
Abstract
Diabetic hyperglycemia increases brain damage after cerebral ischemia in animals and humans, although the underlying mechanisms remain unclear. Gender-linked differences in ischemic tolerance have been described but have not been studied in the context of diabetes. In the current study, we used a model of unilateral common carotid artery ligation, combined with systemic hypoxia, to study the effects of diabetes and gender on hypoxic–ischemic (HI) brain damage in the genetic model of Type II diabetes, the db/db, mouse. Male and female, control and db/db, mice were subjected to right common carotid artery ligation followed by varying periods of hypoxia (8% oxygen/92% nitrogen) to assess mortality, infarct volume, and tissue damage by light microscopic techniques. End-ischemic regional cerebral blood flow (CBF) was determined using [14C] iodoantipyrine autoradiography. Glycolytic and high energy phosphate compounds were measured in blood and brain by enzymatic and fluorometric techniques. Gender and diabetes had significant effects on mortality from HI and extent of brain damage in the survivors. Female mice were more resistant than their male counterparts, such that the severity (mortality and infarction size) in the male diabetics > female diabetics ~ male controls > female controls. End-ischemic CBF and depletion of cerebral high energy reserves were comparable among all groups. Surprisingly, female diabetic mice were more hyperglycemic and demonstrated a greater prolonged lactacidosis than the males; however, they were more resistant to damage. The results suggest a unique pathophysiology of hypoxia–ischemia in the female diabetic brain.
Diabetes is a well-known risk factor for stroke and frequently enhances tissue damage once an ischemic insult is underway. Diabetic stroke patients are typically younger than their nondiabetic counterparts, yet mortality is greater and recovery time is prolonged in the diabetic, with the greatest prevalence in Type II diabetics (Jorgensen et al., 1994). Postmenopausal diabetic women represent an at-risk group with a greater incidence of stroke as compared with age-matched men or nondiabetic women (Kuller et al., 1985; Kannel and Thorn, 1994). It has been suggested that the loss of female reproductive steroids in some manner potentiates this devastating neurologic complication of diabetes; however, there are no studies that support or refute a unique pathophysiology of stroke in diabetic women.
The effects of diabetic and hyperglycemic stoke have been confirmed in numerous experimental ischemia models. Increased lactic acid production and tissue acidosis have been most commonly correlated with the more extensive tissue damage in hyperglycemic stroke. However, acutely hyperglycemic nondiabetic rats were more vulnerable than diabetic rats with the same level of glycemia, implying a protective adaptation in the diabetic (Wagner and Lanier, 1994). Again, no such studies have examined the response of the female diabetic brain. Previous work indicates that female db/db mice, a genetic model of noninsulin dependent (Type II) diabetes, have smaller brains than wild type mice and have abnormal cerebral glucose utilization (Vannucci et al., 1997). The defect in the db/db mouse is because of a point mutation in the leptin receptor, resulting in a truncated, nonfunctional protein (Chen et al., 1996; Lee et al., 1996). Thus, the db/db mouse consistently develops severe diabetes, characterized by significant hyperglycemia with hyperphagia, hyperinsulinemia, and obesity by 6 weeks of age (Coleman, 1982). The db/db mouse also does not respond to the administration of exogenous leptin (Campfield et al., 1995).
This study is the first to examine cerebral ischemic outcome and ischemic glucose metabolism in the db/db strain. We examined the hypothesis that diabetic stroke is gender-dependent, and that the mechanism of injury is dependent on altered ischemic glucose metabolism with enhanced acidosis. We focused on fundamental pH-related mechanisms because hyperglycemic acidosis is so well established as an enhancer of histologic damage in every known animal and clinical model of ischemic stroke. The current results indicate that the female diabetic brain actually sustains increased hyperglycemia and prolonged lactic acidosis relative to males, yet is surprisingly resistant to infarction.
MATERIALS AND METHODS
Animals
C57BL/KsJ db/db and sex-matched controls were purchased from Jackson Laboratories (Bar Harbor, Maine, U.S.A.) at 6 weeks of age; hypoxia–ischemia (HI) was induced at 8 to 9 weeks of age. All animals were allowed food and water ad libitum until the day of the experiment. All animal protocols were approved by the Pennsylvania State University College of Medicine Animal Care and Use Committee.
