
Abstract
Clinical experimental stroke induces injurious local brain inflammation. However, effects on the peripheral immune system have not been well characterized. We quantified mRNA and protein levels for cytokines, chemokines, and chemokine receptors (CCR) in brain, spinal cord, peripheral lymphoid organs (spleen, lymph node, blood, and cultured mononuclear cells from these sources), and blood plasma after reversible middle cerebral artery occlusion (MCAO) or sham treatment in male C57BL/6 mice. Middle cerebral artery occlusion induced a complex, but organ specific, pattern of inflammatory factors in the periphery. At both 6 and 22 h after MCAO, activated spleen cells from stroke-injured mice secreted significantly enhanced levels of TNF-α, IFN-γ, IL-6, MCP-1, and IL-2. Unstimulated splenocytes expressed increased chemokines and CCR, including MIP-2 and CCR2, CCR7 & CCR8 at 6 h; and MIP-2, IP-10, and CCR1 & CCR2 at 22 h. Also at 22 h, T cells from blood and lymph nodes secreted increased levels of inflammatory cytokines after activation. As expected, there were striking proinflammatory changes in postischemic brain. In contrast, spinal cord displayed suppression of all mediators, suggesting a compensatory response to intracranial events. These data show for the first time that focal cerebral ischemia results in dynamic and widespread activation of inflammatory cytokines, chemokines, and CCR in the peripheral immune system.
Introduction
Clinical stroke and experimental cerebral ischemia induce local inflammatory processes that undoubtedly contribute to total cerebral injury (Allan and Rothwell, 2003; del Zoppo et al, 2001). Within hours, transcription factors are activated locally in brain tissue (e.g., nuclear factor-κB; O'Neill and Kaltschmidt, 1997) that upregulate proinflammatory genes, including the cytokines tumor necrosis factor α (TNF-α) (Liu et al, 1994), interleukin 1β (IL-1β) (Liu et al, 1993; Wang et al, 1994), IL-6 (Wang et al, 1995a, b), and IL-1 receptor antagonist (IL-1ra) (Wang et al, 1997), and chemokines such as IL-8 (Liu et al, 1993), interferon inducible protein-10 (IP-10) (Wang et al, 1998) and monocyte chemoattractant protein-1 (MCP-1) (Kim et al, 1995; Wang et al, 1995a, b). These factors promote expression of adhesion molecules by vascular endothelial cells that allow infiltration into the brain of blood neutrophils, monocytes, macrophages, and T cells that promote further brain injury (Barone and Feuerstein, 1999). Moreover, inflammatory and antigenic products derived from brain (e.g., myelin basic protein) may leak across a damaged blood brain barrier and produce reciprocal systemic activation.
While postischemic inflammation within brain has been well studied in models of focal stroke, systemic inflammatory responses have been poorly characterized. In patients with stroke, C-reactive protein, white blood cell counts, and plasma IL-6 levels were increased on admission and persisted for > 7 days (Emsley et al, 2003). A later study from this group found a significant correlation in peak plasma IL-6 levels measured within the first week after the stroke with brain infarct volume, stoke severity, and long-term clinical outcome (Smith et al, 2004). Additionally, experimental stroke in mice caused a reduction in immune cells in peripheral lymphoid organs and decreased secretion of TNF-α and IFN-γ that contributed to spontaneous bacterial infections, a leading cause of mortality in stroke patients (Prass et al, 2003). Gendron et al (2002) recently showed that occlusion of the left or right hemispheres caused a reduction in total splenocytes and CD8+ T cells, and increased splenocyte proliferation to mitogens. These results suggest that there may be systemic repercussions in lymphoid organs that occur in response to postischemic inflammation in the brain. However, the relationship between such repercussions and CNS pathology is unclear, both from the perspective of the brain and the peripheral immune system.
To initiate a more comprehensive study of this problem, the present study quantified mRNA and protein levels for cytokines, chemokines, and chemokine receptors (CCR) in brain, spinal cord, peripheral lymphoid organs (spleen, lymph nodes, and blood), and blood plasma 6 and 22 h after middle cerebral artery occlusion (MCAO) in C57BL/6 mice. We found that in addition to previously described inflammatory changes in the brain, stroke induced a complex, but organ specific, pattern of inflammatory factors in the periphery as early as 6 h after occlusion. These findings indicate that the drastic inflammatory changes occurring in the damaged brain are dynamically reflected in the peripheral immune organs.
Materials and methods
Animals
The study was conducted in accordance with National Institutes of Health guidelines for the use of experimental animals, and the protocols were approved by the Institutional Animal Care and Use Committee. Age-matched, sexually mature male mice (C57BL/6J; Charles Rivers; body weight 20 to 25 g) were used in all experiments.
