
Abstract
Cerebral inflammation involves molecular cascades contributing to progressive damage after traumatic brain injury (TBI). The chemokine CC ligand-2 (CCL2) (formerly monocyte chemoattractant protein-1, MCP-1) is implicated in macrophage recruitment into damaged parenchyma after TBI. This study analyzed the presence of CCL2 in human TBI, and further investigated the role of CCL2 in physiological and cellular mechanisms of secondary brain damage after TBI. Sustained elevation of CCL2 was detected in the cerebrospinal fluid (CSF) of severe TBI patients for 10 days after trauma, and in cortical homogenates of C57Bl/6 mice, peaking at 4 to 12 h after closed head injury (CHI). Neurological outcome, lesion volume, macrophage/microglia infiltration, astrogliosis, and the cerebral cytokine network were thus examined in CCL2-deficient (−/−) mice subjected to CHI. We found that CCL2−/− mice showed altered production of multiple cytokines acutely (2 to 24 h); however, this did not affect lesion size or cell death within the first week after CHI. In contrast, by 2 and 4 weeks, a delayed reduction in lesion volume, macrophage accumulation, and astrogliosis were observed in the injured cortex and ipsilateral thalamus of CCL2−/− mice, corresponding to improved functional recovery as compared with wild-type mice after CHI. Our findings confirm the significant role of CCL2 in mediating post-traumatic secondary brain damage.
Introduction
Following focal traumatic brain injury (TBI), secondary cellular and molecular processes, including inflammation, excitotoxicity, and oxidative stress, may contribute to delayed neuronal loss (Morganti-Kossmann et al, 2001, 2007). Hematogenous macrophages migrate across the injured blood–brain barrier and accumulate in the lesioned parenchyma, releasing a plethora of inflammatory cytokines which sustain immune responses and contribute to both secondary tissue damage and repair (Morganti-Kossmann et al, 2001; Popovich et al, 1999). Due to their ability to induce directional migration of leukocytes, chemokines are considered essential mediators of post-traumatic neuroinflammation.
The potent chemotactic capacity of CC ligand-2 (CCL2; formerly monocyte chemoattractant protein-1, MCP-1) for monocytes, macrophages, and microglia has been well-established by means of transgenic mice, which demonstrate mononuclear-rich infiltrates into the targeted central nervous system (CNS) (Chen et al, 2003; Fuentes et al, 1995). In contrast, genetic deletion of CCL2 results in macrophage recruitment deficiencies in animal models of stroke and multiple sclerosis, implying a unique, non-redundant role for this chemokine in inflammatory neuropathologies (Huang et al, 2001; Hughes et al, 2002; Lu et al, 1998). Experiments using gene-knockout mice have also showed the involvement of the CCL2 receptor CCR2 in mediating macrophage accumulation in spinal cord injuries (Ma et al, 2002).
Predominantly produced in the CNS by astrocytes (Berman et al, 1996; Glabinski et al, 1996), CCL2's expression is rapidly elevated in response to stab-wound, aspiration, and diffuse axonal injury (Glabinski et al, 1996; Hausmann et al, 1998; Muessel et al, 2000; Rancan et al, 2001). CCL2 overexpression exacerbated ischemic brain injury in mice (Chen et al, 2003), whilst increased production of this chemokine has been associated with neurological dysfunction after traumatic axonal injury in rats (Rancan et al, 2001). However, upregulation of CCL2 has not been investigated thoroughly in human TBI, nor has the impact of CCL2 elevation been examined in terms of tissue degeneration and functional outcomes after experimental TBI (Semple et al, 2009).
This study thus explored the role of CCL2 as a potential mediator of secondary damage after TBI using two approaches. First, we investigated CCL2 levels in the cerebrospinal fluid (CSF) of patients over 10 days after severe TBI. Second, a mouse model of focal TBI was used to examine alterations in cerebral cytokine production, lesion volume, neuronal loss, macrophage accumulation, astrocyte activation, and neurological recovery in CCL2-knockout (−/−) mice as compared with that in wild-type (wt) controls. Our findings confirm a non-redundant role of CCL2 in the pericontusional accumulation of macrophages, associated with improvement in neurological dysfunction and brain tissue damage in CCL2−/− mice after experimental TBI. As such, further studies to therapeutically inhibit this chemokine to reduce TBI-mediated secondary brain injury are warranted.
Materials and methods
CCL2 Measurements in the CSF of TBI Patients
Patients included in this study (n = 21) were admitted either to the Department of Trauma Surgery, University Hospital Zurich, Switzerland (n = 8 males; mean age 33 years) or the Department of Trauma Surgery, Alfred Hospital Melbourne, Australia (n = 10 males and 3 females, mean age 36 years). All patients suffered from severe TBI, defined as a Glasgow coma scale score < 8 upon hospital admission and morphological abnormalities on primary computed tomography scan. All patients received intraventricular catheters for continuous intracranial pressure monitoring, and CSF was drained when intracranial pressure exceeded 15 mm Hg. This did not interfere with patient management, and received approval from the Zurich University Hospital Ethics Board and the Alfred Human Research Ethics Committee.
CSF samples were collected daily from admission (day 0) to day 9 after injury, and were centrifuged (1,000 r.p.m., 10 mins, 4°C) then aliquoted for storage at −80°C. Control CSF from non-traumatized patients without infections, autoimmune, cancerous, or ischemic disease (n = 15) were collected by diagnostic lumbar puncture or ventriculoperitoneal shunt. Analysis of CCL2 in CSF samples was performed by using commercially available ELISA kits, with a lower detection limit of 5.0 pg/mL (R&D Systems, Minneapolis, MN, USA), and data are expressed as pg/mL CSF.
Mouse CHI Model
Mice deficient in CCL2 (B6.129S4-Ccl2tm1Rol/J) on a C57Bl/6 background (Lu et al, 1998) were purchased from Jackson Laboratory (Bar Harbor, ME, USA). A homozygote breeding colony was established at the Alfred Medical Research and Education Precinct (AMREP) Animal Centre, Melbourne, Australia. C57Bl/6 mice were used as wt controls. Experimental closed head injury (CHI) was performed as previously detailed (Bye et al, 2007; Chen et al, 1996; Stahel et al, 2000), and approved by the AMREP Animal Ethics Committee. Male mice (12 to 15 weeks, 27 to 34 g) were ether-anesthetized and received unilateral, focal trauma to the left hemisphere by an electric weight-drop device, resulting in skull fracture and underlying cortical contusion. After trauma, the scalp incision was sutured and mice were allowed to recover on heat pads. Sham-operated mice underwent anesthesia and surgery without CHI.
Assessments of Functional Impairment
Neurological severity score (NSS):
A 10-point Neurological Severity Score (NSS), described previously in detail, was used to assess post-traumatic neurological impairments (Beni-Adani et al, 2001; Flierl et al, 2009; Stahel et al, 2000). One point was given for failure of an individual task, while no points were given for succeeding. Testing was conducted by an investigator masked to treatment and strain, initially at 1 h after injury, daily for 7 days, then twice-weekly to 4 weeks.
Ledged beam test:
A ledged beam test, described previously by our group in this model (Bye et al, 2007), was used to examine hemiparesis after trauma. Mice underwent three consecutive trials at times corresponding to NSS assessment, and scores were averaged after masked analysis of videotape recordings.
Tissue Collection and Processing for Immunohistochemical Analysis
For immunological analysis of the brains, mice (n = 6 to 7 per strain) were killed at 2, 4, 12, 24 h, and 4 and 7 days after CHI, while sham-operated mice (n = 3 per strain) were killed at 2 h after sham operation. Mice were ether-anesthetized before decapitation and fresh brains were frozen in TissueTek OCT (Sakura, Torrance, CA, USA) for storage at −80°C. Consecutive coronal cryosections (12 μm) were collected for each brain.
A separate, additional cohort of mice was killed at later time points: 7, 14, and 28 days after CHI (n = 6 to 7 per strain), and 28 days after sham operation (n = 4 per strain). These mice were terminally anesthetized with urethane (10 mL/kg of a 25% w/v, intraperitoneal) and perfused transcardially with 4% paraformaldehyde. Brains were post-fixed in Bouin's solution and then 70% ethanol, and paraffin-embedded serial coronal sections (5 μm) were cut. Approximately 300 sections from Bregma+ 0.5 to −4.0 mm were collected per brain to ensure inclusion of the entire lesioned cortex.
