
Abstract
Chronic cerebral hypoperfusion causes white-matter lesions (WMLs) with oxidative stress and cognitive impairment. However, the biologic mechanisms that regulate axonal plasticity under chronic cerebral hypoperfusion have not been fully investigated. Here, we investigated whether L-carnitine, an antioxidant agent, enhances axonal plasticity and oligodendrocyte expression, and explored the signaling pathways that mediate axonal plasticity in a rat chronic hypoperfusion model. Adult male Wistar rats subjected to ligation of the bilateral common carotid arteries (LBCCA) were treated with or without L-carnitine. L-carnitine-treated rats exhibited significantly reduced escape latency in the Morris water maze task at 28 days after chronic hypoperfusion. Western blot analysis indicated that L-carnitine increased levels of phosphorylated high-molecular weight neurofilament (pNFH), concurrent with a reduction in phosphorylated phosphatase tensin homolog deleted on chromosome 10 (PTEN), and increased phosphorylated Akt and mammalian target of rapamycin (mTOR) at 28 days after chronic hypoperfusion. L-carnitine reduced lipid peroxidation and oxidative DNA damage, and enhanced oligodendrocyte marker expression and myelin sheath thickness after chronic hypoperfusion. L-carnitine regulates the PTEN/Akt/mTOR signaling pathway, and enhances axonal plasticity while concurrently ameliorating oxidative stress and increasing oligodendrocyte myelination of axons, thereby improving WMLs and cognitive impairment in a rat chronic hypoperfusion model.
Introduction
Cerebral white matter constitutes 50% of human brain mass and contains abundant axons and oligodendrocytes. 1 Cerebral white matter is damaged by a variety of neurologic disorders including ischemic stroke, and white-matter lesions (WMLs) have been associated with cognitive impairment. 2 Oligodendrocytes are lipid rich and exhibit low levels of antioxidant enzymes, which reduce their ability to cope with the increased generation of reactive oxygen species during ischemic insults. 3 Axons are vulnerable to ischemia, and are also damaged by axon—oligodendrocyte interactions after ischemia. 4 Permanent occlusion of both common carotid arteries can induce chronic cerebral hypoperfusion in experimental animals, which results in WMLs in the corpus callosum.5, 6, 7 Various pathogenic factors are implicated in WMLs, including oxidative stress, inflammatory reactions, and endothelial injuries. 6
L-carnitine therapy is required for children with an inborn error of metabolism as well as chronic debilitating diseases including cancer, diabetes mellitus, and chronic kidney disease.8, 9 In such chronic diseases, L-carnitine has a pivotal role in suppressing inflammatory reactions, oxidative stress, and apoptosis. 9 L-carnitine has also been proven beneficial in ischemic heart disease and peripheral artery disease.10, 11 Continuous supplementation with L-carnitine increases carnitine content, stimulates pyruvate oxidation, and thereby remodels the myocardium and improves cardiac dysfunction. 12 However, little information is available on the effect of L-carnitine on cerebral ischemia.13, 14, 15 To date, the effect of L-carnitine on WMLs induced by chronic cerebral hypoperfusion is essentially unknown.
Axonal outgrowth and plasticity are critical processes during brain repair after stroke injury, and related to improvements in neurologic deficits after stroke.16, 17 Stroke induces axonal outgrowth, and emerging data indicate that the phosphoinositide 3-kinase/Akt/glycogen synthase kinase-3β signaling pathway enhances axonal outgrowth after stroke. 18 To date, the mechanisms that regulate axonal plasticity after chronic hypoperfusion have not been extensively studied. In the present study, we used a rat chronic hypoperfusion model to determine whether L-carnitine exerts a neuroprotective role in WMLs and enhances axonal plasticity, and investigated the signaling pathways that mediate axonal plasticity.
Materials and methods
Chronic Cerebral Hypoperfusion
All animals used in the present study were acquired and cared for in accordance with the guidelines published in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The Animal Care Committee of Juntendo University approved all animal protocols used in the present study, and efforts were made to minimize the number of animals used and their suffering. Adult male Wistar rats (9 to 11 weeks old) weighing 280 to 320 g were purchased from Charles River Institute (Kanagawa, Japan). Chronic cerebral hypoperfusion was induced by ligation of both common carotid arteries (LBCCA), as described previously.4, 5, 6 Rats were anesthetized with 1.0% to 2.0% isoflurane in 30% O2 and 70% N2O. Through a midline incision, the bilateral common carotid arteries were carefully separated from the cervical sympathetic and vagal nerves, and ligatured permanently. During this procedure, the body temperature was maintained at 37.0±0.5°C. Rats at baseline (before LBCCA) or 7, 14, 21, and 28 days after LBCCA were reanesthetized with 1% isoflurane, 70% N2O:30% O2, and transcardially perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde. The brain was dissected out immediately, postfixed in 4% paraformaldehyde for 48 hours, and stored in 30% sucrose in 0.1 mol/L PBS. For immunohistochemistry, 20-μm-thick free-floating coronal sections of rat brain were prepared for staining.
Drug Administration and Classification
Rats were randomly divided into the following groups: (1) L-carnitine group: post-LBCCA, rats were treated per os daily with 600 mg/kg (in 0.7 mL saline) L-carnitine (Sigma Aldrich, Inc., St Louis, MO, USA) using an oral gavage tube, until euthanasia; 19 (2) vehicle group: rats received per os administration of saline, volumes similar to the L-carnitine group, using a gavage tube; and (3) control sham-operated group: rats underwent the protocol described for the vehicle group, but were spared LBCCA.
Measurement of Cerebral Blood Flow
Cerebral blood flow (CBF) was measured at a left temporal window, using laser Doppler flowmetry (Laser Tissue Blood Flow Meter FLO-C1; Omega Wave, Inc., Tokyo, Japan). A flat rectangular sheet-shaped probe (7.5 mm in length and 1.0 mm in depth) was positioned between the temporal muscle and the lateral aspect of the skull, according to a previously published method. 20 Craniotomy was not required and CBF was monitored continuously in 3 to 5 minutes sessions before, immediately after, and 7, 14, 21, and 28 days after LBCCA. Reproducibly recorded CBF velocities were obtained.