Unilateral cerebral hypoxia–ischemia
Hypoxia–ischemia was induced in male and female, control and db/db, mice by a modification of the procedure developed for the immature rat (Rice et al., 1981; Vannucci et al., 1996b; O'Donnel et al., 1996). On the morning of the experiment, animals were anesthetized with halothane (4% in 70% N2O:30% O2) and a small incision was made in the neck. The right carotid artery was exposed and double-ligated with 4-O surgical silk, the incision was sutured, and the animals were allowed to recover with access to food and water for 3 hours. Hypoxia was induced by placing each animal in 500-mL glass jars partially submerged in a temperature-controlled water bath. Preliminary studies determined that maintaining the water bath at 35.5°C maintained the animals' core body temperature at 37.5°C to 37.7°C throughout the hypoxic interval. Two animals from each group—that is, wild type female (wt ♀), diabetic female (db/db ♀), wild type male (wt ♂), diabetic male (db/db ♂)—were subjected to HI in a paired fashion, for a total of 20 to 25 animals/group. A humidified gas mixture of 8% O2/balance N2 was delivered for specific intervals ranging from 15 to 30 minutes. At the end of the hypoxic interval, animals were allowed to recover in room air for 30 minutes, then returned to their cages with free access to food and water. Initial mortality studies involved a 30-minute hypoxic exposure for all groups. At 24 hours of recovery from 30 minutes of H/I, surviving animals were re-anesthetized with halothane, decapitated, and brains were processed for infarction as described below. Subsequent studies used shorter hypoxic exposures as described.
Histology
Brains were removed and cut coronally into four 2-mm sections in a precooled brain slicer. Slices were incubated in a 2% solution of TTC (2,3,5-triphenyltetrazolium chloride) (Sigma, St. Louis, MO, U.S.A.) for 30 minutes at 37°C and then transferred to a 4% buffered formaldehyde solution (Bederson et al., 1986). Forty-eight hours later, the brain slices were photographed and analyzed for infarct size using an image analysis system (NIH Image 1.64). The hemispheric infarct area in each section was calculated by subtracting the area of normal, TTC-stained tissue in the hemisphere ipsilateral to the ligation from the contralateral nonischemic area to generate the infarct fraction (%), as described by Swanson et al. (1990).
In an additional group of animals subjected to either 17 minutes (males) or 22 minutes (females) of HI, brains were frozen in isopentane (–40°C) at 2 days of recovery, and 16 (μm cryosections were taken at 9 regular intervals from striatum to posterior hippocampus. Sections were stained with hematoxylin and eosin, photographed, and evaluated for difference in hemispheric area as an indication of edema and infarct fraction, as for the TTC-stained brains.
Cerebral blood flow
End-ischemic regional cerebral blood flow (CBF) was measured in a separate set of animals using [14C]iodoantipyrine ([14C]IAP), as previously described in the rat (Alkayed et al., 1998), and for use in the mouse (Sawada et al., 2000) with minor modifications. Mice were anesthetized with 1% to 1.2% halothane in an oxygen-enriched facemask, arterial and venous femoral catheters were inserted, and the right common carotid artery was exposed and ligated. Animals were placed in restraining devices (PlastiLabs, Lansing, MI, U.S.A.), loosely shrouded to minimize noise, and allowed to recover from anesthesia for 90 minutes. Rectal temperature was controlled at 37°C + 0.5°C with a heating lamp. Hypoxia–ischemia then was induced by placing the restrained animal in a barrel chamber equilibrated with 8% O2/balance N2. At the end of the hypoxic interval, arterial blood pressure, pH, Pco2, and Po2 were measured. A total of 4 (μCi [14C]IAP in 81 (μL isotonic saline was then infused intravenously over 45 seconds; freely flowing arterial blood was collected at 5-second intervals. The mouse was decapitated 45 seconds after the infusion start, the brain was removed, frozen in isopentane, and stored at −80°C. Twenty-micrometer-thick cryosections were thaw-mounted on glass coverslips and apposed to film (Kodak SB-5; Rochester, NY, U.S.A.) with 14C standards for 1 week. Blood radioactivity was determined by liquid scintillation spectroscopy after decolorization with 0.2 mL of tissue solubilizer (Soluene-350; Packard Instruments, Downers Grove, IL, U.S.A.). Autoradiographic images representing 7 coronal levels (–2, −1, 0, 1, 2, 3, and 4 mm from bregma) were digitized, and regional CBF was determined by image analysis software (Inquiry; Loats Associates, Westminster, MD, U.S.A.). Rates of regional CBF were calculated by the Kety–Schmidt modification of the Fick principle, as previously described (Jay et al., 1988; Sawada et al., 2000).
