, monoclonal hamster anti-MCP-1 (Pharmingen, San Diego, CA, U.S.A.) (1:500 dilution) with biotinylated goat anti-hamster antiserum and Vectastain ABC-Kit as the detection system were used. For double staining of neurons with anti-MCP-1 and anti-MAP-2 (Sigma, Munich, Germany), the sections were stained with rabbit anti-MCP-1 antibody as described above. As detection antibody, a donkey–anti-rabbit antiserum carrying Cy3 as fluorescent tag (Dianova, Hamburg, Germany, dilution: 1:500) was used. Sections then were incubated with goat anti-MAP-2 antibody (dilution 1:250, Sigma, Munich, Germany). Detection of these primaries antibodies was performed using DTAF-labeled mouse anti-goat secondary antibody (Dianova, Hamburg, Germany).
Western blot analysis
Axotomized and control facial nuclei were punched out at −20°C and immediately homogenized in 80 μL ice-cold lysis buffer (50 mmol/L Hepes, pH 7.5, 150 mmol/L NaCl, 1.5 mmol/L MgCl2, 5 mmol/L EGTA, 10% Glycerin, 1% Triton, 1 mmol/L Na3VO4, 10 μg/mL aprotinine, 10 μg/mL leupeptine, 1 mmol/L phenylmethylsulfonyl) by sonication. Lysates then were centrifuged at 12,000x g for 10 minutes at 4°C and the supernatants were transferred to new tubes. Samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by immunoblotting. For Western blotting, 20 μg protein lysate was loaded per lane and resolved by 7.5% SDS-PAGE, transferred onto Immobilon membrane (polyvinylidene difluoride, Millipore, Eschborn, Germany), and immunostained with a polyclonal antibody directed against CCR2 (Santa Cruz, Heidelberg, Germany). Detection was performed by incubating the membrane using appropriate secondary anti-rabbit antibodies coupled to horseradish peroxidase (DAKO, Carpinteria, CA, U.S.A.), followed by addition of ECL (Amersham, Arlington Heights, IL, U.S.A.). Blots were exposed to x-ray films for 1 to 10 minutes.
RESULTS