Water Maze Task
The water maze task was performed to evaluate deficits in learning and spatial memory induced by cerebral chronic hypoperfusion, using the method described previously. 6 A circular transparent acrylic platform with a radius of 20 cm was placed in a 150-cm diameter circular pool filled with water (20 cm depth) set at 22±1°C, with the top surface of the platform positioned 3 cm below the surface of the water. The water in the pool was made opaque with milk so that the rats were unable to see the underwater platform. Rats were released facing a wall from a standard point, and the time taken to escape to the platform was recorded as the escape latency. The rats performed five trials per day, with a constant intertrial interval of 30 minutes, for 3 consecutive days before LBCCA. The escape latency was analyzed before LBCCA and 7, 14, 21, and 28 days after LBCCA.
Rotarod Performance Test
The rotarod performance test was performed to assess motor dysfunction induced by chronic cerebral hypoperfusion. After 3 consecutive days of training before the operation, rotarod performance was evaluated at 7, 14, 21, and 28 days after LBCCA. The speed was slowly increased from 10 to 40 r.p.m. (0.005 to 0.08g) over a period of 4 minutes. The trial ended when the animal falls off the rungs. The maximum duration on the device was recorded with three rotarod measurements. Rotarod test data are presented as percentages of the maximal duration, compared with the internal baseline control.
Immunohistochemistry and Double Immunofluorescence
Immunohistochemistry was performed on brain sections, as previously described. 18 After incubation in 0.3% H2O2 followed by 10% block ace in 0.1 mol/L PBS, coronal sections of the corpus callosum were immunostained overnight at 4°C using a mouse monoclonal antibody against 4-hydroxy-2-nonenal (HNE, a marker of lipid peroxidation; dilution, 40:1; Japan Institute for the Control of Aging, Japan), a mouse monoclonal antibody against 8-hydroxy-deoxyguanosine (8OHdG, a marker of oxidative DNA damage; dilution 100:1; Japan Institute for the Control of Aging), a rabbit polyclonal antibody against glutathione-S-transferase-pi (GST-pi, a marker of oligodendrocytes; dilution, 1,000:1; Chemicon International, Inc., Temecula, CA, USA), and ss-DNA (a marker of apoptotic cells; dilution, 100:1; Dako Corporation, Carpentaria, CA, USA) staining. The sections were then treated with secondary antibodies (dilution, 300:1; Vectastain; Vector Laboratories, Burlingame, CA, USA). Immunoreactivity was subsequently visualized using the avidin-biotin complex method (Vectastain, Vector Laboratories) and developed with diaminobenzidine. For double immunofluorescence staining, mouse anti-SMI31 (a marker of phosphorylated high molecular weight neurofilament, pNFH; 1:100; Covance, Emeryville, CA, USA), mouse anti-SMI32 (a marker of nonphosphorylated NFH, 1:100; Covance), and rat anti-myelin basic protein (MBP; 1:100; Millipore, Philadelphia, PA, USA) were used. After washing with PBS three times, rat coronal sections were incubated overnight at 4°C with the primary antibodies listed above, and subsequently with Cy3 or FITC-conjugated secondary antibodies for 2 hours at room temperature.
Electron Microscopy
Anesthetized rats were fixed by cardiac perfusion with 2% paraformaldehyde/2% glutaraldehyde buffered with 0.1 mol/L phosphate buffer. Brain tissues were quickly excised and subsequently immersed in the same fixative overnight at 4°C. Using Rodent Brain Matrices (ASI Instruments, Warren, MI, USA) 1-mm-thick brain slices were made. The samples were postfixed with 2% OsO4 in 0.1 mol/L phosphate buffer for 2 hours at 4°C in the dark, block-stained in 1% uranyl acetate for 1 hour, dehydrated with a graded series of aqueous alcohol solutions, and embedded in Epon 812 (TAAB, Barks, UK). Silver sections were cut with an ultramicrotome (Leica UC6; Leica Microsystems, Vienna, Austria), stained with uranyl acetate and lead citrate, and observed with an electron microscope (HT7700; Hitachi, Tokyo, Japan).
Western Blot
All procedures were performed at 4°C. To prepare samples for western blot, corpus callosum tissues were extracted from rat brains, and transferred into 700 mL homogenization buffer (0.25 mol/L sucrose, 0.15 mol/L KCl, 10 mmol/L Tris-Cl, pH 7.5, 1 mmol/L EDTA, 0.5% fatty acid-free BSA) containing Protease Inhibitor (Calbiochem, San Diego, CA, USA), and homogenized with 3 × 10 strokes using a Dounce tissue grinder (Wheaton Scientific, Millville, NJ, USA). The homogenates were centrifuged 10 minutes at 700 g. The supernatant was collected and filtered through layers of cheesecloth and centrifuged 15 minutes at 12,000 g. The final supernatant was referred to as cytosol and used for western blotting. 21 Western blots were performed according to published methods. 6 Briefly, equal amounts of total protein for each sample were loaded on 10% SDS-polyacrylamide gels. After electrophoresis, the proteins were transferred onto nitrocellulose membranes, and the blots were subsequently probed with the following primary antibodies: mouse anti-SMI 31 (1:2,000; Covance), rabbit anti-phosphorylated Akt (Ser 473, 1:1,000; Cell Signaling Technology, Danvers, MA, USA), rabbit anti-Akt (1:2,000; Cell Signaling Technology), rabbit anti-phosphorylated phosphatase and tensin homolog deleted on chromosome 10 (PTEN, Ser 380/Thr 382/383, 1:1,000; Cell Signaling Technology), rabbit anti-PTEN (1:2,000; Cell Signaling Technology), rabbit anti-phosphorylated mammalian target of rapamycin (mTOR, Ser 2448, 1:1,000; Cell Signaling Technology), rabbit anti-mTOR (1:1,000; Cell Signaling Technology), rat anti-MBP (1:2,000; Millipore), rabbit anti-carnitine palmitoyltransferase 1A (CPT1A, 1:1,000; Millipore) and CPT2 (1:1,000; Millipore), and mouse monoclonal anti-β-actin (1:10,000; Abcam, Cambridge, MA, USA). Protein levels of PTEN, Akt, mTOR, and β-actin were used as internal controls. Western blots were performed from at least three individual experiments.