Ischemic Model
Focal cerebral ischemia was induced by 90 mins of reversible MCAO of the right hemisphere under halothane anesthesia, as previously described (McCullough et al, 2003; Sawada et al, 2000). In brief, mice were anesthetized with 1.5% to 2.0% halothane in O2-enriched air. The common carotid artery was exposed and the external carotid artery was ligated and cauterized. Unilateral MCA occlusion was performed by inserting a 6 to 0 silicone coated, nylon monofilament surgical suture with heat-blunted tip into the internal carotid artery via the external carotid artery stump. The tip was positioned at a distance of 6 mm beyond the internal carotid/pterygopalatine artery bifurcation, and occlusion was confirmed by a laserdoppler flow (LDF; Moor Instruments) probe positioned over the ipsilateral hemisphere at the mid ear-to-eye distance. The suture was then secured in place, and the animal was awakened and assessed for intraischemic neurologic deficit, that is, the presence or absence of forelimb weakness; torso turning to the ipsilateral side when held by tail; circling to affected side; inability to bear weight on affected side; or spontaneous locomotor activity or barrel rolling. Any animal without a visible deficit was excluded from the study. At end-ischemia (90 mins), the animal was briefly reanaesthetized and reperfusion was initiated by filament withdrawal. Our MCAO studies involved three separate experiments, each involving a minimum of three replicate mice per group.
Isolation of Mononuclear Cells from Spleen, Lymph Nodes, and Blood
Spleen and inguinal LN were isolated from sham and MCAO mice and a single-cell suspension was prepared by passing the tissue through a 100 μm nylon mesh screen. The cells were washed using RPMI and the red cells lysed using red cell lysis buffer (8.3 g NH4Cl in 0.01 mol/L TRIS-HCl, pH 7.4) and incubated for 8 mins. The cells were then washed twice with RPMI, counted and resuspended in stimulation medium containing 10% FBS for cytokine detection by CBA and enzyme-linked immunosorbent assay (ELISA). For real-time polymerase chain reaction (PCR), splenocytes were pelleted, snap-frozen, and stored at −80°C until tested.
Cardiac blood was collected in 3 mg/ml EDTA. Cells were then pelleted and the supernatant (plasma) was collected and stored at −80°C until tested for cytokines by CBA and ELISA. Red cell lysis buffer was added to the cell pellet and incubated for 10 mins. Cells were washed twice using RPMI, counted and resuspended in stimulation medium containing 10% FBS for CBA and ELISA assays.
Preparation of Spinal Cord for Polymerase Chain Reaction
Spinal columns were dissected out of the mice and the cords were purged using a 10 cm3 syringe containing RPMI. The cords were then snap frozen using methylbutane over dry ice and stored at −80°C until further testing with reverse transcription (RT)-PCR.
Terminal Histopathology
The brains were harvested after 22 h of reperfusion and sliced into five 2-mm-thick coronal sections for staining with 1.2% triphenyltetrazolium chloride (TTC, Sigma, St Louis, MO, USA) in saline as previously described (McCullough et al, 2003). Infarction volume was measured using digital imaging (MTI Series 68 Video Camera) and image analysis software (Sigma Scan Pro, Jandel). The area of infarct was measured on the rostral and caudal surfaces of each slice and numerically integrated across the thickness of the slice to obtain an estimate of infarct volume in each slice.
Cytokine Determination by Cytometric Bead Array
Spleen, lymph node and blood mononuclear cells were cultured in a 24-well flat bottom culture plate with 5 μg/ml plate-bound anti-CD3 and 2 μg/ml anti-CD28 antibodies at 4 × 106 cells/well in stimulation medium containing 10% FBS for 24 h. Supernatants were then harvested and stored at −80°C until tested for cytokines. Also, plasma was collected and frozen at −80°C until tested for cytokines. The mouse inflammation CBA (Cytometric Bead Array) kit was used to detect IL-12p40, TNF-α, IFN-γ, MCP-1, IL-10 and IL-6 simultaneously (BD Bioscience, San Diego, CA, USA). Briefly, 50 μl of sample was mixed with 50 μl of the mixed capture beads and 50 μl of the mouse PE detection reagent. The tubes were incubated at room temperature for 2 h in the dark, followed by a wash step. The samples were then resuspended in 300 μl of wash buffer before acquisition on the FACScan. The data were analyzed using the CBA software (BD Biosciences). Standard curves were generated for each cytokine using the mixed bead standard provided in the kit and the concentration of cytokine in the supernatant was determined by interpolation from the appropriate standard curve.