Glycolytic intermediates and high-energy phosphate
Additional cohorts of mice were briefly anesthetized with halothane at the end of HI or at 4 hours of recovery and decapitated directly into liquid nitrogen. Each brain then was removed from the skull in a cold box (–20°C), and 60 to 100 mg of tissue was dissected from the middle cerebral artery territory, powdered under liquid nitrogen, and weighed as previously described (Vannucci et al., 1997). Perchloric acid extracts were prepared from each powder as previously described (Vannucci and Duffy, 1974). Blood samples also were obtained and extracted in 0.5 mol/L perchloric acid. Concentrations of glucose, pyruvate, lactate, phosphocreatine (P~Cr), ATP, ADP, and AMP were analyzed by specific enzymatic and fluorometric techniques (Lowry and Passoneau, 1972; Vannucci and Duffy, 1974). Intracellular pH was derived from measured phosphocreatine (P~Cr), creatine (Cr), ATP and ADP concentrations, and the calculated observed equilibrium constant for the creatine kinase reaction (Kobs,ck) according to the equation: Kobs,ck = [P~Cr][ADP][H+]/[ATP][Cr], as previously described (MacMillan and Siesjo, 1972). In control animals, mean Kobs,ck was derived with and assumed intracellular pH of 7.00 (Duffy et al., 1975), and the equation then solved for [H+] using the measured metabolite concentrations. The use of the creatine kinase equilibrium to estimate pH
Statistical analysis
Mortality data were analyzed using Fisher's exact test for individual effects of gender and diabetes on mortality. Interaction of gender and diabetes effects on mortality was tested using the Breslow–Day test. Infarct fraction, biochemical data, and regional CBF were analyzed with a two-way analysis of variance. Significance was established at P < 0.05.
RESULTS
Initial experiments assessed the effect of diabetes on mortality from severe (30 minute) HI. Preischemic blood glucose, mortality, and infarct volume in the survivors are depicted in Table 1. Both male and female db/db mice were significantly hyperglycemic relative to normoglycemic controls. Blood glucose levels in the diabetic females were greater than in the male diabetics. Circulating insulin levels, although not measured in this study, have been shown to be comparable in male and female db/db mice at 3 weeks of age (Timers et al., 1990) and slightly greater in male versus female at 8 weeks of age (E.M. Gibbs, personal communication). Both male gender and diabetes significantly increased mortality, P < 0.05 for each; however, there was no significant interaction of gender and diabetes on mortality. There was no mortality in male or female mice subjected to hypoxia without carotid artery ligation (n = 5 per group, data not shown). Thirty minutes of HI produced significant damage to parietal cortex, striatum, and hippocampus of the survivors. Diabetic males sustained significantly greater infarcts relative to nondiabetic males, but this was not statistically significant in the females (P = 0.1), relative to their respective controls. Diabetic females were significantly more protected than their male counterparts.
Mortality and infarct volume in control and diabetic mice

All animals were exposed to 30 minutes of hypoxia–ischemia as described in Methods. Infarct volume was determined in the survivors as follows: n = 14, male wild type (♂ wt); 5 male diabetic (♂ db/db); 22 female wild type (♀ wt); 12 female control (♀ db/db) from TTC-stained sections as a percentage of the contrlateral hemisphere.