TUNEL Staining
For identification of dying cells, terminal deoxynucleotidyltransferase-mediated dUTP nick 3′-end labeling (TUNEL) was performed as previously described in detail by our group (Bye et al, 2007). All TUNEL-positive cells in the injured cortex were counted on each stained section (8 to 10 sections per brain, 500 μm apart). Cell counts were summed per brain and expressed as group means for analysis.
Hematoxylin and Eosin (H&E)
To assess the lesion core and pericontusional tissue damage, 12 to 14 sections per brain (250 μm apart) were stained with hematoxylin and eosin (H&E). Due to differences in tissue processing between fresh-frozen and paraffin-embedded brains, no direct comparisons were made between early (2, 4, 12, and 24 h; 4 and 7 days) and later time points (7, 14, and 28 days after CHI). Paraffin embedding was adopted as we found this technique to better preserve tissue morphology as degeneration progressed over time. Note that a 7-day time point was included in both cohorts to ensure data reproducibility.
Immunohistochemistry
Immunohistochemistry was performed with sections 500 μm apart throughout the lesioned brain, with 8 to 10 sections per brain analyzed for each antibody. This allowed for the visualization and quantification of infiltrating macrophages/microglia (monoclonal rat anti-mouse F4/80; Serotec, Raleigh, NC, USA), activated astrocytes (rabbit polyclonal glial fibrillary acidic protein (GFAP); Dako, Carpinteria, CA, USA), neuronal loss (monoclonal anti-mouse NeuN; Chemicon, Billerica, MA, USA), and localization of the chemokine receptor CCR2 (goat polyclonal CCR2; Abcam, Cambridge, MA, USA). Briefly, antigen retrieval was performed on paraffin sections by microwave treatment in 0.1% sodium citrate buffer (for NeuN and CCR2) or digestion with proteinase-K (20 mg/mL in 10 mmol/L Tris (pH 8.0), for F4/80). Sections were blocked in 4% milk in phosphate-buffered solution and serum before primary antibody incubation: overnight at 4°C for F4/80 and CCR2, or 1 h at room temperature for NeuN and GFAP. The following biotinylated secondary antibodies were applied for 1 h, respectively: goat anti-rat IgG, goat anti-rabbit IgG, horse anti-mouse IgG, or horse anti-goat IgG (Vector Laboratories, Burlingame, CA, USA). Positive staining was detected using the Vectastain ABC kit (Vector Laboratories) and nickel-enhanced 3,3′-diaminobenzidine tetrahydrochloride. Each experiment included negative control sections on which primary antibodies were omitted.
Fluorescence double labeling allowed the localization of CCR2 in the brain, following the protocol described above, except for the use of a rabbit anti-goat AlexaFluor-488 detection antibody (Invitrogen, Carlsbad, CA, USA). Sections were then incubated with NeuN or GFAP antibodies, which were detected with goat anti-mouse or goat anti-rabbit AlexaFluor-594, respectively. Fluorescent images were captured and overlaid using the Olympus Cell Imaging software. Cellular coexpression of F4/80 with CCR2 was assessed differently, since the F4/80 antigen was susceptible to the microwave treatment required for CCR2 immunohistochemistry. Thus F4/80 staining was performed first and images of the secondary antibody (goat anti-rat AlexaFluor-594) were captured. The mounting medium was then removed and CCR2 staining was performed. Fluorescent images of the same sections were captured and overlaid with Adobe Photoshop 7.0, using significant landmarks (i.e., the section edge) to ensure accurate overlay positioning.
Quantification of Lesion Volume and Neuronal Loss
For measurement of the cortical lesion, images of H&E-stained sections were captured using an Olympus BX50 microscope. The Olympus Cell Imaging software was used to outline the damaged tissue area, defined by decreased H&E staining intensity and the presence of pyknotic and/or small nuclei. The total lesion volume throughout the brain was calculated using Cavalieri's formula, volume = ΣA × ISF × t, where A is lesion area, t is section thickness, and ISF is the inverse of the sampling fraction.
NeuN immunohistochemistry was chosen to complement H&E staining on tissue collected at 7, 14 and 28 days after CHI for assessment of cortical neuronal loss. The cortical regions completely lacking NeuN-labeled cells were outlined on images and the enclosed area was measured as above. The volume of neuronal loss was calculated using the Cavalieri formula.
Quantification of Immunolabeled Cells
As F4/80 is a marker for both infiltrating macrophages and activated resident microglia, a distinction was made according to cell morphology differences as described (Bye et al, 2007). Due to the vast number of densely compacted cells within the lesioned cortex, particularly at later times after injury, assessment of total cell numbers was deemed unfeasible. Thus, the spread of macrophage/reactive microglia accumulation around the lesion was assessed at 4, 7, 14, and 28 days by measuring the area of F4/80-positive staining on each section and using Cavalieri's formula to determine the F4/80-positive volume (mm3). Semi-quantitative cell counts were performed at the peak of macrophage infiltration (4 and 7 days after CHI) by capturing five frames per section (1,730 × 1,303-μm images; 8 to 10 sections per brain, × 10 magnification) spanning the lesioned hemisphere. A 400 × 400-μm grid was overlaid on each frame and F4/80-positive cells were counted within five squares systematically selected out of a possible 12 squares (total cells per mm2).
Due to their extensive distribution, GFAP-stained astrocytes were quantified by densitometric analysis using ImageJ software (National Institutes of Health, USA, v1.40). GFAP-staining intensity was examined within select regions of interest (ROIs), including cortex (within 0.35 mm2 area) and dorsolateral thalamus (1.10 mm2). Data were expressed as ‘% ROI GFAP-positive,’ corresponding to % GFAP-staining intensity of the contralateral hemisphere ROI, averaged for three sections per brain and expressed as group means. The same method was used to measure NeuN-staining intensity within the dorsolateral thalamus. All quantification of lesion volumes and immunolabeled cells was performed blinded to strain.
Cytokine and Chemokine Measurement in the Injured Cortex
For cytokine analysis, mice were killed at 2, 4, 12, and 24 h after CHI or 2 h after sham operation (n = 5). Punches (5 mm diameter) of cortical tissue comprising the lesion were homogenized in chilled extraction buffer containing Tris–HCl (50 mmol/L, pH 7.2), NaCl (150 mmol/L), 1% Triton X-100, and 1 μg/mL protease inhibitor cocktail (Complete tablet; Roche Diagnostics, Basel, Switzerland), agitated (90 mins, 4°C) and then centrifuged (12,000 g, 15 mins), and frozen at −80°C. Total protein concentrations were determined using the Bradford Assay (Bio-Rad Laboratories, Hercules, CA, USA).
The level of 18 cytokines/chemokines was determined in 200 μg total protein of punch homogenates using a Bio-Plex Mouse Cytokine Array (Bio-Rad Laboratories) as per manufacturers' instructions. CCL2 and macrophage inflammatory protein-2 (MIP-2) levels were measured using commercially available ELISA kits (R&D Systems), with minimal detection levels of < 2 and < 1.5 pg/mL, respectively.
Data Analysis
Statistical analysis was performed using Sigma Stat 2.03 (SPSS, Chicago, IL, USA) or SPSS 15.0 for Windows (SPSS). All data are expressed as mean ± s.e.m. CCL2 in patient CSF was analyzed by one-way, repeated-measures analysis of variance (ANOVA) using transformed (ln) data, and Mann–Whitney rank sum tests for comparisons with control values. Two-way RM ANOVA evaluated the NSS and ledged beam test. Immunohistochemical and cytokine measurements were analyzed by two-way ANOVA and P-values for overall effects of time or strain are reported in full to three decimal places. Tukey's post hoc test was used for comparisons between and within individual strains and time points, reported as P < 0.05 or < 0.001. Statistically significance was considered at the 5% level.
Results
Elevated CCL2 in the CSF of Severe TBI Patients
CCL2 concentration was measured by ELISA in CSF collected over 10 days from severe TBI patients (n = 21), and compared to control CSF from healthy uninjured patients (n = 15; Figure 1A). CCL2 production was highest in TBI patients on the day of admission (day 0; 18,960 ± 5,326.04 pg/mL) and remained significantly elevated on day 1 and 2 as compared with subsequent measurements up to day 9 (P < 0.001). CCL2 levels in TBI patients decreased to a plateau from day 3 onwards, of approximately 3,000 pg/mL, which, however, remained significantly higher than control levels (717.01 ± 71.5 pg/mL) at all times (P < 0.001).