Image Acquisition and Quantification
For histologic experiments, the number of stained cells and the density of axons were analyzed in three coronal sections at 200 μm intervals in each rat brain. Two images in the corpus callosum per section were acquired, and ss-DNA-positive cells were counted in the entire area of the corpus callosum. A confocal microscope (LEICA TCS SP5 II, Leica Microsystems) with a 40 × objective was used to acquire the images. Images were analyzed using the National Institutes of Health Image analysis program version 1.43, as described previously. 17 Briefly, the pNFH+-MBP+ areas were split into green and red channels, and images showing common positive areas in both channels were obtained. The intensity levels were then digitally adjusted with pixel intensity thresholds (black pixels represent pNFH+-MBP+ immunoreactive areas). The total area of black pixels was divided by the total area in each image to estimate the degree of axonal plasticity and myelination after LBCCA. Electron microscopy image analysis involved estimating the number of myelinated axons in the corpus callosum. Only myelinated axon profiles with diameters greater than 0.5 μm were included in the counts. 22 Thirty to forty axons were randomly counted in predefined bilateral areas of the corpus callosum in each rat brain. All in vivo experiments and measurements were performed in a blinded and randomized manner.
Statistical Analysis
Values presented in this study are expressed as mean±standard error. All statistical analyses were performed using the Statistical Package for the Social Sciences (version 15; SPSS Inc., Chicago, IL, USA). Student's t-test and one-way ANOVA with Bonferroni post hoc tests were used when comparing two groups and greater than two groups, respectively. Chi-square tests were used for comparisons of the number of axons according to the axonal diameter among groups. A probability value of <0.05 was considered as significant.
Results
Temporal Changes in Cerebral Blood Flow after Ligation of Both Common Carotid Arteries
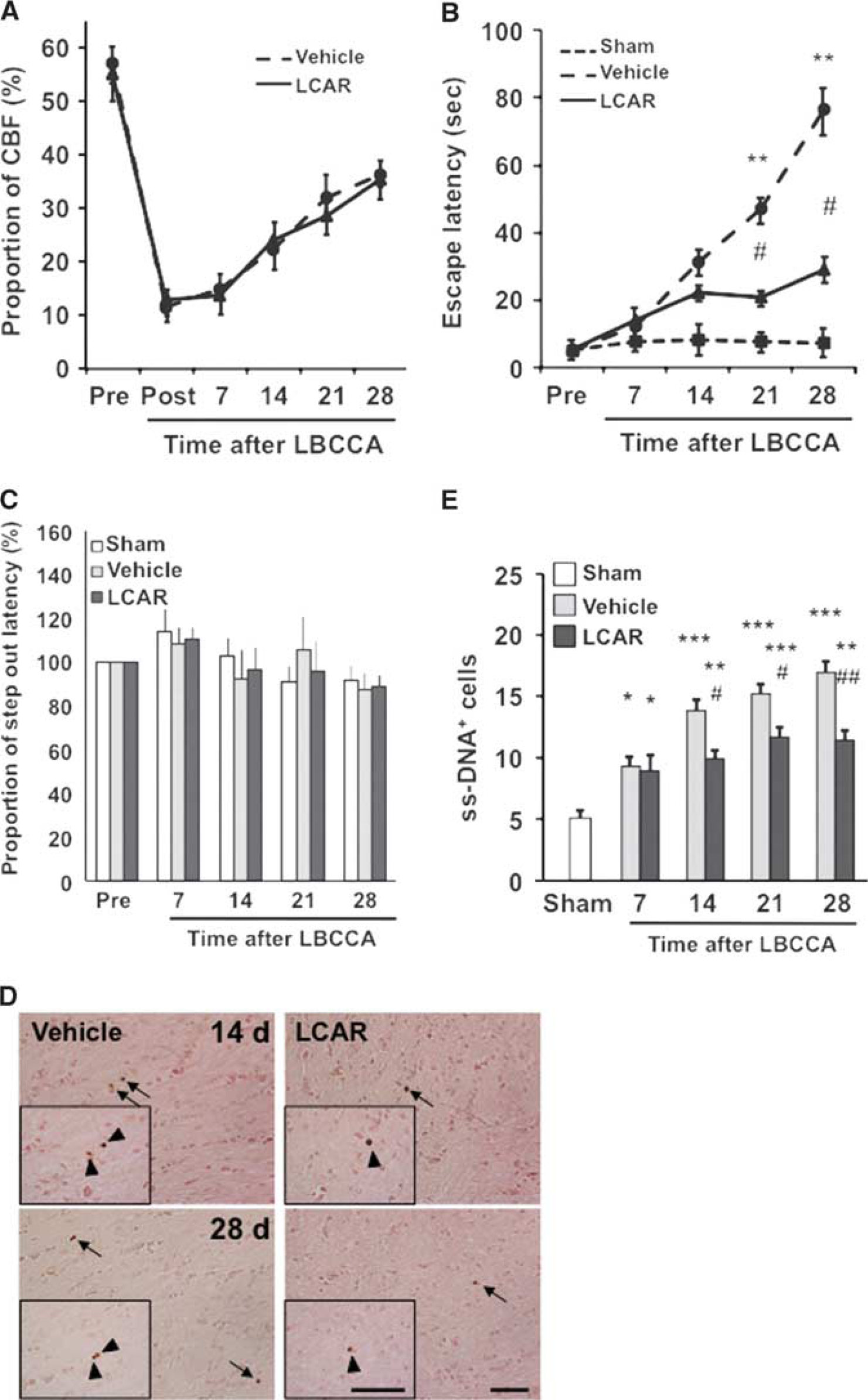
Figure 1A shows the temporal changes in CBF in each group. After LBCCA, CBF gradually increased in the vehicle- and L-carnitine-treated rats up to 28 days after LBCCA. There were no significant differences in CBF between the vehicle and L-carnitine-treated rats.