When mortality in controls and diabetics was analyzed separately there was a weak gender effect (P = 0.16, controls; P = 0.19, diabetics), but when controls and diabetics were analyzed together there was significant evidence of a gender effect (P = 0.038). wt, wild type; db/db, Type II diabetes.
Diabetes effect, P < 0.05.
Gender effect, P < 0.05.
To further investigate the effects of diabetes within each gender, cohorts in the second experiment were subjected to a duration of HI sufficient to produce significant damage but lower mortality. To assure a 75% survival, a 17-minute HI duration was required in the male and a 22-minute HI duration in the female. We quantified brain damage in each experimental group (n = 3 per group) at 48 hours of recovery. Hemispheric swelling, as an indication of edema, and mean hemispheric infarct areas are depicted in Fig. 1. Brains of both male and female diabetic mice demonstrated significant edema in the ipsilateral hemisphere relative to their nondiabetic controls. Male mice suffered larger infarcts than females regardless of diabetic state. Therefore, ischemic damage in the male diabetic brain was more severe, despite the shorter duration of HI, and was not linked to differences in edema.
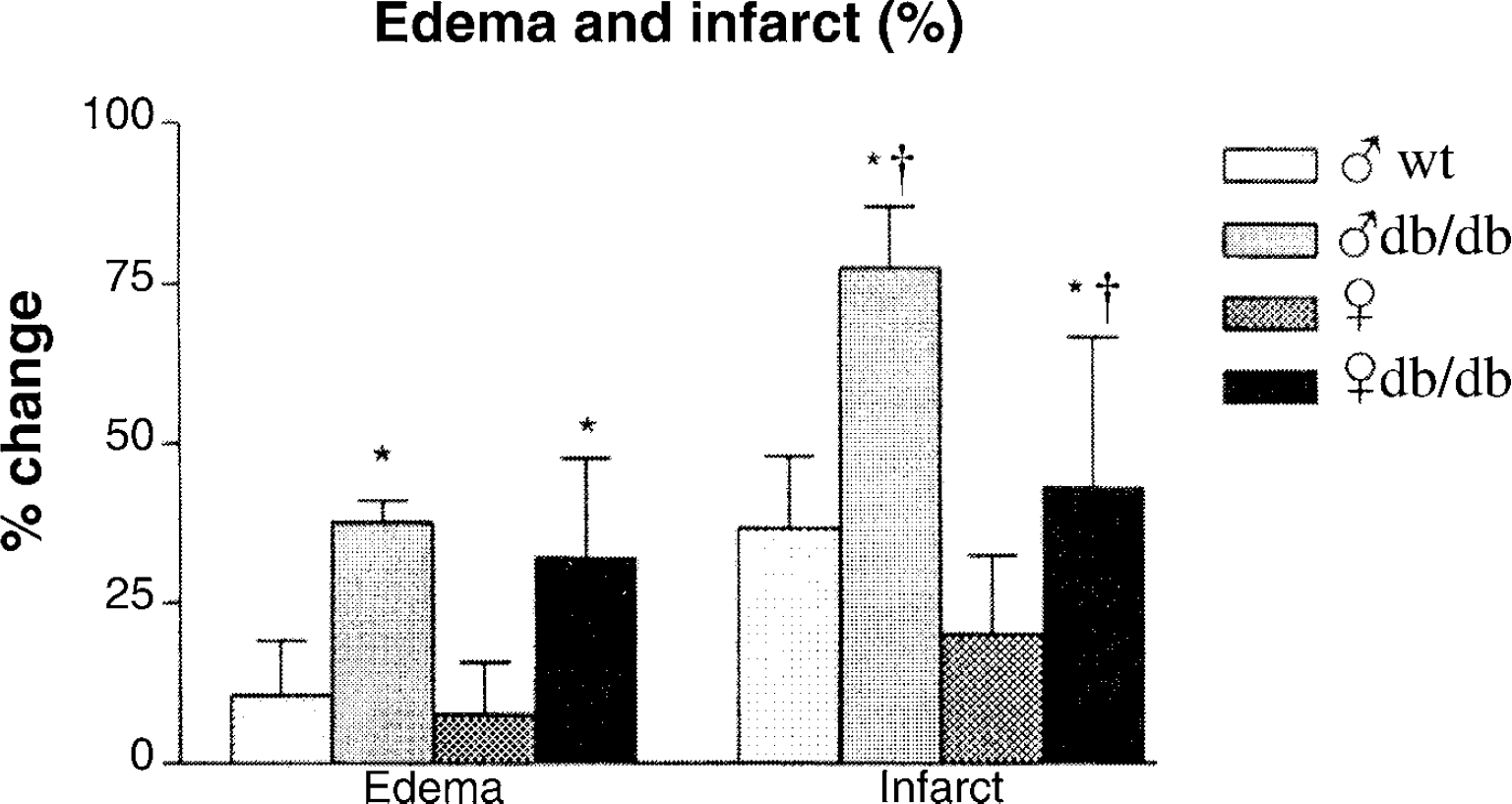