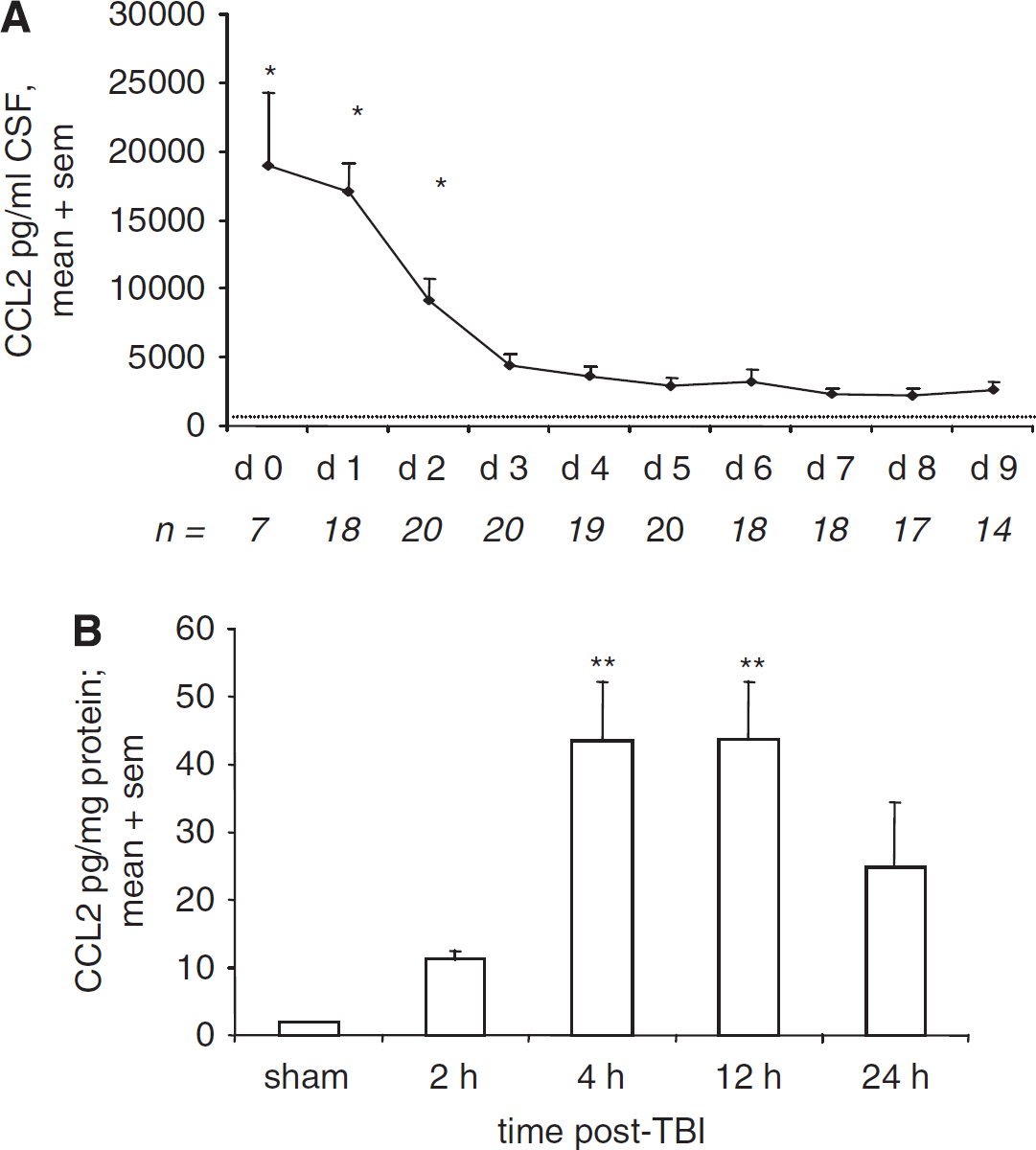
The time profiles of CCL2 production after TBI in humans and mice. CSF from severe TBI patients (n = 21) was collected by drainage of the intraventricular catheter from day 0 (day of admission) to day 9, and CCL2 levels were measured by ELISA (
Elevated CCL2 in the Injured Mouse Cortex After CHI
CCL2 measured in lesioned cortex homogenates over 24 h in C57Bl/6 mice showed an early elevation in chemokine concentration (Figure 1B). Increased CCL2 was evident by 2 h, with a marked peak 40-fold over sham levels by 4 and 12 h (P < 0.01). This elevation was transient, as by 24 h after CHI, CCL2 concentration was no longer different from sham levels (P = 0.185).
Improved Neurological Recovery in CCL2−/− mice After CHI
The abundant macrophage infiltration previously observed by our group in this model (Bye et al, 2007) may be mediated by cerebral elevation of CCL2, contributing to tissue loss and neurological deficit. Therefore, we investigated whether CCL2 deletion would affect behavioral outcomes after CHI (Figure 2). Both CCL2−/− and wt mice showed similarly high NSS scores at 1 h after CHI (P = 0.347), indicating similar trauma severity. Regardless of strain, spontaneous recovery of function was observed over time (P < 0.001). Overall, CCL2−/− mice (n = 8) had lower NSS scores as compared with wt (n = 9; P < 0.010), particularly at 1, 7, 10, 14, 17, 21, and 24 days after CHI (P < 0.05). As expected, both wt and CCL2−/− sham-operated mice showed normal function, reflected by low NSS scores (n = 4 to 5; no difference between strains, P = 0.863).
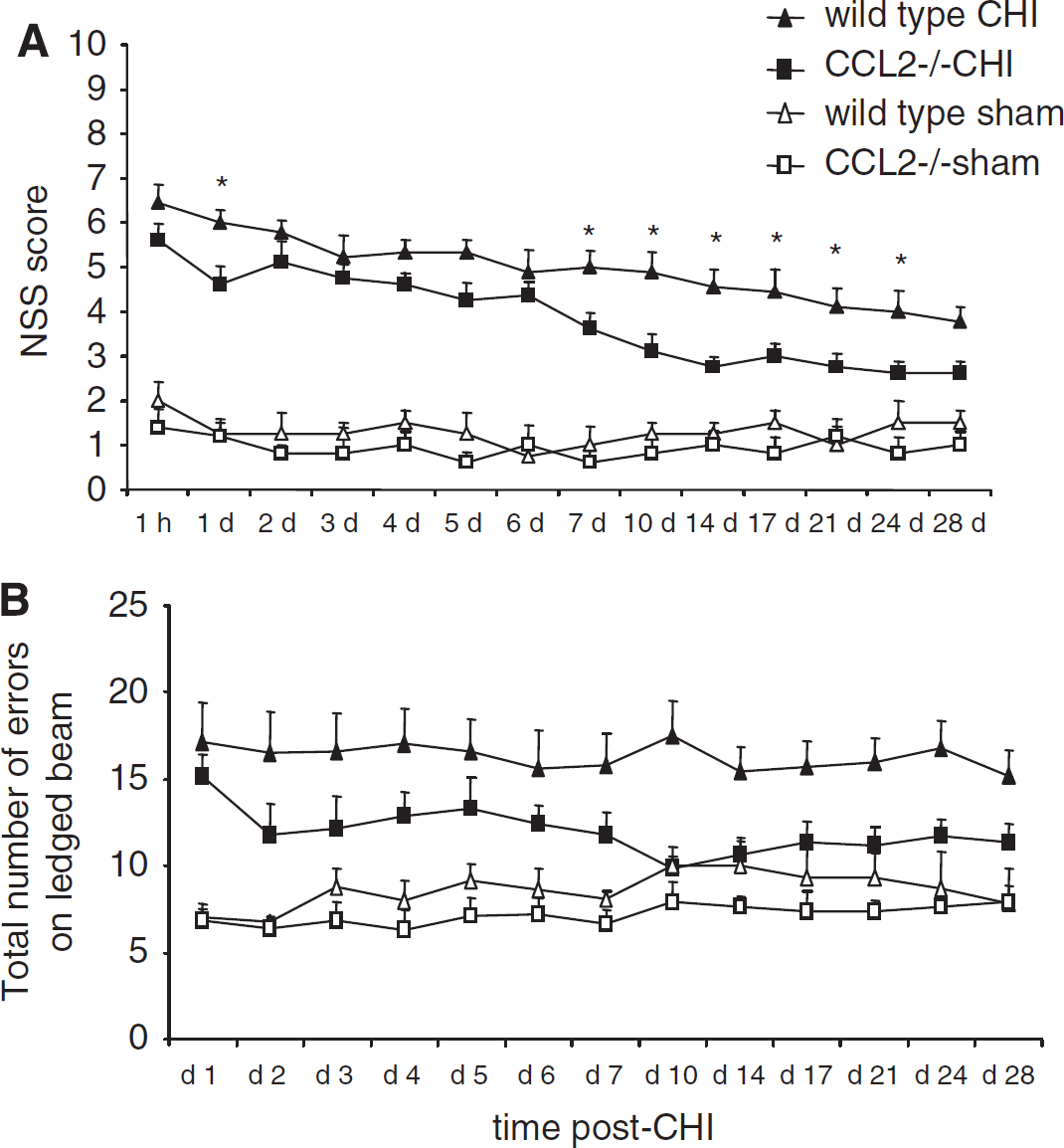
CCL2−/− mice have improved functional recovery after CHI. The NSS reflects motor function, alertness, and behavior, with a score of 10 indicating the most severe dysfunction (
The same cohorts of mice were assessed on the ledged beam test to specifically detect unilateral limb motor dysfunction. Compared with wt, CCL2−/− mice tended to make fewer errors overall by limbs contralateral to the injured cortex (right side; P = 0.115; Figure 2B). Post hoc analysis showed significant difference between the strains only at 10 days after CHI (P < 0.05). As with the NSS, no differences were evident among sham-operated mice (P = 0.949). Furthermore, a similar lower number of errors were made by limbs ipsilateral to the injury (left side; range: 4.8 to 7.9 errors) by all mice regardless of strain or treatment (P = 0.830; data not shown).