Enzyme-linked Immunosorbent Assay for Detection of Interleukin 1β and Interleukin 2
Plasma and culture supernatants from anti-CD3/CD28 antibody-activated spleen, lymph node and blood mononuclear cells were obtained as above. In total, 96-well plates were coated with 100 μl of anti-mouse IL-1β or IL-2 capture antibody (4 μg/ml) in 1 × PBS or sodium bicarbonate coating buffer. Plates were incubated at 4°C overnight, washed with buffer (1 × PBS/0.05% Tween-20), and treated with blocking buffer (1 × PBS, 2% BSA) for 2 h at room temperature. Plates were then washed and 100 μl of sample or standard was added to each well. Interleukin 1β plates were incubated at room temperature for 2 h whereas IL-2 plates were incubated at 4°C overnight. Plates were then washed and 100 μl of biotinylated cytokine-specific antibody was added. Interleukin 1β plates were incubated at room temperature for 2 h while IL-2 plates were incubated at room temperature for 45 mins. Plates were then washed and 100 μl of 1:250 (IL-1β) or 1:400 (IL-2) diluted HRP was added. Plates were incubated at room temperature for 30 mins followed by a wash step. This was followed by addition of 100 μl TMB chromogen (KPL Cat #52-00-2). The color was allowed to develop for approximately 30 mins, and the reaction stopped by adding 100 μl stop solution (KPL Cat #50-85-05). Optical density was then measured at 450 nm.
Ribose Nucleic Acid Isolation and Reverse Transcription-Polymerase Chain Reaction
Total RNA was isolated from brains and spinal cords using the RNeasy mini kit protocol (Qiagen, Valencia, CA, USA) and then converted to cDNA using oligo dT, random hexamers and Superscript RT II enzyme (Invitrogen, Grand Island, NY, USA). Real-time PCR was performed using Quantitect SYBR Green PCR master mix (Qiagen) and primers (synthesized by ABI). Reactions were conducted on the ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) to detect mRNA quantified as relative units (RE). Primer sequences for the following genes are:

Statistical Analyses
Means ± s.d. of cytokine and chemokine concentrations and RE of chemokines and CCR were calculated for groups of MCAO versus sham treated mice and differences were evaluated for significance (P < 0.05) using Student's t-test.
Results
Physiologic Measurements and Histology
Intraoperative rectal temperature was controlled in all animals (36.9°C ± 0.6°C and 36.7°C ± 0.6°C in MCAO and shams, respectively). Occlusion was confirmed in all MCAO animals, and intraischemic LDF was 13% ± 1% and 11% ± 2% of baseline signal in the 6 and 22 h MCAO groups. Infarction at 22 h was present in all animals, and damage was consistent with previous work in this model (cortex: 48% ± 10% of contralateral cortex; striatum: 76% ± 12% of contralateral striatum; total: 47% ±10% of contralateral hemisphere).
Middle Cerebral Artery Occlusion-Induced Changes in Cytokines and Chemokines in Brain and Spinal Cord
As expected, there were striking differences in cytokines, chemokines, and chemokine receptor levels in postischemic brain (Figure 1). At 6 h, ipsilateral cortex and striatum showed pronounced increases in expression of inflammatory cytokines (TNF-α, IL-1β, IL-6, Figure 1A) and chemokines (RANTES, IP-10, MIP-2, Figure 1C), as well as noninflammatory factors (TGF-β1, IL-10, IL-13, Figure 1B), but not IFN-γ or FoxP3 (Figures 1A and 1B). In most instances, there was already substantial basal expression of CCR, and MCAO further enhanced expression of only CCR3 (right hemisphere only) and CCR8 (both hemispheres) (Figure 1D). After 22 h of reperfusion, ipsilateral tissue showed nearly the same exact pattern but generally lower levels of expression of cytokines and chemokines, with the exception of IL-6 (Figure 1A) and MIP-2 (Figure 1C), which were notably increased. However, more widespread changes in expression of CCR were evident at 22 h, including five-fold additional increases in intensity of CCR1 and CCR2 (Figure 1C), and a 40-fold increase in intensity of CCR5 (Figure 1D), and lower but significant changes in CCR3, CCR7, and CCR8 (Figure 1D).