Effects of L-carnitine on physiologic parameters and cell viability. (
Effects of L-Carnitine on Learning Memory and Motor Function
Before LBCCA, the majority of the rats escaped within 10 seconds after training in the Morris water maze task. The escape latencies of the sham-operated group were within 10 seconds up to 28 days after LBCCA. The escape latency of the vehicle group gradually increased from 7 to 28 days after LBCCA. At 21 and 28 days after LBCCA, the escape latency of the vehicle group significantly increased compared with the sham-operated group (P<0.01, Figure 1B). The L-carnitine-treated rats exhibited shorter latencies than the vehicle-treated group. In particular, at 21 and 28 days after LBCCA the escape latency was significantly different between the vehicle and L-carnitine groups (P<0.05, Figure 1B). In the rotarod test, the step out latency did not change after LBCCA, and there were no significant differences among the sham-operated, vehicle-treated, and L-carnitine-treated rats at each time point (Figure 1C). These data suggest that L-carnitine improves cognitive dysfunction induced by chronic cerebral hypoperfusion in the Morris water maze task, while motor function in the rotarod test was not impaired after chronic cerebral hypoperfusion.
L-carnitine Sustained Cell Viability in Corpus Callosum
The ss-DNA staining experiments indicated that ss-DNA+ cells were substantially increased from 7 to 28 days after LBCCA from baseline. However, L-carnitine significantly reduced the number of ss-DNA+ cells at each time point after LBCCA compared with the vehicle group (P<0.05, Figures 1D and 1E).
L-carnitine Enhances Axonal Plasticity after Chronic Hypoperfusion
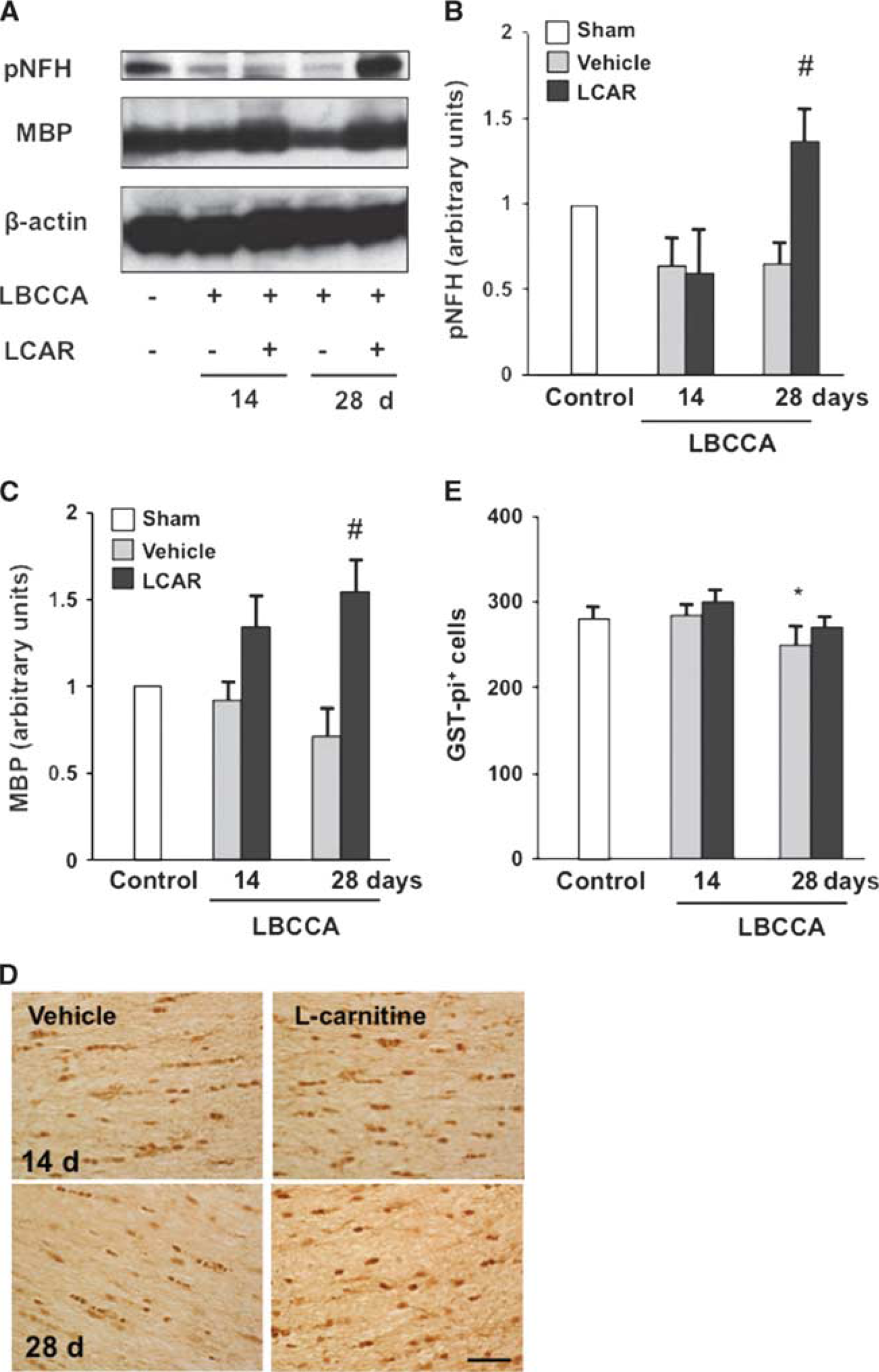
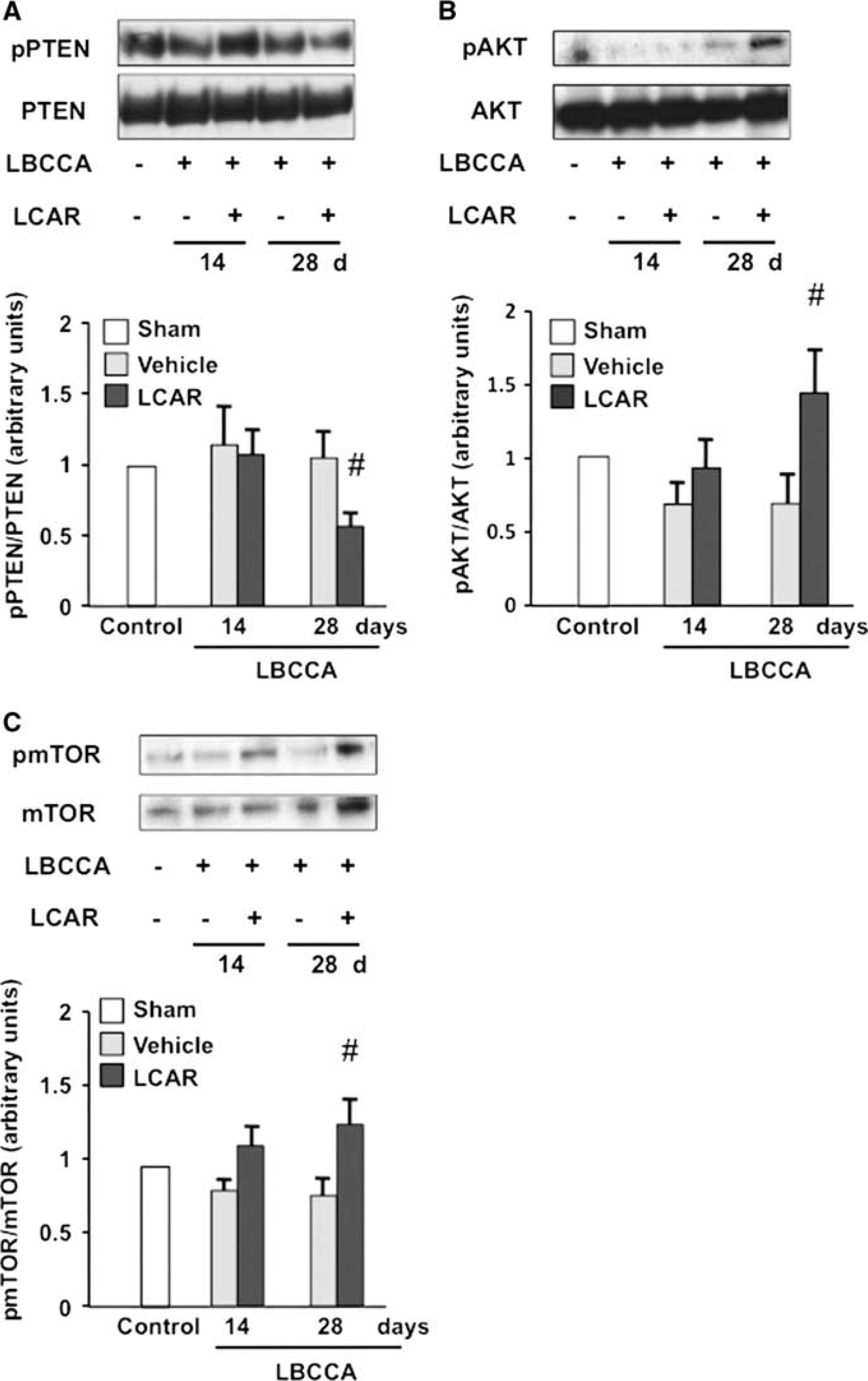
The pNFH is an abundant architectural cytoskeletal element in axons, participates in axonal growth, and regulates synaptic function.23, 24 On western blot analysis, pNFH density decreased at 14 and 28 days after LBCCA in the vehicle-treated rats. However, L-carnitine significantly increased the density of pNFH compared with the vehicle-treated rats at 28 days after LBCCA (P<0.05, Figures 