CCL2 Deficiency had no Effect on Lesion Volume or Cell Death Early After Injury
Lesion volume was measured on H&E-stained sections to examine potential differences in post-traumatic tissue loss. No differences were found between the size of lesions in wt and CCL2−/− mice up to 7 days after CHI (P = 0.275; Figure 3A). In general, lesion volumes were consistent up to 12 h, with a significant reduction of approximately 65% evident over time, regardless of strain, between 12 h and 7 days after CHI (P < 0.05).
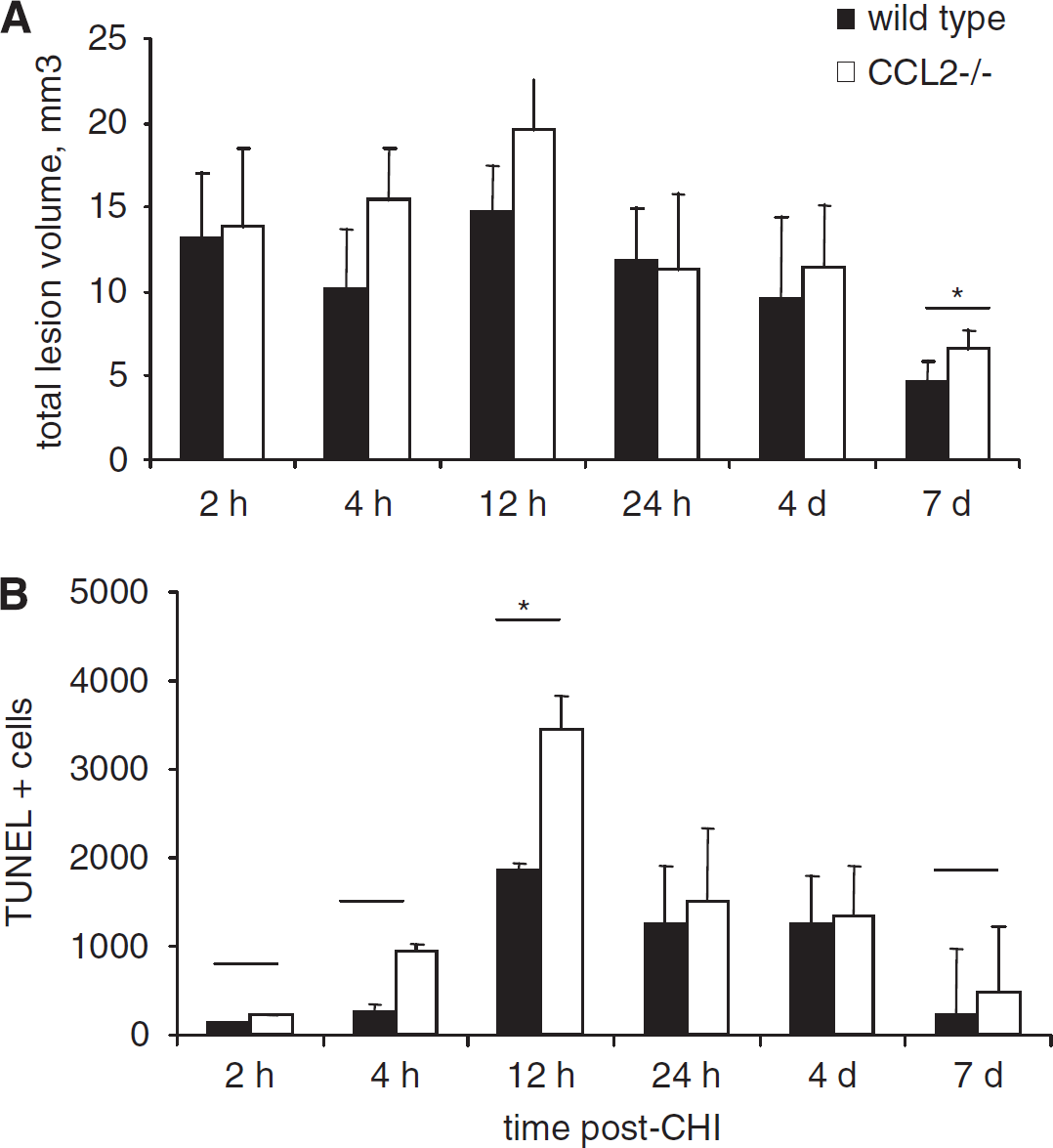
Lesion volume and cell death in wt and CCL2−/− mice over the acute time course. No differences were detected in total lesion volume between wt and CCL2−/− brains, as assessed using H&E-stained sections over 7 days after CHI (
TUNEL staining was performed on adjacent sections to determine the contribution of cell death to lesion enlargement. Cell counts in the injured hemisphere from 2 h to 7 days after CHI showed the typical time course of cell death observed previously in this model. Minimal TUNEL-positive staining was detected at 2 and 4 h, before rapid and maximal elevation at 12 h (P < 0.05, 12 versus 2 h, 4 h, and 7 days; Figure 3B). Although a trend toward greater TUNEL-positive cell numbers in CCL2−/− mice was noted, this difference from wt mice did not reach significance (P = 0.059). The similar extent of cell death in wt and CCL2−/− mice corroborates comparable lesion volumes within 1 week after injury.
Reduced Lesion Volumes in CCL2−/− Mice Over a Delayed Time Course After Injury
Contrary to the acute post-traumatic period, analysis of lesion progression over an extended time course (7 to 28 days) showed an overall significant reduction in H&E lesion volumes in CCL2−/− mice as compared with that in wt mice (P = 0.047; Figure 4A). This is particularly notable at 28 days (Figure 4B and 4C), by which time the lesions in chemokine-deficient mice were 50% smaller compared with that in wt mice (P < 0.05). Interestingly, whilst CCL2−/− lesions remained consistent from 7 to 28 days, in wt mice they tended to increase over time, indicating ongoing secondary damage. Despite progressive tissue loss, few TUNEL-positive cells were detected in either strain after 7 days of CHI and thus cell death was not quantified at these times. Variation in lesion volume magnitude between the earlier and later time courses, 2 h to 7 days (fresh frozen) versus 7 to 28 days (paraffin), can be attributed to the different tissue-processing techniques used, as paraffin embedding, while preserving tissue morphology, involves considerable dehydration of tissue.