Male control (♂ wt) and db/db (♂ db/db) mice were subjected to 17 minutes hypoxia–ischemia; female control (♀ wt) and db/db (♀ db/db) mice were subjected to 22 minutes hypoxia–ischemia, n = 3 per group. At 48 hours of recovery, brains were frozen and 16-μm cryosections were taken at 9 regular intervals from the striatum to posterior hippocampus and stained with hematoxylin and eosin. Sections were photographed and analyzed with NIH Image 1.64. Edema was determined by the percent increase of the ipsilateral/contralateral area and the percentage of infarction was determined by the following equation: (size of the contralateral hemisphere – area of normal tissue in the ipsilateral hemisphere)/contralateral hemisphere (Swanson et al., 1990). *Diabetes effect, P < 0.05. †Gender effect, P < 0.05.
We next examined end-ischemic CBF (Fig. 2) under these HI conditions. Physiologic parameters such as arterial blood pressure and blood gases were the same in all groups. All animals became significantly hypotensive by the end of the HI insult, but there were no significant differences among the groups in either basal mean arterial blood pressure (MABP, 115 to 120 mm Hg) or end-ischemic MABP (75 to 80 mm Hg). Neither gender nor diabetes affected end-ischemic CBF (Fig. 3), suggesting equivalence of the ischemic insult in diabetics and nondiabetics in either sex.