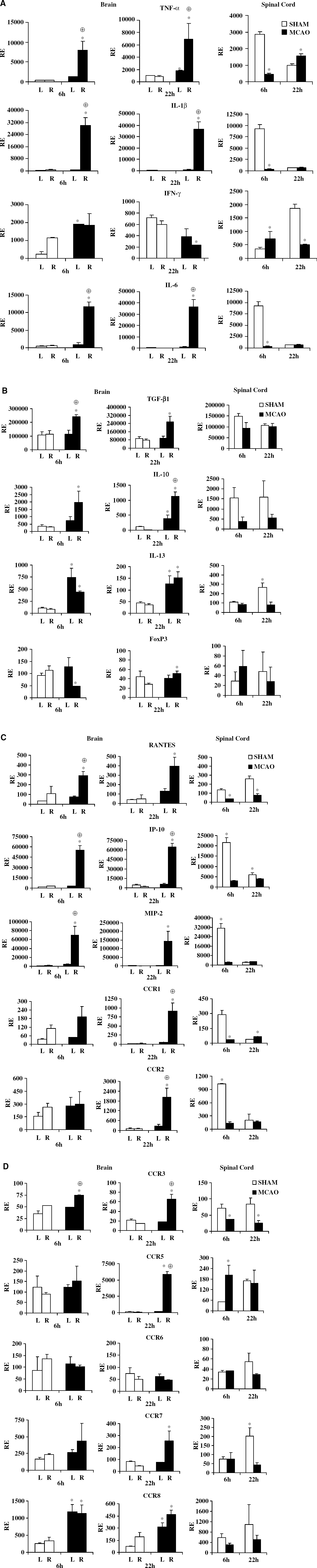
Effects of stroke on expression of cytokines and chemokines/receptors in CNS tissue. Brains and spinal cords were collected from sham and MCAO-treated mice 6 and 22 h after occlusion, and mRNA prepared from ipsilateral (right) and contralateral (left) hemispheres of brain and spinal cords tissues for RT-PCR analysis. Relative expression (RE) of message levels are presented for (
Changes in expression of inflammatory factors were not limited to the brain. In spinal cord, many mediators were decreased at 6 h MCAO (relative to sham), with a striking reduction in mRNA for most of the same inflammatory cytokines (TNF-α, IL-1β, IL-6) and chemokines (IP-10, MIP-2) as were increased in injured ipsilateral brain tissue (Figures 1A and 1C). The exception was an increase in expression of IFN-γ in spinal cord. Interestingly, there was a similar reduction of expression at 6 h of CCR1 and CCR2, but enhanced expression of CCR5 (Figures 1C and 1D). By 22 h after occlusion, expression of most mediators and CCRs was reduced or normalized, with the exception of TNF-α and CCR1. These findings suggest an early compensatory effect within the uninvolved spinal cord tissue in mice with MCAO that was attenuated by 22 h after occlusion.
Changes in Peripheral Cytokine Levels Induced by Stroke
Although induction of inflammatory factors has been documented in stroke-injured brain tissue, little is known about these factors in the circulation or peripheral immune organs. Previous reports noted a significant increase in IL-6 in blood plasma from patients with stroke (Emsley et al, 2003; Smith et al, 2004). We confirmed plasma elevation of IL-6 in MCAO mice at both 6 and 22 h after occlusion, as well as IFN-γ and MCP-1 at the 6 h time point (Figures 2A and 2B). Other cytokines and chemokines tested in blood plasma were unchanged at either time point.
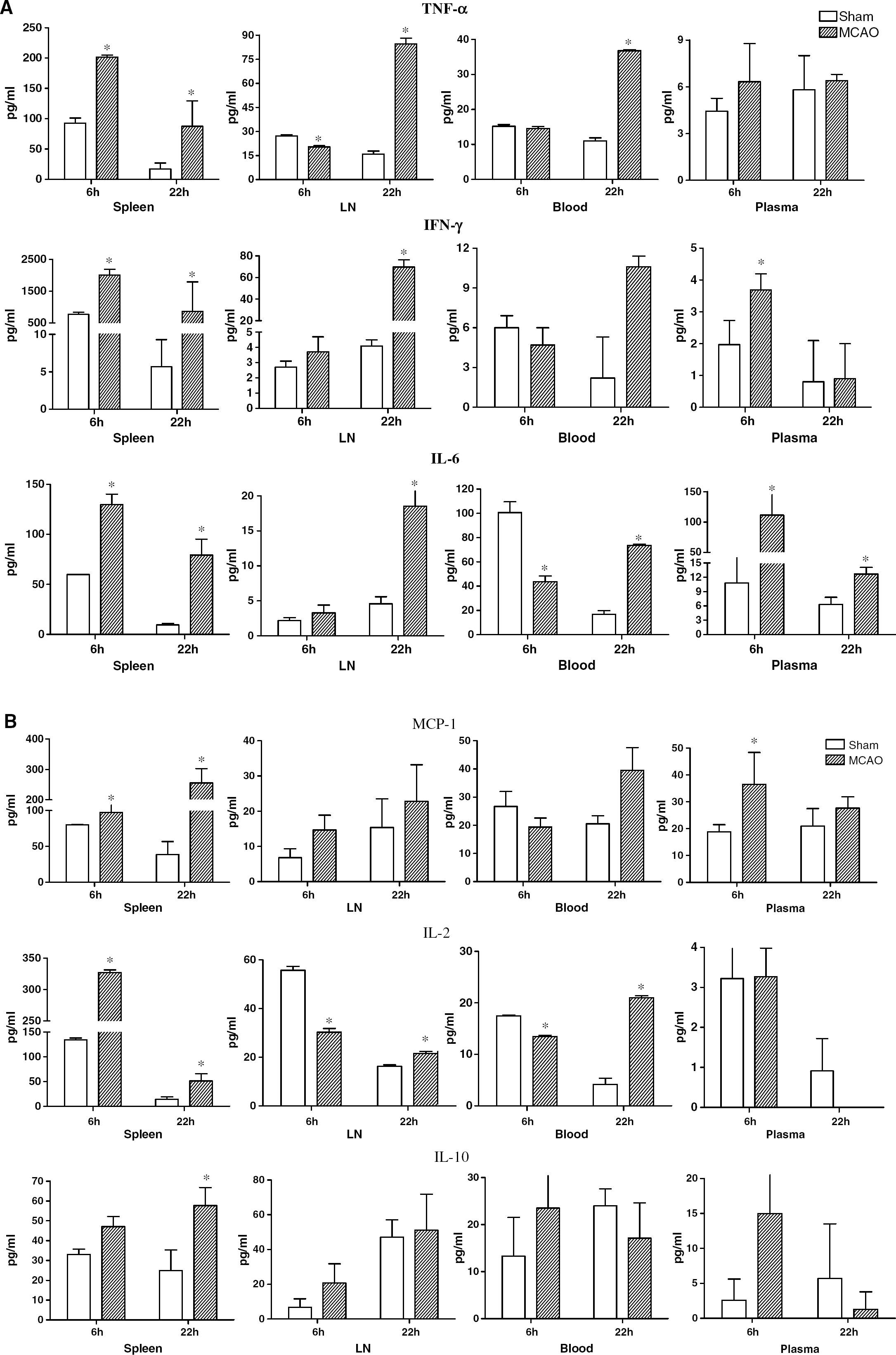