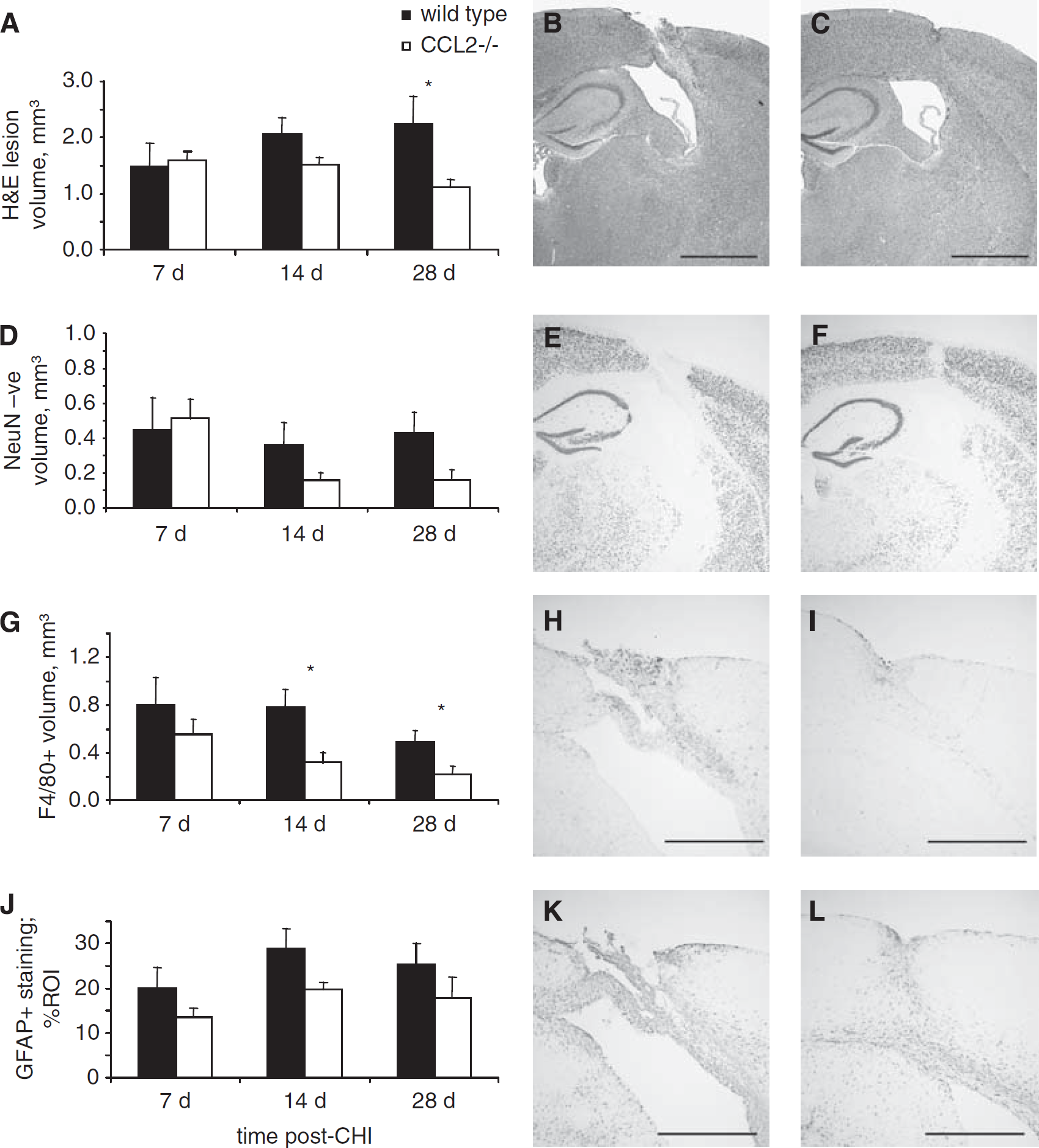
Immunohistochemical analysis of the injured hemisphere in wt and CCL2−/− mice over the delayed time course. Total lesion volumes in wt and CCL2−/− mice was assessed using H&E-stained sections (
In addition, immunohistochemistry for the neuronal marker NeuN was performed to specifically quantify cortical neuronal loss (Figure 4D). At 7 days, a similar volume of cortical neuronal loss was observed in wt and CCL2−/− mice. Whilst unchanged in wt mice at 14 and 28 days, neuronal loss appeared reduced by approximately 50% in CCL2−/− mice; however, this did not quite reach statistical significance (P = 0.088; Figure 4E and 4F).
Reduced Macrophage Accumulation in CCL2−/− Mice is Delayed After Injury
Studies by our group and others have described the time course of macrophage infiltration after CNS injury, peaking at 3 to 5 days (Schnell et al, 1999; Morganti-Kossmann et al, 2001; Bye et al, 2007). The spread of F4/80-positive cells in the injured hemisphere was evaluated to determine potential differences in macrophage infiltration/microglia activation between the strains. Contrary to our expectations, CCL2−/− and wt mice had a similar degree of accumulated macrophages and reactive microglia around the lesion site at 4 and 7 days after CHI, as evident by the volume of F4/80-positive cells, 2.34 ± 0.79 versus 4.15 ± 1.13 mm3 at 4 days, and 1.22 ± 0.53 versus 1.52 ± 0.29 mm3 at 7 days, in wt and CCL2−/− mice, respectively (P = 0.161). This was consolidated by cell counts, which found 127.72 ± 43.40 versus 153.80 ± 39.60 F4/80-positive cells at 4 days, and 91.26 ± 27.63 versus 110.27 ± 22.68 cells at 7 days, in wt and CCL2−/− mice, respectively (P = 0.430).
We next examined the spread of F4/80-positive cells around the lesion up to 28 days, based on a previous study showing reduced macrophage infiltration into CCL2−/− brains only at 2 to 3 weeks after stroke (Hughes et al, 2002). Consistent with this, we demonstrated a clear reduction in F4/80-positive cell volume in CCL2−/− mice as compared with that in wt mice over the later time course (P = 0.007 overall). This was particularly notable at 14 and 28 days after CHI, when the F4/80-positive volume was lower by approximately 46% and 27%, respectively, in CCL2−/− mice (P < 0.05; Figure 4G). Counts of total F4/80-positive cells performed at 28 days confirmed this volume reduction, with 907.14 ± 218.78 cells in wt as compared with 308.57 ± 115.46 cells in CCL2−/− mice (P < 0.05; Figure 4 H and 4I).
Reduced Astrogliosis in CCL2−/− Mice After CHI
Activation of astrocytes associated with proliferation, hypertrophy, and elevated GFAP expression is considered a hallmark of post-traumatic neuroinflammation. Widespread astrogliosis was observed over the later time course throughout the injured hemisphere, including the cortex, corpus callosum, hippocampus, and thalamus. When GFAP immunoreactivity was evaluated in the injured cortex (Figure 4J, 4K, and 4L), we found a significant reduction in CCL2−/− mice by approximately 30% as compared with that in wt mice between 7 and 28 days (P = 0.026).
Reduced Neuronal Loss and Inflammation in CCL2−/− Mice Ipsilateral Thalamus
In addition to cortical damage resulting from CHI, tissue degeneration was also observed at later time points in distal subcortical thalamic regions. The decrease in healthy neurons was thus quantified as the loss of intensity of NeuN staining within the ipsilateral thalamus ROI and was expressed as a percentage of the contralateral thalamus ROI (Figure 5A). At 7 days after CHI, a similar loss of NeuN staining was noted in the injured thalamus of wt and CCL2−/− mice, being 26.51 ± 2.81% and 20.40 ± 4.32%, respectively. By 28 days, whilst the loss of NeuN intensity remained consistent (24.38 ± 5.52%) in the CCL2−/− group, wt animals showed increased neuronal loss (42.61 ± 6.02%; Figure 5B and 5C). Overall, this difference between the strains was significant (P = 0.032).