mRNA regulation for MCP-1, RANTES, and IP10 in response to peripheral nerve injury
Transection of the hypoglossal and facial nerves was used as a model for remote brain injury. In situ hybridizations with sense and antisense probes for the chemokines MCP-1, RANTES, and IP-10 of the brain stem and cerebellum were performed at different time intervals after peripheral nerve cut. In detail, the examined time intervals ranged from 3 hours to 84 days after facial nerve transection and from 3 days to 7 days after hypoglossal nerve transection. A baseline expression of RANTES and IP10 was found in neuronal cells of both nuclei (Fig. 1, facial N., hypoglossal N.) as well as in the brainstem and cerebellum. In contrast, no specific signal for MCP-1 could be detected in uninjured intact brain.
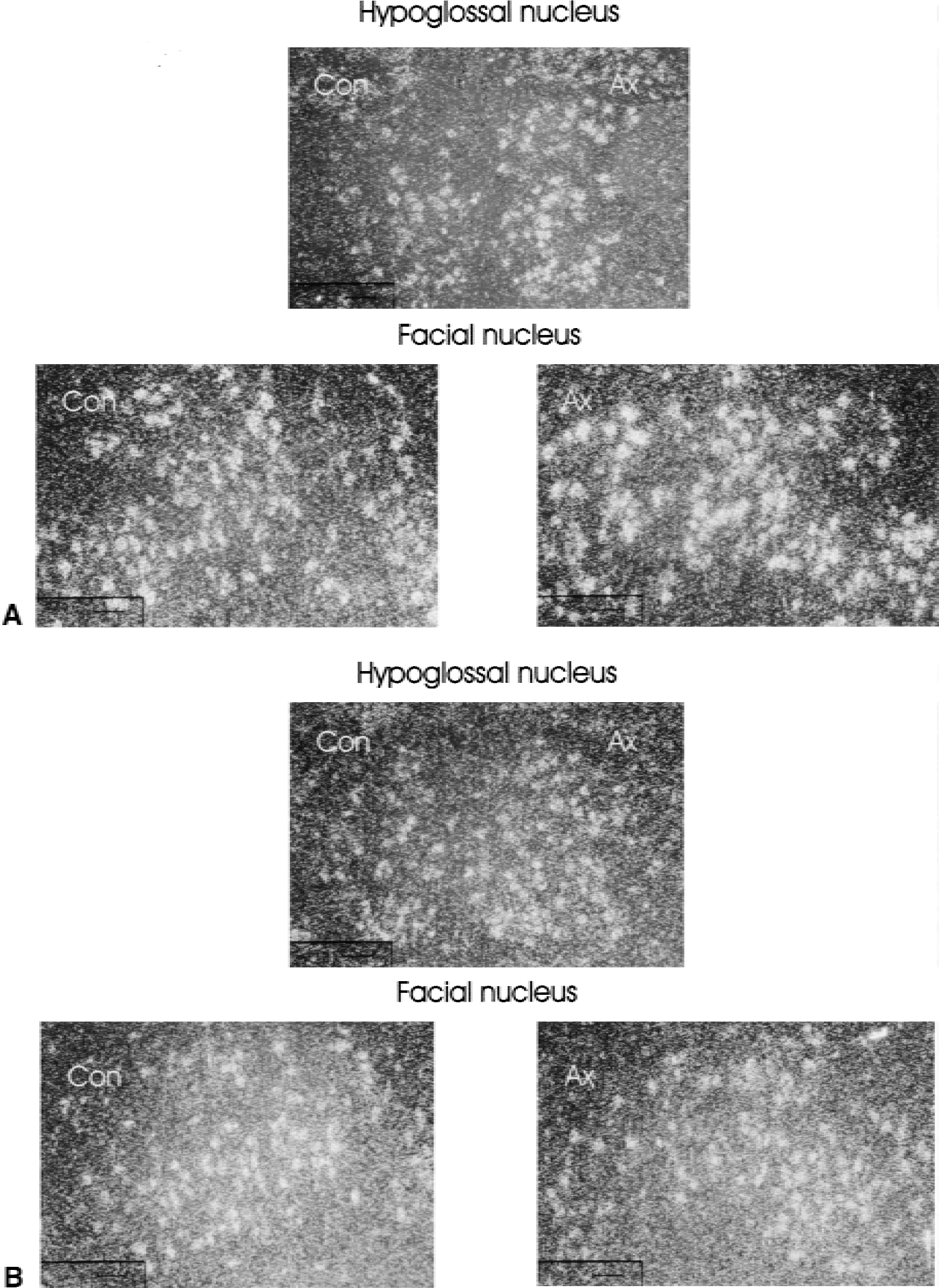
In situ hybridization with probes for IP10
Beginning 6 hours after peripheral facial nerve transection, autoradiographic silver grains indicating the presence of MCP-1 mRNA accumulated in neurons of the corresponding nuclei (Fig. 2). This positive signal was confined to subpopulations of neurons. Quantification of MCP-1–positive neurons in the facial nucleus revealed a maximum of positive cells at day 3 postaxotomy (83%, Fig. 3). At day 84, MCP-1–positive cells were no longer detectable (Fig. 3). Up-regulation for MCP-1 was also observed after hypoglossal nerve transection analyzed at day 3 and day 7 (data not shown). Similar results for neuronal MCP-1 expression were obtained for the facial nerve after nerve crush. Up-regulation of neuronal MCP-1 was not species specific. Thus, axotomized mouse facial nerve nuclei showed a similar MCP-1 expression kinetic (data not shown). MCP-1 expression after remote lesion was almost restricted to the lesioned neurons. Only few glial cells were occasionally found to express MCP-1 at later time points beginning at 14 days postaxotomy (Fig. 2).