Effects of stroke on cytokines secreted from stimulated splenocytes, lymph node cells and blood cells, and from blood plasma. Spleens, lymph nodes, blood, and blood plasma were collected 6 h and 22 h after vascular occlusion and immune cells were stimulated for 48 h with plate-bound anti-CD3/CD28 antibodies. Supernatants and blood plasma were evaluated for levels of secreted factors, including (
We envisioned that ischemia might also induce cytokine changes in distant peripheral immune cell populations. Thus, mononuclear cells were isolated from various organs 6 and 22 h after MCAO or sham treatments, and cytokines were assessed by CBA and ELISA in supernatants of cultures stimulated for an additional 24 h with plate-bound anti-CD3/CD28 antibodies. The most striking and consistent changes induced in the stroke mice versus shaminjured mice were observed in the spleen. At both the 6 and 22 h time points, activated spleen cells from stroke-injured mice secreted significantly enhanced levels of the inflammatory factors TNF-α, IFN-γ, IL-6, MCP-1, and IL-2 (Figures 2A and 2B), with increased secretion of the anti-inflammatory factor, IL-10, only at the 22 h time point. Levels of IL-12p40 were low and did not change significantly at either time point after occlusion (not shown). Moreover, unstimulated spleen tissue from stroke mice had increased expression of message for MIP-2, CCR2, CCR7, and CCR8 at the 6 h time point, and MIP-2, IP-10, CCR1 and CCR2 at the 22 h time point (Figure 3). Similar increases in secretion of TNF-α, IL-6, IL-2, and IFN-γ (LN only) were observed only at the 22 h time point in activated lymph node and blood mononuclear cells (Figures 2A and 2B). Interestingly, IL-1β was not detected at either time point in any of the peripheral lymphoid organs or in plasma (not shown), suggesting that the source of this cytokine was solely from injured brain. These data show that focal cerebral ischemia produced local inflammatory effects within the recovering brain, and distal effects in lymphoid organs. In contrast, early suppression of inflammatory mediators was observed in spinal cord.