2A and 2B). A significant decrease in phosphorylated phosphatase and PTEN at 28 days after LBCCA was observed in L-carnitine-treated rats compared with vehicle-treated rats (P<0.05, Figure 3A), together with an elevation of phosphorylated Akt and phosphorylated mTOR (P<0.05, Figures 3B and 3C). Our data suggest that L-carnitine enhances pNFH expression related to regulation of the PTEN/Akt/mTOR pathway after chronic cerebral hypoperfusion.
L-carnitine increases phosphorylated high molecular weight neurofilament (pNFH) and myelin basic protein (MBP) after chronic hypoperfusion. (
L-carnitine regulates phosphorylation of PTEN, AKT, and mTOR in the corpus callosum of chronic hypoperfusion rats. (
L-carnitine Reduces Lipid Peroxidation and Oxidative DNA Damage in Oligodendrocytes
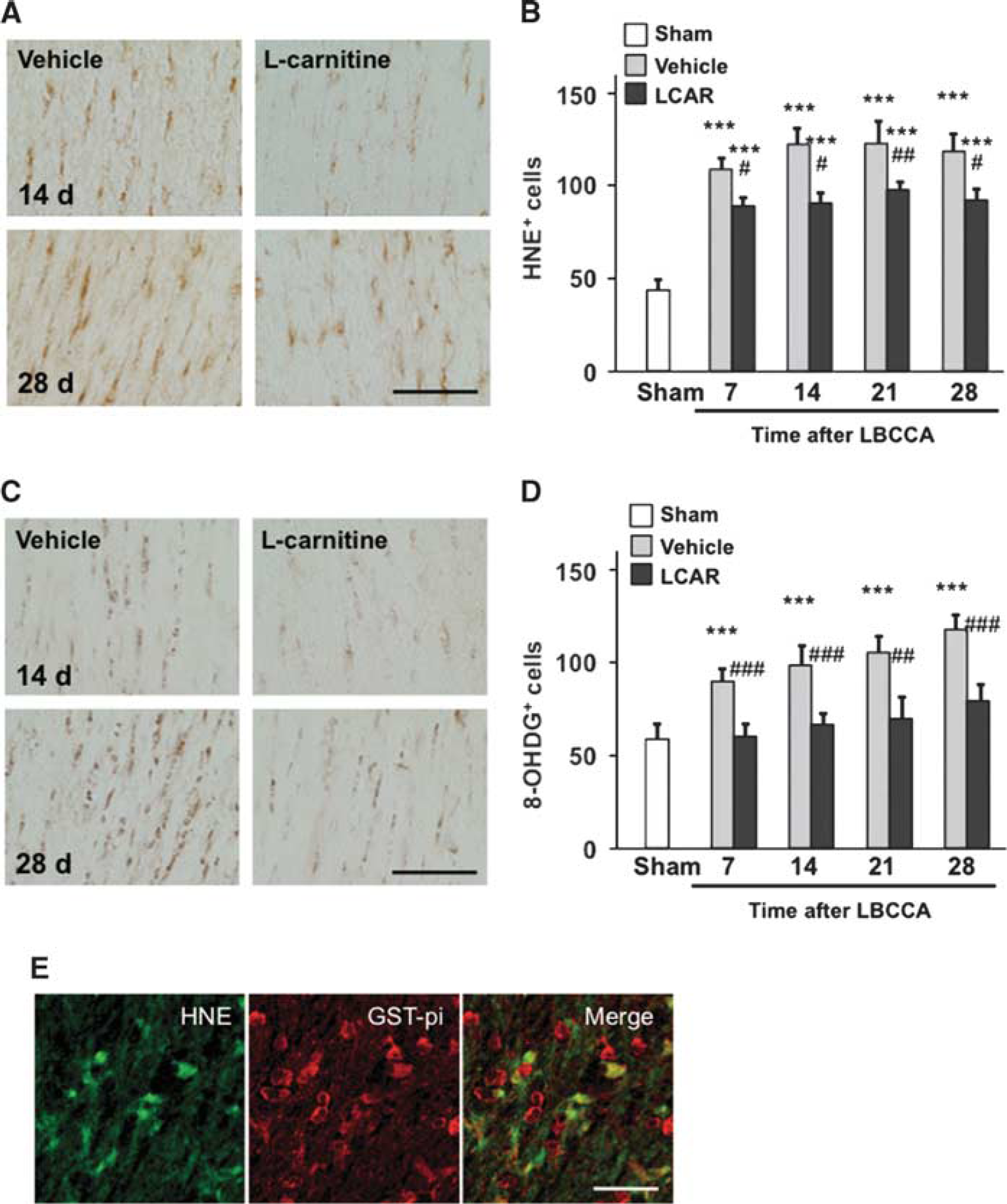
MBP is a marker of mature oligodendrocytes. Our data show that MBP levels gradually decreased in vehicle-treated rats and increase in L-carnitine-treated rats from baseline after LBCCA. At 28 days after LBCCA, L-carnitine significantly increases MBP levels compared with vehicle-treated rats (P<0.05, Figures 2A and 2C). The number of oligodendrocytes in vehicle-treated rats significantly decreased from baseline at 28 days after LBCCA, while the number of oligodendrocytes in L-carnitine-treated rats was not substantially altered (P<0.05, Figures 2D and 2E). To examine oxidative stress in WMLs after LBCCA, lipid peroxidation and oxidative DNA damage were assessed using HNE and 8OHdG staining, respectively. In the vehicle-treated group, HNE+ cells gradually but substantially increased at 7, 14, 21, and 28 days after LBCCA compared with the sham-operated group (P<0.001, Figures 4A and 4B). In the L-carnitine-treated group, HNE+ cells also increased at each time point after LBCCA. However, HNE+ cells were significantly decreased at 7, 14, 21, and 28 days after LBCCA compared with the vehicle-treated group (P<0.05, Figures 4A and 4B). In the vehicle-treated group, 8OHdG+ cells were substantially increased at 7, 14, 21, and 28 days compared with the sham-operated group (P<0.001, Figures 4C and 4D). L-carnitine significantly