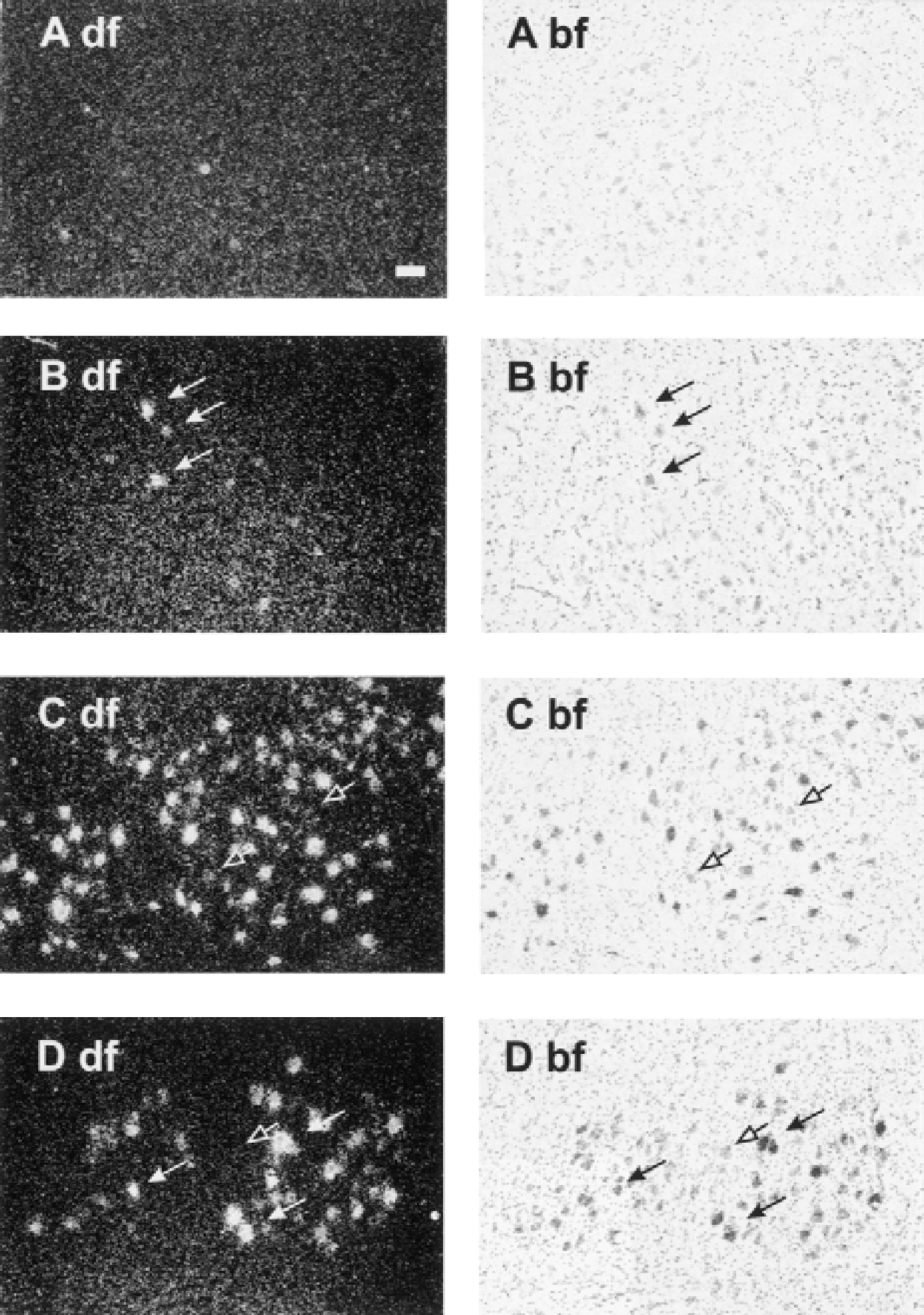
Time course of the expression of MCP-1 mRNA during the first week in response to facial nerve axotomy. Note that there is no detectable MCP-1 expression in the uninjured brain areas. Even 6 hours after axotomy some neurons start to get strongly positive for MCP-1. Left side (df): dark field; right side (bf): bright field (
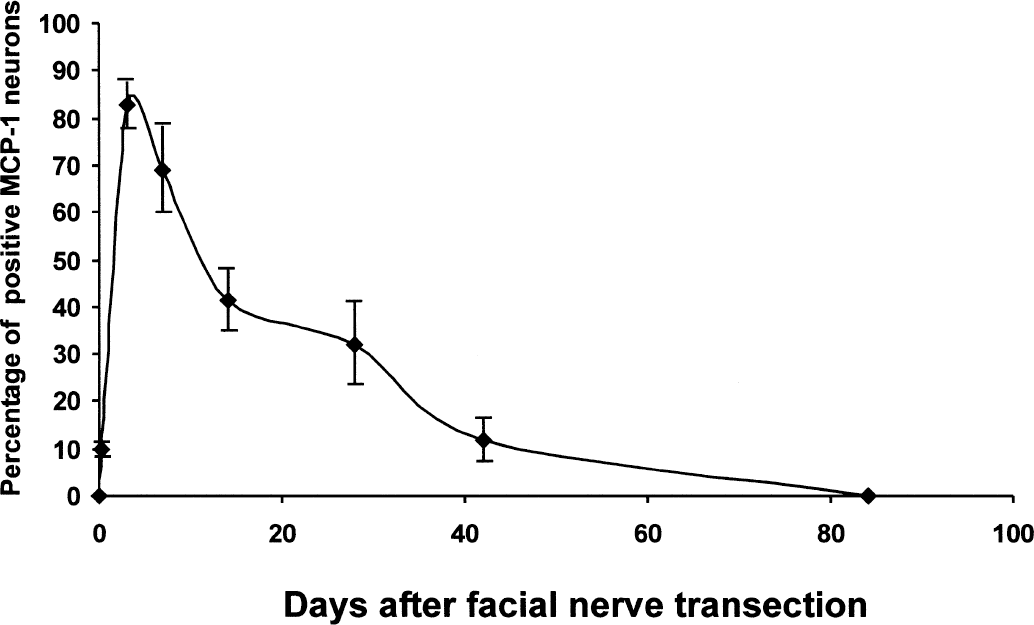
Percentage of MCP-1 expressing neurons 6 hours, 3, 7, 14, 21, 42, and 84 days after facial nerve transection. For each time point, six brain slices of three animals each have been examined. Ten percent of all motoneurons were positive for MCP-1 after 6 hours. After 84 days, no cellular signal for MCP-1 was detectable. Data represent means and SD of 18 analyzed samples per time point.
The expression pattern as well as the intensity of signals obtained with RANTES or IP10 antisense probes in the injured area of the facial nucleus remained unchanged during the regeneration period in comparison with the contralateral side (data not shown). Controls using sense probes were negative for all investigated chemokines (data not shown).
Immunohistochemistry for MCP-1 in response to facial nerve axotomy
Facial nuclei were stained with anti-MCP-1 antibody three days after axotomy. Immunoreactivity for MCP-1 was confined to motoneurons (Fig. 4), which confirmed the data obtained by ISH experiments. The contralateral nucleus showed no specific staining with anti-MCP-1 antibodies. Similar to the ISH, immunohistochemistry marked only subpopulations of neurons of the lesioned nucleus (Fig. 4A). This signal could be blocked after preabsorption of the primary antibody with MCP-1 protein (data not shown). The emergence of neurons labeled for MCP-1 protein in response to nerve axotomy resembled the time course of the expression found for mRNA. Colocalization of MCP-1 with MAP2 confirmed the axotomized motoneurons as cellular source of MCP-1 expression (Fig. 4B).