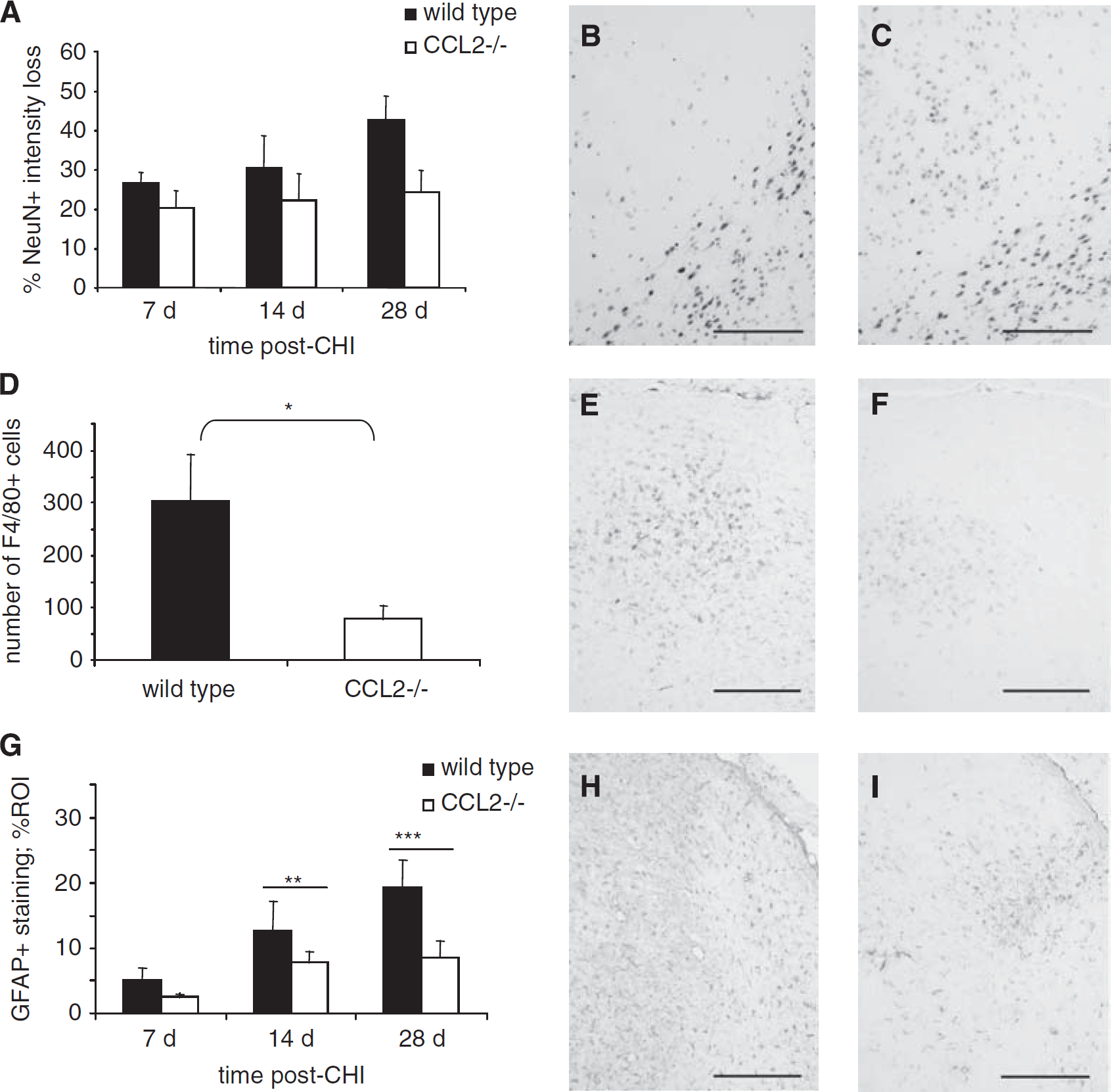
Neuronal loss and glial activation in the ipsilateral thalamus of wt and CCL2−/− mice. Neuronal loss in the ipsilateral dorsal thalamus was determined as % NeuN-positive staining of the uninjured contralateral thalamus (
Intense microglia activation was also detected in the dorsal ipsilateral thalamus after injury (Figure 5D, 5E, and 5F). Cell counts showed fewer F4/80-positive microglia in the thalamus of CCL2−/− mice (79.29 ± 90.08 cells) as compared with that in wt controls (302.71 ± 24.14; P < 0.05) at 28 days. Furthermore, abundant GFAP-positive reactive astrocytes were identified in this region, particularly within the thalamic nuclei, which also showed neuronal loss and microglial staining (Figure 5H and 5I). Overall, thalamic GFAP intensity was lower in CCL2−/− mice from 7 to 28 days after CHI as compared with wt mice (P = 0.026; Figure 5G). Whilst GFAP intensity remained relatively consistent over the time course in CCL2−/− mice, astrocyte activation in wt mice increased steadily (overall effect of time, P < 0.001), peaking at 28 days approximately 50% higher than CCL2−/− mice.
The CCR2 Receptor is Expressed in a Subpopulation of Pericontusional Macrophages
CCR2 is the primary receptor through which CCL2 mediates the chemotaxis of monocytic cells (Kuziel et al, 1997). Double immunofluorescence for CCR2 in combination with NeuN, GFAP, or F4/80 was applied on sections at the peak of macrophage infiltration (4 days) to identify the cellular localization of CCR2 after injury. Despite previous studies showing the expression of CCR2 on neurons and glia (Banisadr et al, 2002; Stamatovic et al, 2005), we found no overlap of CCR2 with either NeuN or GFAP staining (Figure 6). The receptor was however detected on a small subset of F4/80-positive amoeboid macrophages bordering the lesion core. Due to the small number of CCR2-positive cells, no quantification was made in wt or CCL2−/− mice.
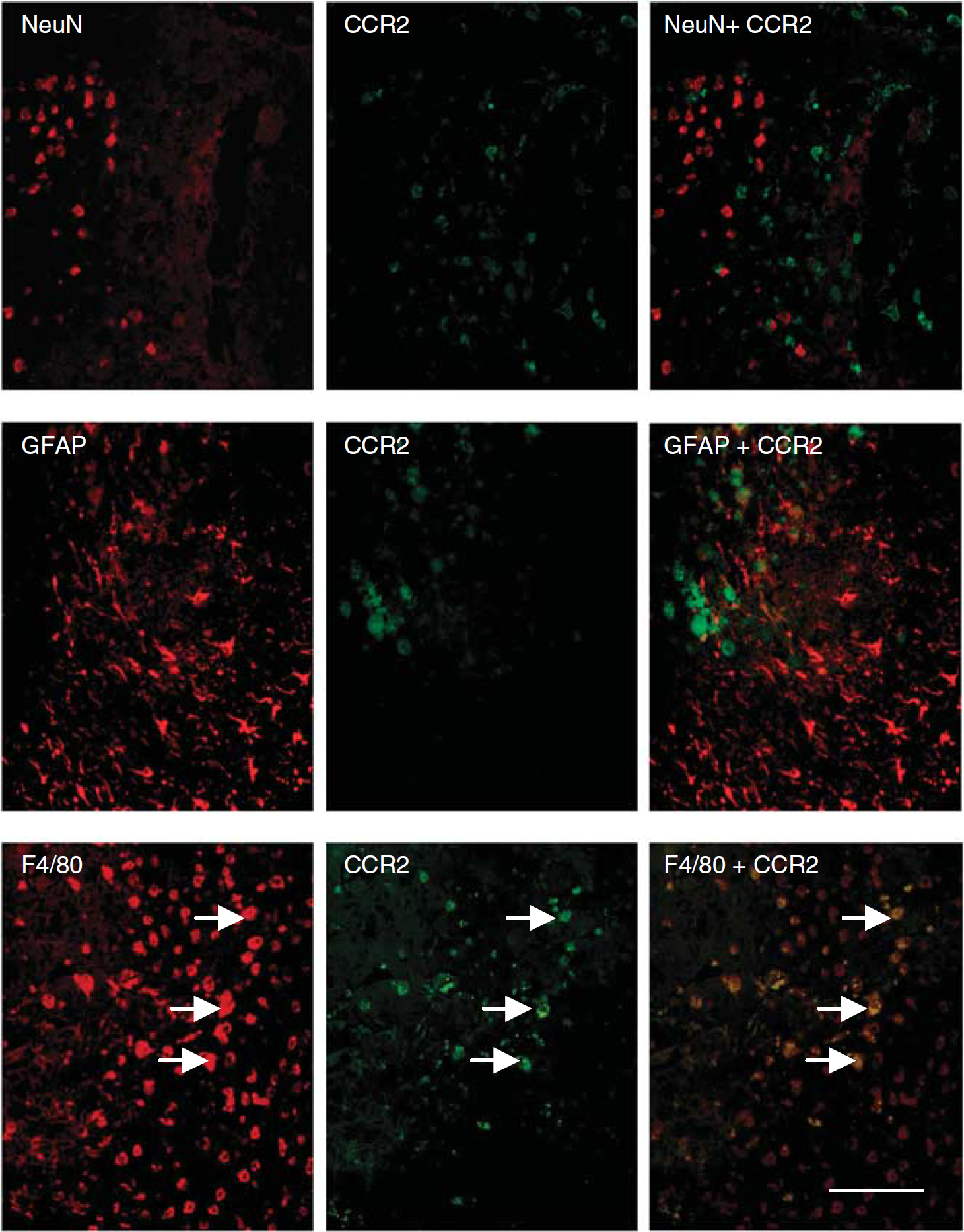
Localization of CCR2 receptor on F4/80-positive amoeboid macrophages. Double-labeling immunofluorescence of CCR2, in combination with NeuN, GFAP, or F4/80 illustrates the cellular localization of CCR2 at 4 days after CHI in wt mice. In the pericontusional cortex adjacent to the lesion core, no colocalization of NeuN or GFAP was detected with CCR2 staining (upper and middle panels). In contrast, CCR2 expression was evident within a subset of F4/80-positive amoeboid macrophages accumulating in the lesion (lower panels; arrows). Scale bar = 100 mm.