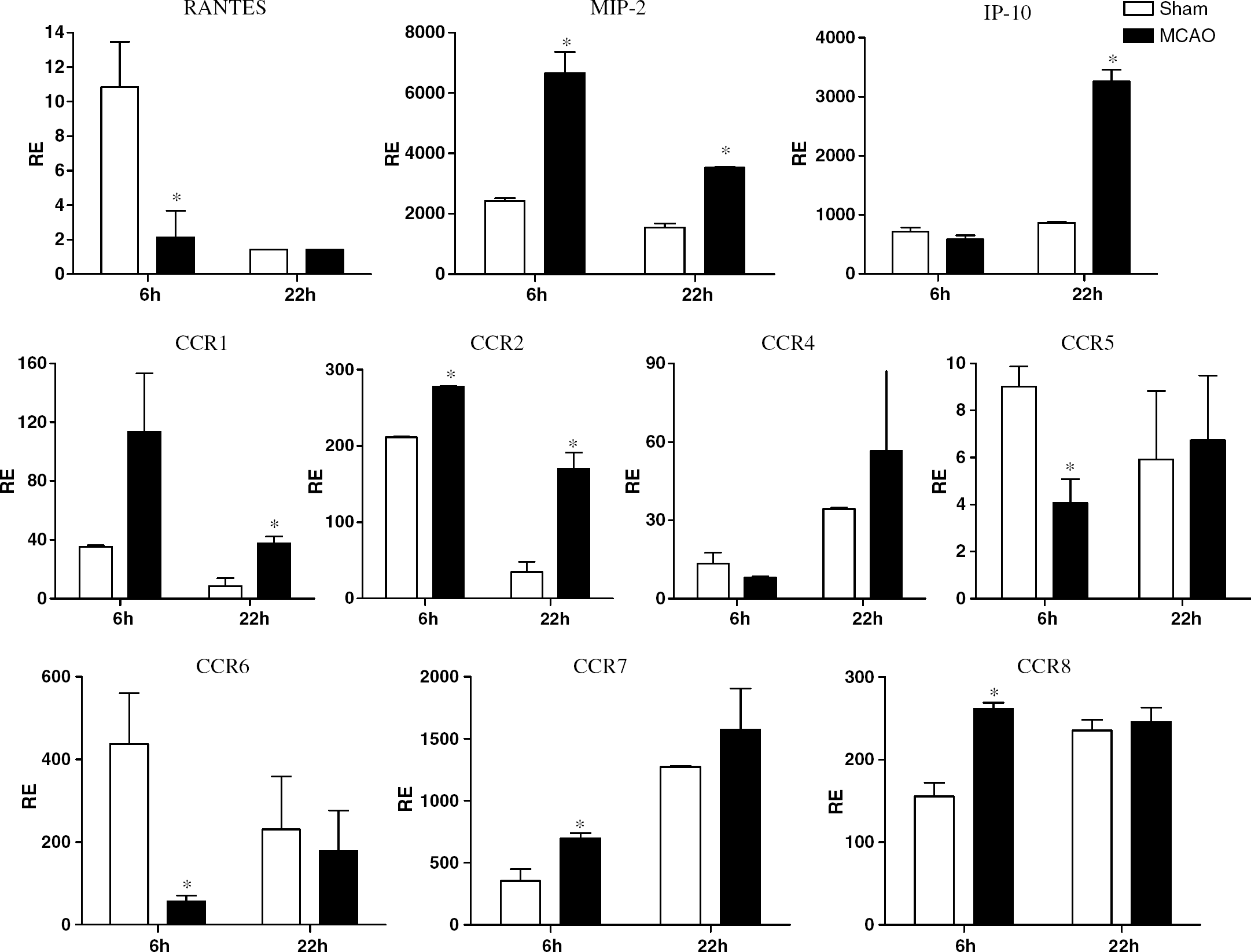
Effects of stroke on expression of chemokines/receptors in spleen tissue. Spleens were collected from sham and MCAO-treated mice 6 and 22 h after occlusion and mRNA evaluated by RT-PCR for expression of chemokines and chemokine receptors. * indicates a significant difference in expression in stroke mice versus sham-treated mice.