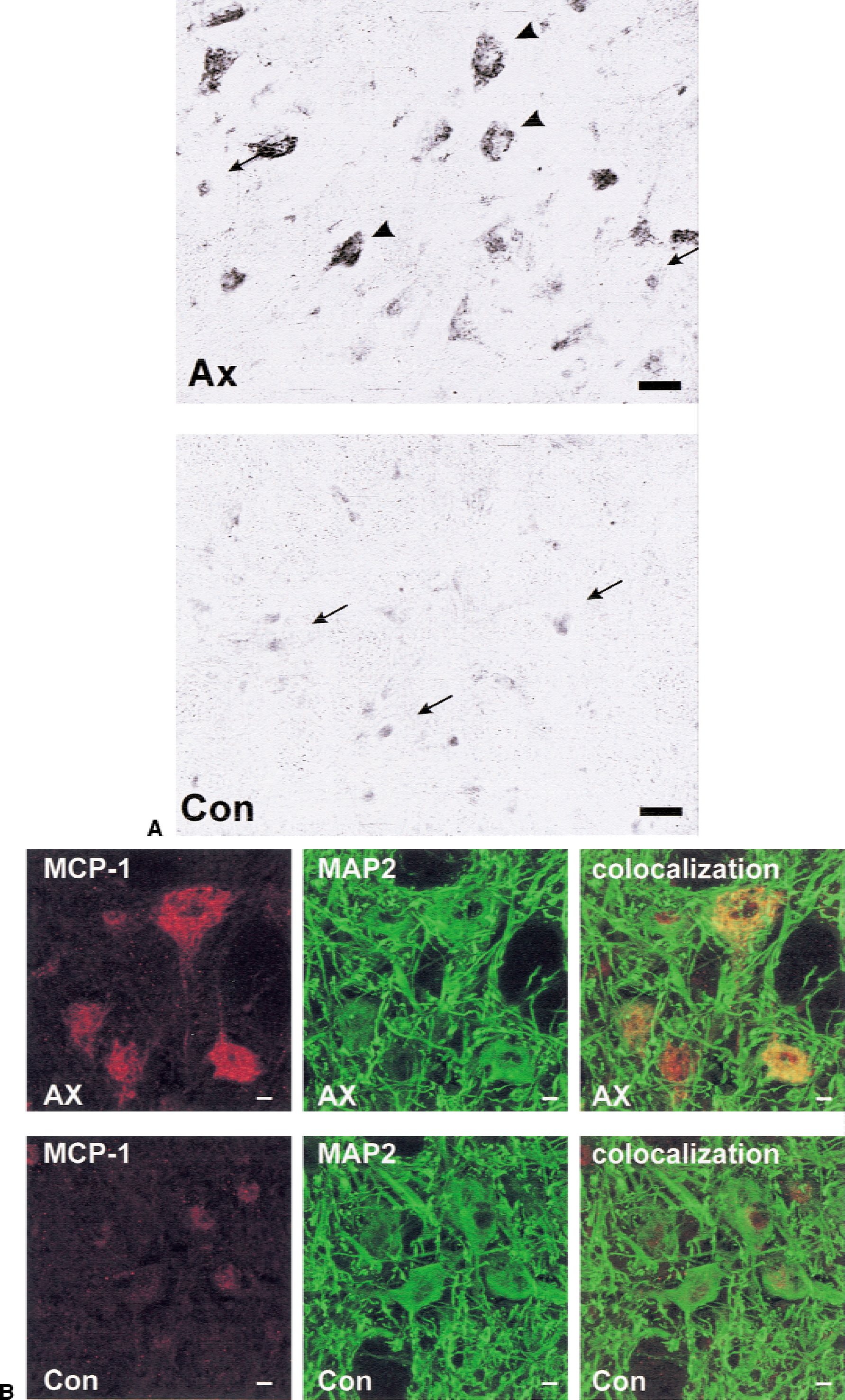
MCP-1 immunoreactivity in the facial nucleus three days after operation on the axotomized (Ax) and the uninjured control side (Con) using an anti-MCP-1 polyclonal antibody
Western blot analysis for CCR2 in response to facial nerve axotomy
To investigate if the MCP-1 receptor, CCR2, was as equally regulated as its ligand, the authors analyzed specific mRNA and protein of the isolated facial nerve nucleus from 6 hours to 84 days after remote facial nerve axotomy. The RT-PCR for the CCR2 mRNA revealed only a low level expression in the facial nucleus (data not shown). CCR2 protein was detected by Western blot analysis (Fig. 5). No significant difference in CCR2 protein expression could be found at all time points investigated.