reduced the number of 8OHdG+ cells compared with the vehicle-treated group, and there were no significant differences in the number of 8OHdG+ cells between the sham-operated group and the L-carnitine-treated group (P<0.01, Figures 4C and 4D). Previously, we showed that HNE+ and 8OHdG+ cells in the corpus callosum were colocalized with GST-pi+ cells, a marker of oligodendrocytes. 6 In the current study, HNE+ cells were colocalized to GST-pi+ oligodendrocytes (Figure 4E) after LBCCA.
carnitine reduces oxidative stress in oligodendrocytes after chronic hypoperfusion. (
L-carnitine Enhances Myelin Sheath Growth and Facilitates Myelination of Axons
To examine alterations in axon—oligodendrocyte interactions after LBCCA, we measured the diameter of myelin sheaths using double immunofluorescence staining. The diameter of MBP+ oligodendrocyte processes decreased at 28 days after LBCCA in vehicle-treated rats from baseline levels (P<0.05, Figure 5A). However, L-carnitine substantially increased the diameter of MBP+ myelin sheaths compared with vehicle-treated rats at 28 days (P<0.001, Figure 5A). To analyze myelin sheath thickness in more detail, we used electron microscopy. After LBCCA, myelin sheath thickness in vehicle-treated rats gradually decreased over time from baseline levels (Figure 5B). However, L-carnitine-treated rats exhibited substantially increased myelin sheath thickness after LBCCA (Figure 5B). Myelin sheaths in L-carnitine-treated rats were significantly thicker than those in sham-operated and vehicle-treated rats at 28 days after LBCCA (P<0.001, Figure 5C). Concurrently, pNFH+ axons were well myelinated by MBP+ oligodendrocytes in the corpus callosum of L-carnitine-treated rats (P<0.05, Figure 5D). Thus, axonal damage and oxidative damage in oligodendrocytes were induced after chronic cerebral hypoperfusion. The aforementioned data suggest that continuous treatment with L-carnitine suppresses oxidative stress in oligodendrocytes after LBCCA, and enhances oligodendrocyte marker expression as well as axonal plasticity, thereby facilitating myelination of axons in the chronic stage of chronic cerebral hypoperfusion.