Discussion
It is now well established that the initial insult from stroke is followed by an early induction of inflammatory cytokines and chemokines that attract mononuclear cells and granulocytes, which cause further damage to the ischemic and surrounding areas of brain tissue. The results presented above confirm and extend these previous observations in brain and are the first to document the additional rapid and profound effects of focal cerebral ischemia on systemic immune responses in lymphoid organs. As early as 6 h after cerebral vascular occlusion, activated splenocytes released significantly elevated levels of inflammatory cytokines on stimulation through the T-cell receptor (TCR). In addition, unstimulated splenocytes expressed increased message levels for the chemokine, MIP-2, and CCR, and blood plasma had increased levels of several inflammatory cytokines, including IL-6 that had been reported previously in stroke patients (Emsley et al, 2003; Smith et al, 2004). Later, 22 h after stroke induction, T cells from spleen as well as blood and lymph nodes secreted increased levels of inflammatory cytokines and IL-10 (spleen only) after activation. In contrast, spinal cord tissue from mice with cerebral vascular occlusion had reduced levels of inflammatory factors, suggesting a compensatory response to brain injury.
There is consensus that the major cytokine ‘players’ that contribute to postischemic inflammation include IL-1, TNF-α, and possibly IL-6 (Zheng and Yenari, 2004). Interleukin 1 and TNF-α are pleiotrophic factors that can be detected as early as 1 h after onset of stroke, even before significant neuronal death (Allan and Rothwell, 2001), but later may have neuroprotective effects. Both cytokines promote early-stage inflammation by increasing expression of chemotactic factors and adhesion molecules by vascular endothelium leading to early infiltration of monocytes and macrophages (within 18 h), neutrophils (within 48 h) and T lymphocytes (within 72 h) (Stevens et al, 2002). These early inflammatory factors in combination are neurotoxic, and also induce the production of additional cytokines and chemokines by other brain cells. In the damaged brain, IL-1 and TNF-α are largely produced by activated microglial cells, but may also be secreted by other brain cells, including astrocytes, endothelial cells, and neurons (del Zoppo et al, 2001), and later by infiltrating mononuclear cells from blood. Studies of IL-6 have produced conflicting results. Interleukin 6 and IL-6R expression paralleled cell death by neurons after ischemic insult (Vollenweider et al, 2003), and peak levels of IL-6 in plasma from stroke patients had prognostic value in predicting long-term outcome (Smith et al, 2004). However, mice deficient in IL-6 did not have more severe cerebral damage after stroke than wild-type mice (Clark et al, 2000), suggesting that this cytokine may not be critical for stroke pathogenesis.
Other less well-studied proinflammatory chemokines have also been reported in stroke-damaged brain tissue, including IL-8, MIP-2, and MCP-1 (for attracting neutrophils and monocytes to the site of damage), and IP-10, MIP-1α, and RANTES, along with their respective CCR (Cartier et al, 2005), particularly CXCR3 (that binds IP-10) and CCR5 (that binds MIP-1α/β, RANTES, and MCP-2). Chemokines induce neuronal death either directly through neuronal receptors or indirectly via microglial activation and killing, and previous work indicates these factors are induced after MCAO, for example, IP-10, CXCR3 (Wang et al, 2000), and CKR5 (Spleiss et al, 1998). Furthermore, intraventricular MIP-1α injection enhances cortical infarction in mice, while pharmacological chemokine receptor antagonists reduce damage (Takami et al, 2001, 2002). In contrast, the anti-inflammatory and neuroprotective factors, IL-10, TGF-β, and IL-1ra, were produced in synchrony with the first wave of inflammatory cytokines (Allan and Roth well, 2001; Stoll, 2002). It is noteworthy that we detected selectively enhanced expression within the postischemic right brain hemisphere of TNF-α, IL-1β, IL-6, TGF-β1, IL-10, RANTES, IP-10, MIP-2, and various CCR at both 6 and 22 h after vascular occlusion, a pattern that is entirely consistent with previously published literature.
In stark contrast, the pattern of cytokine expression in spinal cord tissue from stroke-damaged mice was nearly the reverse image of changes observed in the damaged right hemisphere, suggesting that cytokine expression is organ-specific within the CNS. Whether the apparent immunosuppression in spinal cord represents a compensatory response to intracranial events remains to be shown. However, previous studies in rat show neuronal degeneration of lumbar sacral spinal cord after permanent MCAO, accompanied by enhanced tissue immunolabeling for c fos and OX-42 (a microglial marker), and increased local levels of TNFα and IL-1β (Fu et al, 2004; Wu and Ling, 1998a, b). Disagreement between these earlier findings and the present ones may be related to the greater severity of a permanent MCAO model, species differences (rat versus mouse), or tissue sampling (lumbar-sacral cord as compared with the total cord sample in our study). In this regard, it is noteworthy that Smith et al (2004) found a correlation between peak levels of plasma IL-6 and stroke size, severity, and long-term outcome. These data would support the possibility that less severe focal ischemia might induce less pronounced changes in systemic cytokine secretion.