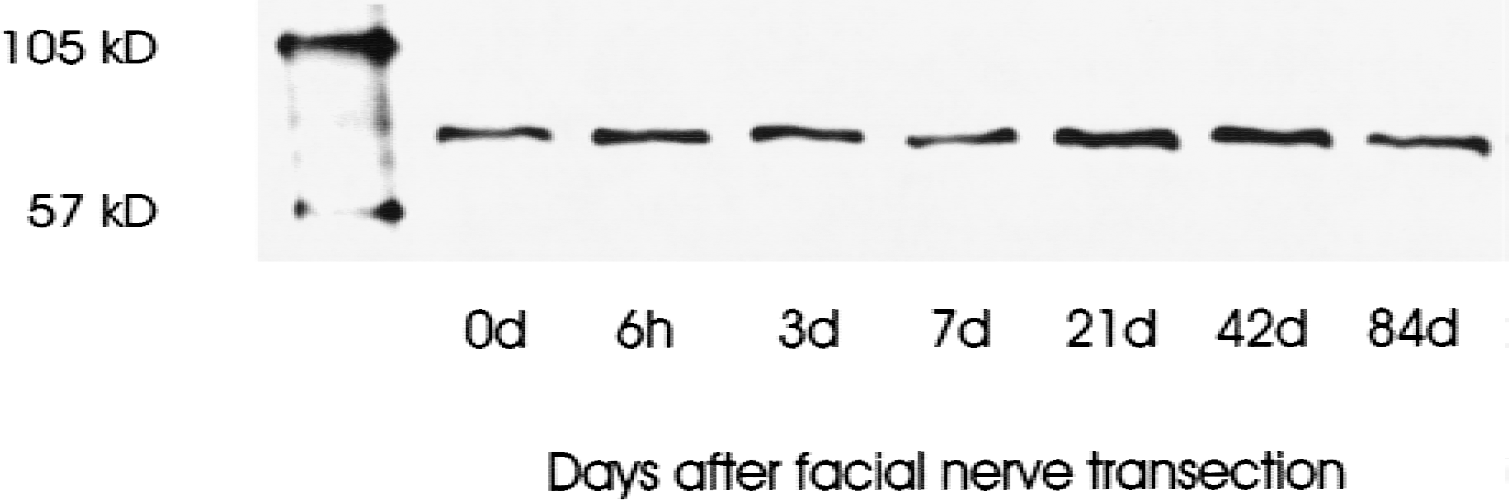
Time course for the expression of the MCP-1 receptor CCR2 in the facial nucleus by Western blot analysis 0 to 84 days after nerve transection using an anti-CCR2 polyclonal antibody.
DISCUSSION
In a number of brain injury models, microglia and T-cells seem to be attracted by neurons (Troost et al., 1989). The authors could demonstrate that the potent chemoattractant, MCP-1, is produced by neurons beginning within 6 hours after peripheral nerve axotomy. MCP-1 expression peaked at day 3 postoperation and up-regulation could be observed for up to several weeks after the operation. The expression of the receptor for MCP-1, CCR2, was not affected by axotomy. In contrast to MCP-1, IP10 and RANTES, two chemokines that are known to be expressed by or to act upon neurons (Karpus and Ransohoff, 1998; Kennedy et al., 1998), did not change their mRNA level in response to injury. Interestingly, the authors did not find any evidence for RANTES or IP10 protein expression or up-regulation in immunohistochemical or Western blot analyses (data not shown), indicating translational regulation of these proteins in neurons. These data argue against an active role of neuronal RANTES and IP10 expression in the healthy and injured CNS.
MCP-1 expression has been described in a number of different models, including cerebral cortical lesions (Grzybicki et al., 1998; Hausmann et al., 1998), multiple sclerosis (Simpson et al., 1998), and ischemia (Gourmala et al., 1997). Early expression of this chemokine in response to brain injury was reported to be confined to astrocytes, whereas, depending on the lesion model, MCP-1 in macrophage/microglial response might be up-regulated at later time points (Gourmala et al., 1997). The current findings show only a limited number of glial cells, preferentially located in the periphery of the facial nucleus, to become transiently positive for MCP-1. After mild remote lesions, such as a nerve crush, neurons seem to be the major source of MCP-1 expression. Although neuronal expression in response to brain injury was also shown for IP-10 in a model of ischemia, it is restricted to the first 6 to 12 hours after artery occlusion (Wang et al., 1998). In the peripheral nerve injury models, IFNγ was not induced nor was STAT1 tyrosine-phosphorylated. As the IFNγ signal transduction pathway is involved in IP10 expression (Ohmori and Hamilton, 1995), this may explain the missing up-regulation of IP10. Such differences in neuronal chemokine expression may represent an early response of injured neurons and may help immune cells to distinguish between different types of brain lesion.