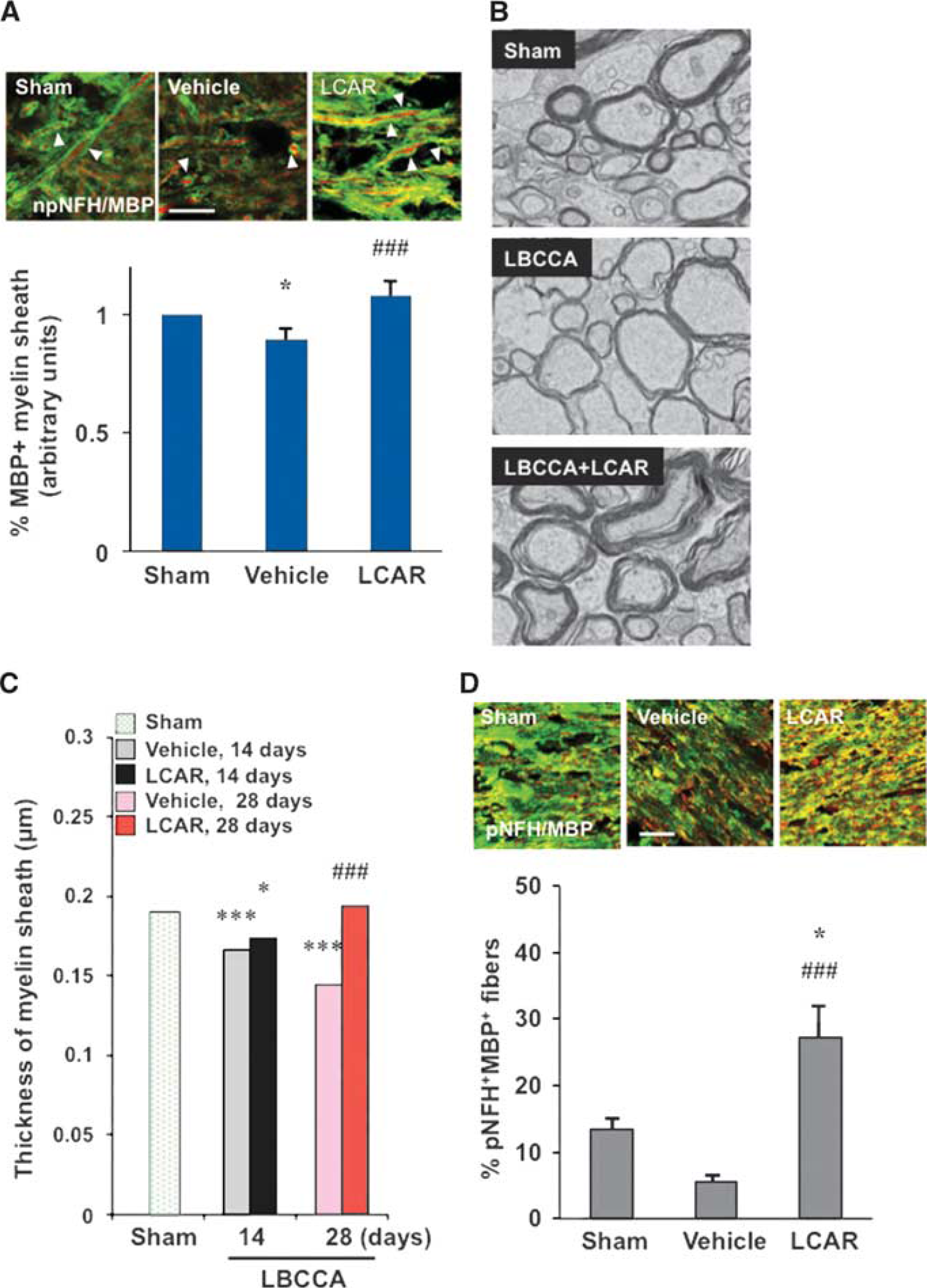
L-carnitine increases myelin sheath thickness after chronic hypoperfusion. (
Improvement of Carnitine Homeostasis
To examine mitochondrial membrane transport function, CPT1A and CPT2, inner and outer mitochondrial membrane transporter enzymes, were analyzed using western blots. After LBCCA, protein levels of CPT1A and CPT2 decreased in vehicle-treated rats. L-carnitine significantly increased CPT1A and CPT2 levels at 28 days after LBCCA compared with vehicle-treated rats, indicating that L-carnitine improves impairment of mitochondrial membrane function induced by chronic cerebral hypoperfusion (P<0.05, Figures 6A–6C). Thus, it is suggested that improved mitochondrial membrane function may be associated with increased axonal plasticity and myelination after L-carnitine treatment after LBCCA.
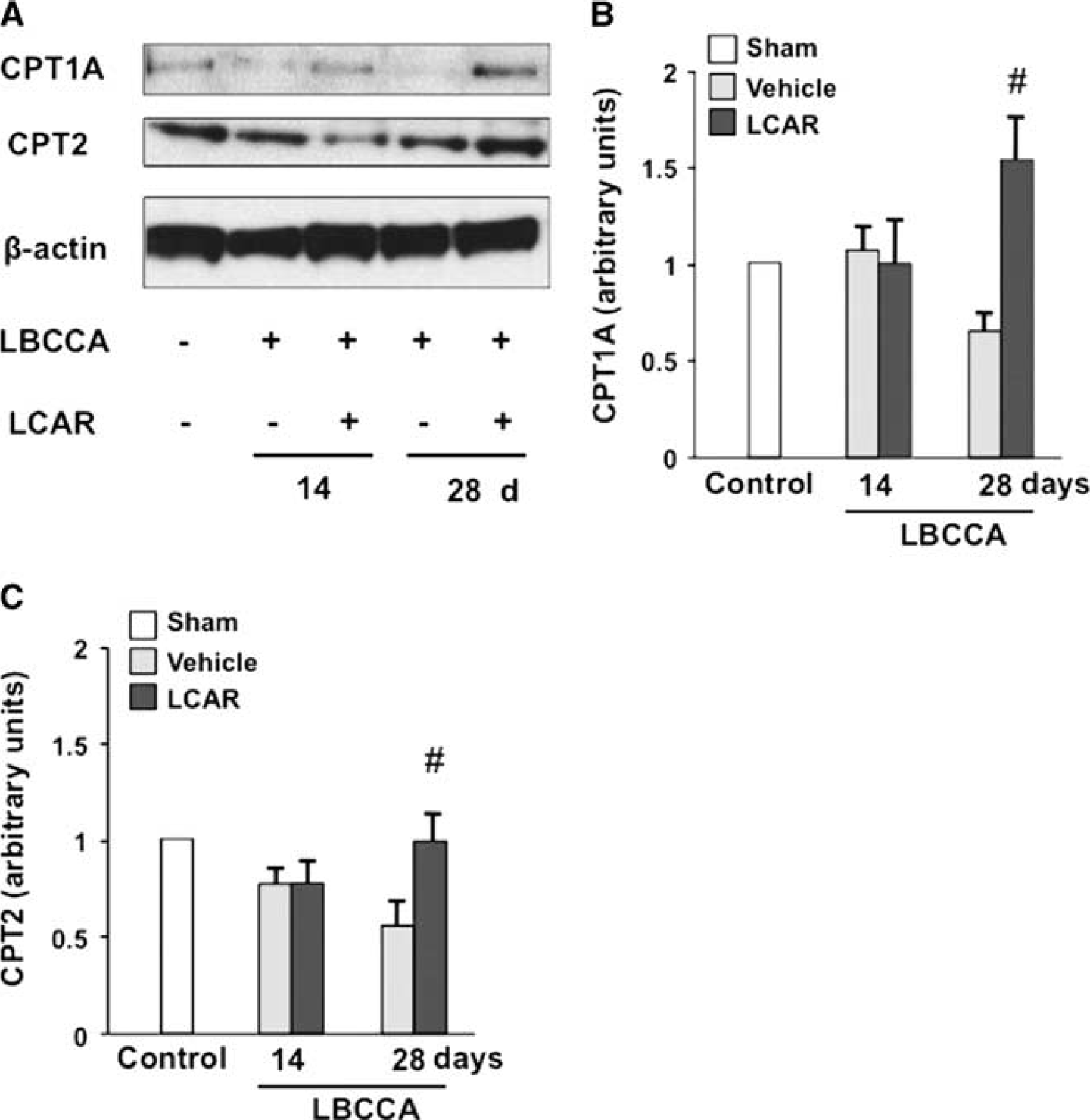
L-carnitine improves carnitine palmitoyltransferase levels after chronic hypoperfusion. (
Discussion
To the best of our knowledge, this is the first study to examine the effect of L-carnitine against WMLs under chronic hypoperfusion. The principal findings presented here were that, in a rat cerebral chronic hypoperfusion model, L-carnitine: enhanced pNFH levels related to the regulation of the PTEN/Akt/mTOR pathway; suppressed lipid peroxidation products and oxidative DNA damage; sustained cell viability; stimulated myelin sheath growth; facilitated myelination of pNFH+ axons; improved mitochondrial membrane function in the corpus callosum; and improved spatial learning and memory.