Altered Expression of Inflammatory Cytokines in CCL2−/− Mice
To elucidate the potential mechanisms underlying the histological and cellular differences identified in traumatized CCL2−/− mice, we assessed an array of inflammatory cytokines in injured cortex homogenates from CCL2−/− and wt mice over 24 h after CHI (Figure 7). Clear differences in the acute inflammatory response were evident. Of the 19 cytokines measured, interleukin-3 (IL-3), IL-4, IL-5, and IL-17 were below detectable levels, in both sham and injured cortex samples (data not shown).
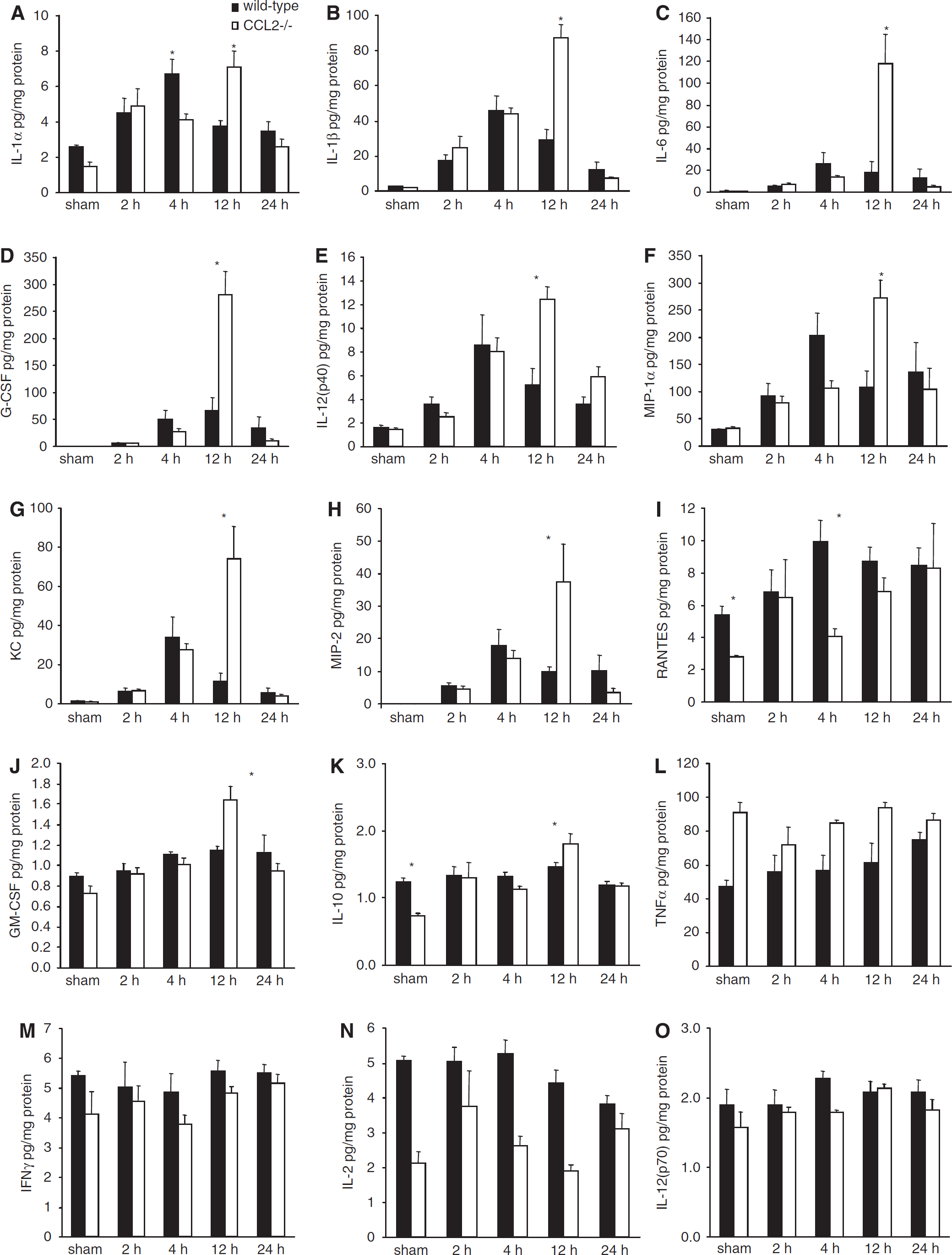
Inflammatory cytokine/chemokine expression in brain homogenates after CHI. The levels of 15 cytokines were detected in the homogenates of lesioned cortex over 24 h after CHI in wt and CCL2−/− mice (n = 5). Production of the cytokines/chemokines IL-1α, IL-1β, IL-6, granulocyte colony-stimulating factor, IL-12(p40), MIP-1α, KC, and MIP-2 (
The most characteristic finding was delayed and exacerbated production of several cytokines in the brains of CCL2−/− mice as compared with wt. The levels of IL-1α, IL-1β, IL-6, granulocyte colony-stimulating factor (G-CSF), and IL-12(p40) (Figure 7A, 7B, 7C, 7D, and 7E) were all significantly induced by CHI compared to sham-operation in both mouse strains (P < 0.001 for each cytokine). Production of the chemokines MIP-1α, keratinocyte-derived chemokine (KC), and MIP-2 (Figure 7F, 7G, and 7H) were also significantly elevated after CHI in both strains (P < 0.001 for each chemokine). Concentrations of these eight inflammatory mediators in wt mice peaked at 4 h before subsequent decrease over 24 h. In contrast, peak expression in CCL2−/− mice was, first, enhanced in respect to the maximal levels in wt mice, and, second, delayed to 12 h after CHI (P < 0.05 overall difference between strains; P < 0.05 wt versus CCL2−/− at 12 h).
Although also elevated in response to CHI (P = 0.049), production of the macrophage chemoat-tractant RANTES (Figure 7I) was lower overall in CCL2−/− mice as compared with that in wt mice (P = 0.022). This was noted in sham-operated CCL2−/− mice, as well as in traumatized CCL2−/− mice at 4 h (P < 0.05).
Production of the cytokines GM-CSF and IL-10 (Figures 7J and 7K) increased over time after CHI (P < 0.001), but at lower concentrations as compared with those of many other cytokines measured. In general, GM-CSF and IL-10 followed a similar time course in wt and CCL2−/− mice, with no significant differences between strains overall (P = 0.256 and 0.965, respectively). However, post hoc analysis showed higher levels of both cytokines in CCL2−/− mice specifically at 12 h after CHI (P < 0.05). Interestingly, the concentration of the multifunctional cytokine tumor necrosis factor-α (TNFα) (Figure 7L) was found significantly higher in CCL2−/− mice as compared with that in wt mice, in both sham-operated and post-CHI groups (P < 0.001).
On the contrary to the aforementioned mediators, other cytokines were unaffected by CHI. Interferon-γ, IL-2, and IL-12(p70) (Figures 7M, 7N, and 7O) did not vary over time after injury, nor did their level change significantly from those in sham-operated mice (P = 0.295, 0.097, and 0.162, respectively). However, concentrations of these cytokines were significantly different between strains independent of time after CHI (P = 0.017, 0.001, and 0.033, for interferon-γ, IL-2, and IL-12(p70), respectively), being lower in CCL2−/− mice as compared with wt.
Discussion
In this study we have explored the relevance of the chemokine CCL2 to post-traumatic inflammation and secondary brain damage progression after TBI. We show for the first time that increased CCL2 levels in the CSF of TBI patients are maximal early after injury, with elevation maintained for over 10 days, perhaps indicative of an ongoing role for CCL2 in delayed inflammatory processes. Another group has recently reported higher CCL2 levels in the serum of patients who died after TBI, suggesting an association with adverse outcome (Rhodes et al, 2009a). Consistent with CCL2 induction, previous work by our laboratory and others has established increased and protracted production of other inflammatory mediators, including IL-6, IL-8, TNFα, and IL-10, in both adult and pediatric TBI patients (Buttram et al, 2007; Morganti-Kossmann et al, 2007).