Recently, chemokine expression studies in the CNS gave new impulses for the examination of direct neuron–microglia interactions in vivo. The CX3C chemokine fractalkine was found to be constitutively expressed by neurons (Harrison et al., 1998). Axotomy of the facial nerve in the periphery led to down-regulation of fractalkine mRNA in facial motor neurons. A presumably secreted, smaller form of fractalkine was detected in Western blot analysis of the axotomized, but not of the intact facial nucleus. In addition, the fractalkine receptor CX3CR1 was shown to be up-regulated in perineuronal microglia in response to nerve transection. This was interpreted as a neuronal signal for microglial cells leading to their activation (Harrison et al., 1998). The onset for this signaling was found to be later than 1 day postaxotomy. MCP-1 expression precedes microglial CX3CR1 expression after peripheral injury and therefore may be the primary chemotactic signal produced by neurons. The early appearance of MCP-1 expression, 6 hours after axotomy, supports the idea that this response is not because of an indirect stimulus of surrounding glial cells but rather a direct effect of the axonal lesion. The peak expression of neuronal MCP-1 coincides with the peak of microglial activation and the time course of MCP-1 expression almost parallels the presence of activated microglia in the regenerating facial nucleus (Schwaiger et al., 1998). In contrast with the regulation of the CX3CR1/fractalkine system, MCP-1 attraction after peripheral nerve axotomy appears to be controlled by secretion of the ligand.
Microglia, as well as blood-derived monocytes/macrophages, memory T-cells, and natural killer cells, has been shown to be attracted by MCP-1 (Matsushima et al., 1989; Yoshimura et al., 1989; Allavena et al., 1994; Carr et al., 1994; Maghazachi et al., 1994). Indeed, MCP-1 and its receptor have been demonstrated to be involved in the pathogenesis of inflammatory brain disease (Ransohoff et al., 1993; Berman et al., 1996; Karpus and Ransohoff, 1998; Kennedy et al., 1998). The authors' observations of the long-lasting expression (up to 42 days postlesion) suggests that MCP-1 expression is likely to influence other inflammatory processes apart from the microglial response. The plateau phase between day 14 and day 28 may contribute to the recruitment of T-cells into the CNS, which is usually found in mice at approximately day 10 after nerve axotomy (Raivich et al., 1998). As neuronal MCP-1 expression can also be observed in mice, MCP-1 secretion therefore may constitute a general adaptation of the injured neurons to recruit immune cells that support neuronal restoration.
The observed neuronal MCP-1 expression further strengthens the view that neurons play an active role in the regulation of inflammatory processes of the brain (Wekerle et al., 2000). The authors recently found that axotomized facial nerve neurons directly interact with encephalitogenic T lymphocytes in the course of adoptive transfer experimental autoallergic encephalomyelitis (tEAE) (Flügel et al., 2000). This did not inflict any harm on the lesioned neurons. T cells, however, were induced to undergo apoptosis, most likely mediated by neuronal FasL. Recently, a protective role has been ascribed to inflammatory cells including monocytes/macrophages and T cells during neuronal degeneration (Schwartz et al., 1999). A possible mechanism, which may be involved in the neuroprotective potential of immune cells, is their capacity to produce neurotrophins (Kerschensteiner et al., 1999). As the current data show, MCP-1 is a likely candidate to initiate the contact between immune cells and neurons.
Furthermore, it is conceivable that MCP-1 expression induced by partial lesion in the periphery (for example, as a nerve crush) may target immune cells bearing harmful agents (for example, HIV) into the CNS in the absence of direct brain lesions. Such a mechanism may go along with AIDS-related brain pathology.
The current data demonstrate for the first time a direct and important molecular link between the nervous and the immune system. The easily induced and long-lasting neuronal production of an immune cell chemoattractant in situ associated with an indirect CNS lesion could be of importance for understanding physiologic and pathologic inflammatory processes in the CNS.
Footnotes
Acknowledgments
The authors thank Anja Wöppel, Maria Koch, and Petra Grämmel for their excellent technical assistance, and Dr. James Chalcroft for his expertise in photographic and digital documentation.