The major finding in this study was the rapid and widespread increase in production of inflammatory factors (TNF-α, IL-6, IL-2, MCP-1, and MIP-2) by basal and activated splenocytes that occurred as early as 6 h after stroke, with similar changes occurring later in the spleen as well as in lymph nodes and blood. Our experiments were performed in male C57BL/6 mice, exclusively, and both genetic strain and sex influence innate immunologic responses to injury. For example, C57BL/6 and Balb/c differ greatly in stress sensitivity and display of dominant immune characteristics (Chiodini and Buergelt, 1993) However, the C57BL/6 strain is a common strain employed in experimental stroke and is well-characterized in terms of lymphoid and leukocyte populations and immunocompetence (Kajioka et al, 2000).
While there were many similarities in the pattern of expression of splenic versus brain cytokines and chemokines, there were also striking differences. In particular, splenic T cells activated with anti-CD3/CD28 antibodies produced a significant increase in IFN-γ (not observed in brain), but no levels of IL-1β (observed only in brain). Others have evaluated the effects of MCAO on lymphoid tissue, demonstrating extensive loss of lymphocytes in spleen and thymus, a shift from T helper cell (Th1) to Th2 cytokine production and increased lymphocyte apoptosis by 12 h of reperfusion (Gendron et al, 2002; Prass et al, 2003). We observed enormous splenic T-cell cytokine/chemokine production that was readily measured by early reperfusion (minimum 6 h). These observations are significant in that they may play a role in the adaptive immune response to stroke. Recent data in humans suggest that T cell mediated immune responses are important to both stroke pathogenesis and outcome (Nadareishvili et al, 2004). Interestingly, induction of T cell tolerance to brain antigens reduced postischemic brain damage (Becker et al, 1997). Moreover, data from neuronal models of traumatic injury suggest that T cells primed to respond to myelin basic protein can enhance recovery (Hauben et al, 2000; Moalem et al, 1999). One possibility that must be considered is that a dysfunctional blood-brain barrier leads to exposure of normally cloistered brain structural elements, initiating autoimmune-like processes and cell-mediated immune defenses in the periphery that may have varying effects on stroke progression. However, autoimmune responses to newly released brain antigens would require days, rather than hours, to manifest.
Given the location of the splenic T cells distant from the site of cerebral stroke, and low level of infiltrating mononuclear cells yet present in the injured brain (Stevens et al, 2002), it is unlikely that inflammatory cells found in the spleen at 6 h represent brain-infiltrating cells that had emigrated out of the damaged brain. However, a second intriguing possibility is that the rapid inflammatory response observed in the spleen and the subsequent spread of activated lymphocytes to lymph nodes and blood resulted from sympathetic neural stimulation initiated from within the damaged brain tissue. Norepinephrine and β2-adrenergic receptor (β2AR) stimulation from infection and injury has been strongly implicated in the regulation of the immune response (reviewed in Kohm and Sanders, 2001), and it is conceivable that brain injury might also transmit sympathetic signals to the spleen. This possible explanation has a number of merits: (1) cytokines such as IL-1β can directly activate sympathetic neurons, which are known to express IL-1R, thus allowing for the possibility that the early induction of IL-1 after stroke might induce efferent sympathetic signals to peripheral lymphoid organs, including spleen; (2) sympathetic stimulation results in the local release of norepinephrine in lymphoid organs including spleen; (3) the β2AR for norepinephrine is selectively expressed by CD4+ T cells and B cells; (4) norepinephrine drives Th1 cell differentiation and secretion of IFN-γ through stimulation of β2AR on naïve CD4 + T cells by augmenting the IL-12 signaling pathway (Swanson et al, 2001); (5) norepinephrine increases the number of circulating lymphocytes, and thus might account for the later appearance of T cells in the lymph nodes and blood, which also produced inflammatory cytokines on stimulation through the TCR.
Lastly, our novel demonstration of a drastic and rapid release of inflammatory cytokines from activated splenocytes and lymphoid tissue may represent only the initial challenge for the peripheral immune system imperiled by stroke. In a murine model similar to the present study, β-adrenoreceptor mediated systemic immunodeficiency was readily manifested at 3 days after cerebral ischemia, characterized by defective IFN-γ production, failure of lymphocyte activation, septicemia, and pneumonia (Prass et al, 2003). We speculate that the ischemic brain uses sympathetic neural signaling to trigger, and ultimately exhaust, endogenous immune defenses against injury, leading to deleterious and organ-specific consequences.
Footnotes
Acknowledgements
The authors thank Ms Eva Niehaus for assistance in preparing and submitting the manuscript.