Although CBF measured by laser Doppler flowmetry does not directly represent tissue flow in white matter, development of WMLs is linked to alterations in CBF.5, 6, 7 In the acute phase, immediately after LBCCA and lasting 2 to 3 days, CBF is substantially reduced and remains very low, which creates hypoxic and ischemic conditions in white matter. The chronic phase lasts 8 to 12 weeks after the acute phase and is characterized by compensatory increases in CBF via collateral vessels. 5 During the chronic phase, inflammatory changes, oxidative stress, and astrogliosis are induced in white matter, and the degree of histologic change in WMLs is consistent with the decreases in CBF. 25 The current data showed that continuous treatment with L-carnitine did not affect CBF but improved WMLs. Thus, it is suggested that L-carnitine exerted direct neuroprotective effects against LBCCA injury.
Stroke induces an acute loss of pNFH+ axons in mammalian brains. 26 Previously, it was reported that pNFH+ axons were increased in the peri-infarct cortex during stroke recovery in a rat model of permanent middle cerebral artery occlusion, and that PTEN/Akt/glycogen synthase kinase-3β signaling promoted axonal outgrowth in cultured cortical neurons after ischemia. 18 In cultured cortical neurons, a local reduction of PTEN and activation of mTOR by miR-17-92 cluster in distal axons enhanced axonal outgrowth. 27 Others have shown that deleting PTEN enhances regeneration of axons in adult corticospinal neurons after spinal cord injury. 28 The PTEN inhibits phosphoinositide 3-kinase/Akt signaling. 29 Activation of Akt and subsequent phosphorylation of mTOR increases axonal outgrowth. 30 In the current study, L-carnitine increased pNFH during the chronic stage of LBCCA injury, which was associated with downregulation of PTEN and activation of Akt and mTOR. Additionally, phosphorylated Akt and mTOR were expressed in pNFH+ axons. Thus, this is the first report to show that L-carnitine enhances pNFH in response to regulation of the PTEN/Akt/mTOR signaling pathway in the corpus callosum of the rat chronic hypoperfusion model.
Loss of oligodendrocytes and demyelination are pathologic hallmarks of ischemic white-matter disease, which is associated with vascular dementia. 31 HNE and 8OHdG proteins accumulate after ischemia.6, 32 MBP+ processes in oligodendrocytes are acutely decreased after hypoxia-ischemia induced white-matter damage, whereas myelin sheath generation and axonal myelination are facilitated during the chronic recovery stage in ischemic rats.18, 33 Our data show that L-carnitine substantially suppresses the production of HNE and 8OHdG in oligodendrocytes after chronic hypoperfusion. Intriguingly, continuous treatment with L-carnitine increases MBP expression and axonal myelination from baseline levels. Although stroke mediates free radical formation and injures oligodendrocytes and axons, the current data suggest that L-carnitine not only exerts an antioxidant effect through the suppression of lipid peroxidation and oxidative DNA damage, but may also facilitate development and myelination of oligodendrocytes in the corpus callosum after chronic cerebral hypoperfusion.
Mitochondrial dysfunction is essential in the pathophysiology of diverse neurologic diseases, including stroke.34, 35 Because reactive oxygen species are mainly generated as byproducts of mitochondrial respiration, mitochondria are thought to be the primary target of oxidative damage. 36 L-carnitine functions in the mitochondria in the transport of long-chain fatty acids into the mitochondrial matrix and facilitates β-oxidation. 37 L-carnitine prevented the reduction of CPT1 and CPT2 after oxygen-glucose deprivation in cultured hippocampal slices. 38 CPT1C regulates dendritic spine maturation and is related to cognitive function in mice. 39 Our data have shown that continuous treatment with L-carnitine ameliorates mitochondrial inner and outer membrane dysfunction after chronic hypoperfusion. The present study is the first to show that impairment of mitochondrial membrane function is implicated as an essential mechanism of WMLs induced by chronic hypoperfusion, and that L-carnitine improves mitochondrial membrane dysfunction, thereby lessening oxidative stress and enhancing axonal plasticity and myelination.
In conclusion, axonal impairment is critically implicated in WMLs induced by chronic cerebral hypoperfusion. The molecular mechanisms underlying axonal plasticity and mitochondrial dysfunction associated with WMLs have previously been unknown. The current study provides evidence that L-carnitine improves mitochondrial membrane dysfunction and alleviates oxidative stress, thereby enhancing axonal plasticity and oligodendrocytic axonal myelination, which is likely to form the basis of the neuroprotective effects of L-carnitine against WMLs and cognitive impairment.
Footnotes
ACKNOWLEDGMENTS
The authors thank Reiko Ishikawa for providing technical assistance.
The authors declare no conflict of interest.