Experimentally, a number of studies have established the upregulation of CCL2 in various brain injury paradigms (Glabinski et al, 1996; Muessel et al, 2000; Rancan et al, 2001). CCL2 was elevated at 6 to 12 h after cortical stab-wound in rats and mice (Berman et al, 1996; Glabinski et al, 1996), and peaked 8 to 12 h after rat lateral fluid percussion injury (Rhodes et al, 2009b). Consistently, we observed maximal CCL2 levels at 4 and 12 h after CHI in wt mice. Despite differences in species, temporal profiles, and sampling (CSF versus brain homogenates), CCL2 production in both TBI patients and CHI mice shows a similar acute upregulation. This suggests that intrathecal chemokine production is one of the initial responses to TBI, triggering subsequent neuroinflammatory cascades.
To understand the functional and pathological role of CCL2 after TBI, we first investigated whether the absence of CCL2 in gene-deficient mice affected neurological deficit. CCL2−/− mice showed improved recovery of neurological function as compared with their wt counterparts over 4 weeks after injury, with significantly lower NSS scores and a trend toward reduced motor dysfunction on the ledged beam test. This improvement may be attributed to reduced macrophage accumulation at later times, corroborating the marked recovery in hindlimb function seen in macrophage-depleted spinal cord-injured rats (Popovich et al, 1999). Together these findings implicate macrophages as effectors of secondary damage contributing to functional loss after CNS injury. Further studies may be required to determine whether CCL2 deficiency has a neuroprotective effect on long-term cognitive recovery, another important outcome parameter in TBI.
Subsequent analysis was undertaken to identify morphological changes, possibly elucidating the underlying mechanisms of improved outcomes observed in CCL2−/− mice. Surprisingly, CCL2 deficiency did not affect lesion volumes acutely after injury. Similarly, the extent of cell death quantified up to 7 days after CHI was comparable in CCL2−/− and wt mice. Consistent with previous studies, maximal TUNEL-positive cells were detected 12 to 24 h after injury, primarily localized pericontusionally (Conti et al, 1998; Stahel et al, 2000); however, few TUNEL-positive cells were detectable at later time points.
Despite the lack of differences in lesion volumes and cell death early after CHI, by 28 days CCL2−/− mice showed a 50% reduction in tissue damage as compared with wt mice. It is conceivable that more extensive lesions seen in wt mice may contribute to sustained motor dysfunction. Our findings support a middle cerebral artery occlusion study, in which CCL2−/− mice developed infarcts 29% smaller than those in wt mice at 14 days, coinciding with reduced macrophage infiltration (Hughes et al, 2002).
As previously reported in focal TBI, a time-dependent accumulation of amoeboid macrophages and reactive microglia was observed within the lesioned cortex (Bye et al, 2007; Schnell et al, 1999). The detection of CCR2 on only a subset of F4/80-positive cells in the brain may indicate internalization of the receptor resulting in its down-regulation on most infiltrated macrophages after CCL2 binding and chemotaxis (Dzenko et al, 2001; Taylaska et al, 2002). The contribution of different subsets of macrophages/microglia in promoting either repair or degeneration is still unclear. The neuroprotection evident in CCL2−/− mice may be attributed to a reduction in the total infiltrate of F4/80-positive cells, or alternatively, reflect the absence of a subset of highly inflammatory macrophages expressing CCR2 (Ly-6Chigh CCR2+ CX3CR2−), thus resulting in an environment more favorable to neuronal survival (Geissmann et al, 2003).
The spread of F4/80-positive cells in the injured cortex was significantly reduced in CCL2−/− mice as compared with that in wt mice at 14 to 28 days after CHI, despite similar cell numbers and volumes at 4 to 7 days. Previously, depletion of hematogenous macrophages in stroke showed that mononuclear phagocytes accumulated in the injured brain are initially resident microglia at 3 to 4 days, and blood-derived macrophages at 6 to 7 days (Schroeter et al, 1997). Similarly, by using GFP-transgenic bone marrow CCL2−/− chimeras, CCL2-deficient mice have been shown to have a normal early microglial response to ischemic injury, but a reduction in F4/80-positive/GFP-positive hematogenous macrophages at 7 days after injury (Schilling et al, 2009). This suggests that mediators other than CCL2 (such as MIP-1α) may promote early microglial activation and migration after brain injury, while CCL2 specifically drives the delayed recruitment of peripheral macrophages. The considerable lag between acute elevation of cerebral CCL2 at mRNA (Gourmala et al, 1997; Hausmann et al, 1998) and protein levels (Bye et al, 2007; Glabinski et al, 1996; Rhodes et al, 2009b), and the delayed patterns of cell accumulation in damaged parenchyma over weeks after injury, needs to be further elucidated.
Parallel with reduced macrophage accumulation, CCL2−/− mice also presented attenuated astrocyte reactivity in the injured cortex as compared with wt mice, consistent with findings after CCL2 inhibition in a rat stab-wound model of brain injury (Ghirnikar et al, 1998). Interestingly, we observed decreased GFAP immunoreactivity in CCL2−/− mice as early as 7 days, when lesion volumes were similar, suggesting that reduced astrogliosis may precede and contribute to reduced neuronal loss in CCL2−/− mice. In addition, the presence of reactive microglia and astrocytes in the ipsilateral thalamus, corresponding to areas of neuronal loss, implicates these inflammatory cells in retrograde degeneration of neurons connecting to the injured cortex (Hermann et al, 2000). Thalamic glial activation was considerably reduced in CCL2−/− mice over the later time course after CHI, consistent with studies reporting attenuated thalamic glial activation in CCL2−/− mice after middle cerebral artery occlusion and visual cortex lesions (Hughes et al, 2002; Muessel et al, 2002). Attenuated astrocytic and microglial responses may simply reflect smaller lesion volumes seen in CCL2−/− mice at these times (Hughes et al, 2002). Alternatively, as expression of CCL2 has been detected in the wt thalamus after cortical injury, the lack of CCL2 in knockout mice may directly attenuate local glial activation (Muessel et al, 2000).
One of the most dramatic differences between CCL2−/− and wt mice was the alteration in cytokine production, preceding leukocyte recruitment. Although multiple cytokines were significantly elevated after trauma in both strains, some showed delayed, exacerbated production in knockout brains. Peaks in IL-1α, IL-1β, IL-6, and G-CSF levels were detected at 12 h in CCL2−/− mice, at higher levels compared with the maximal levels at 4 h in wt mice. Furthermore, significant differences between the sham levels of IL-10 and RANTES in wt and CCL2−/− mice suggests possible compensatory mechanisms in baseline cytokine expression as a result of CCL2 deficiency.
Exacerbated cytokine production in CCL2−/− animals appears paradoxical to the delayed neuroprotection showed by these mice, perhaps indicative of a dual role for this chemokine. Interestingly, TNF−/− mice have improved memory and motor function acutely after TBI as compared with wt controls, but greater tissue loss and neurological impairments by 4 weeks, indicating that this inflammatory mediator is detrimental during the acute phase after TBI, but necessary for long-term recovery (Scherbel et al, 1999). It is feasible that CCL2 may have similarly differential acute and chronic effects in the injured brain, particularly in the context of novel neuroprotective and antiapoptotic functions showed in vitro (Eugenin et al, 2003). Alternatively, another possible contributor to improved outcome in CCL2−/− mice could be the ability of cytokines to trigger neurotrophin synthesis, as shown previously (Kossmann et al, 1997), independent of a reduction in macrophage infiltration. This hypothesis may explain why improvement in neurological dysfunction in CCL2−/− mice was observed from 7 days onwards, while a significant reduction in tissue damage was detected later at 28 days.
In summary, we have demonstrated that CCL2 is significantly elevated in patients' CSF after severe TBI, similar to mice subjected to CHI. The animal data support a primarily deleterious function of CCL2 in the injured brain, as deletion of CCL2 leads to reduction in lesion volume, neuronal loss, macrophage accumulation, and astrocyte activation, corresponding to improved neurological outcome up to 4 weeks after injury. Furthermore, altered production of numerous cytokines in CCL2−/− brains indicates a previously unrecognized role for CCL2 as a modulator of acute CNS inflammation through unknown mechanisms. From this study, we conclude that further preclinical investigations to therapeutically inhibit CCL2-mediated macrophage infiltration in TBI are warranted.