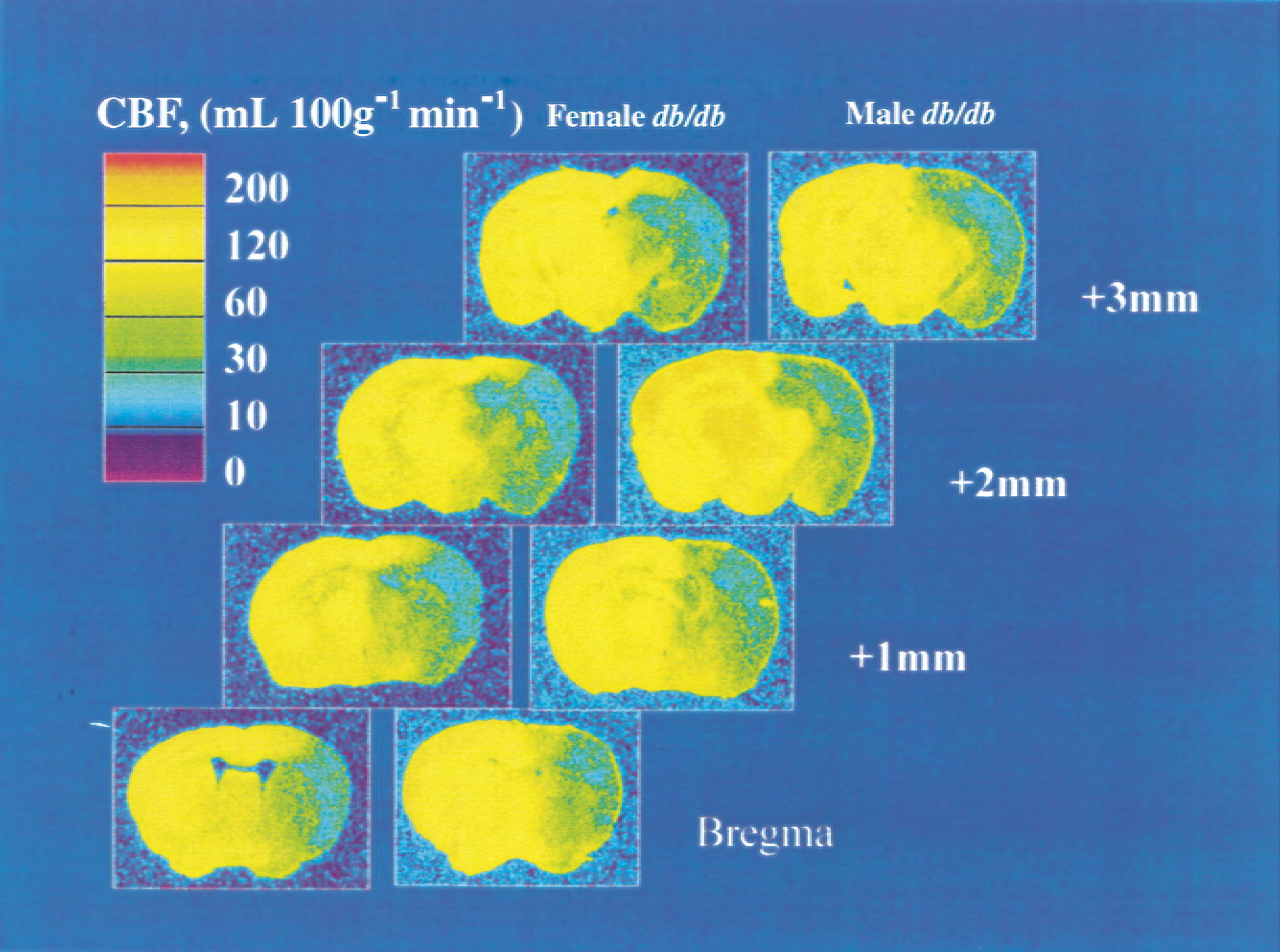
End-ischemic cerebral blood flow (CBF) in diabetic mice. Male mice were subjected to 17 minutes of hypoxia–ischemia, female mice were subjected to 22 minutes of hypoxia–ischemia, and CBF was measured at the end of the hypoxic–ischemic interval by [14C]iodoantipyrine autoradiography. Representative autoradiograms from 1 male diabetic and 1 female diabetic with sections taken at 0, +1, +2, and +3 mm from bregma are shown.
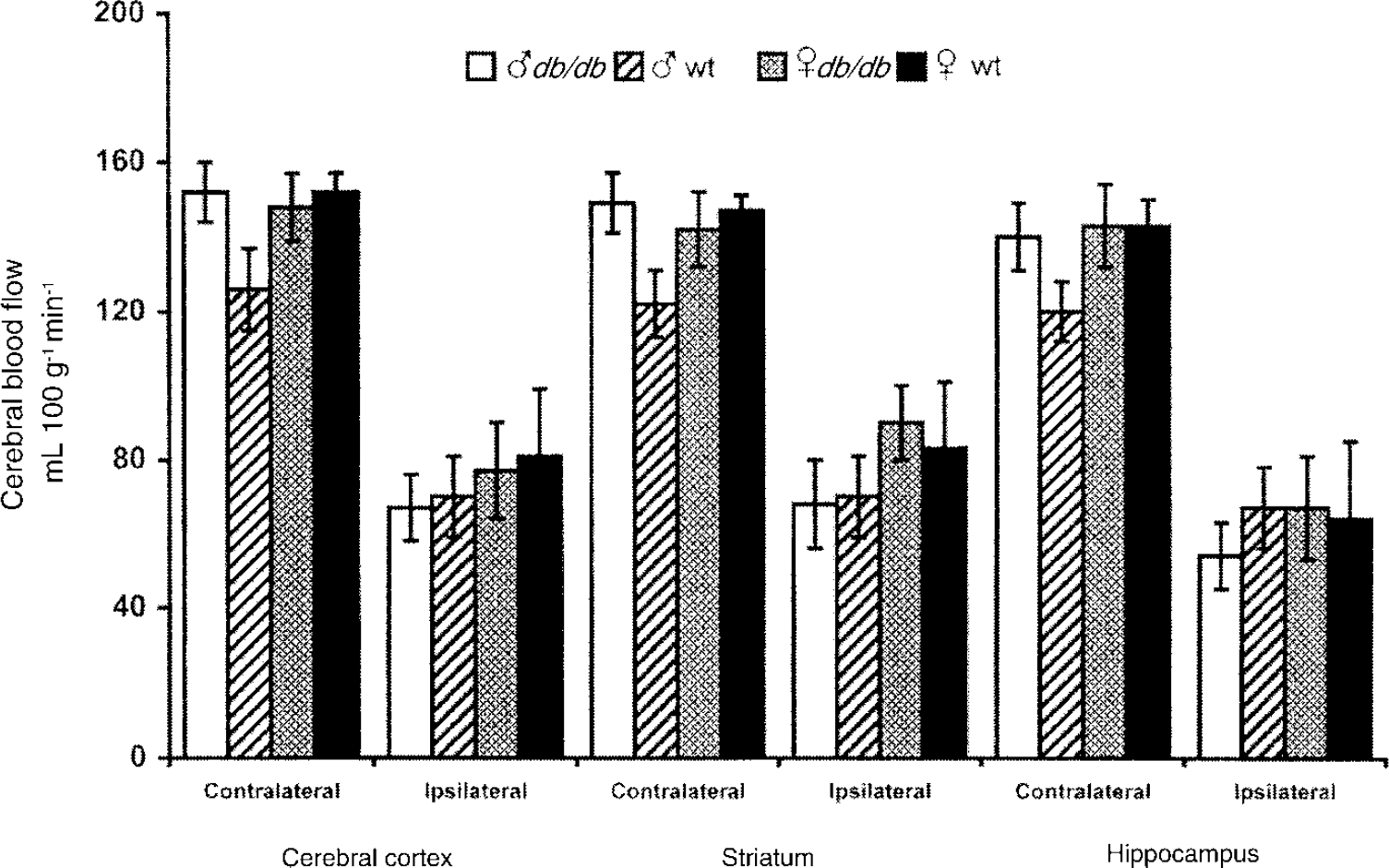
Regional cerebral blood flow (rCBF) within contralateral and ipsilateral cerebral cortex, striatum, and hippocampus for male db/db (♂ wt), male control (♂ db/db), female db/db (♀ wt), and female control (♀ db/db) at 17 minutes (male) and 22 minutes (female) of hypoxia–ischemia as measured by [14C]iodoantipyrine autoradiography. n = 4 per group.
Glycolytic metabolites were determined before, at the end of HI, and at 4 hours of recovery (Table 2). Baseline blood and brain glucose values were greater in the diabetic mice, reflecting profound hyperglycemia. Female diabetic mice demonstrated a greater level of plasma glucose than their male counterparts, but baseline brain glucose values were not different. Blood lactate and brain lactate and pyruvate were comparable in all groups. Blood and brain glucose concentrations did not change during HI, whereas the blood and brain lactate levels increased substantially, especially in the diabetic mice. Initial rise in brain lactate was almost twice as rapid in the diabetic versus wild type mice; lactate accumulation in the latter groups continued in a near linear fashion, whereas the diabetics displayed minimal further accumulation (data not shown). By the end of HI, all groups had brain lactate levels in excess of 17 mmol/L. At 4 hours of recovery, brain glucose exceeded baseline values by 76% to 96% in all groups, despite blood glucose concentrations that were comparable to baseline levels. It appears that the previously high brain lactate during HI was used as a preferential fuel during early recovery, thereby sparing glucose utilization. Lactate levels remained elevated in diabetic mice, with significantly higher lactate in the female diabetics, suggesting a lingering lactacidosis in these mice. There was a significant interaction between diabetes and gender such that the diabetic effect on lactacidosis was greater in the female mice (P < 0.001).
Effect of hypoxia–ischemia on glycolytic intermediates
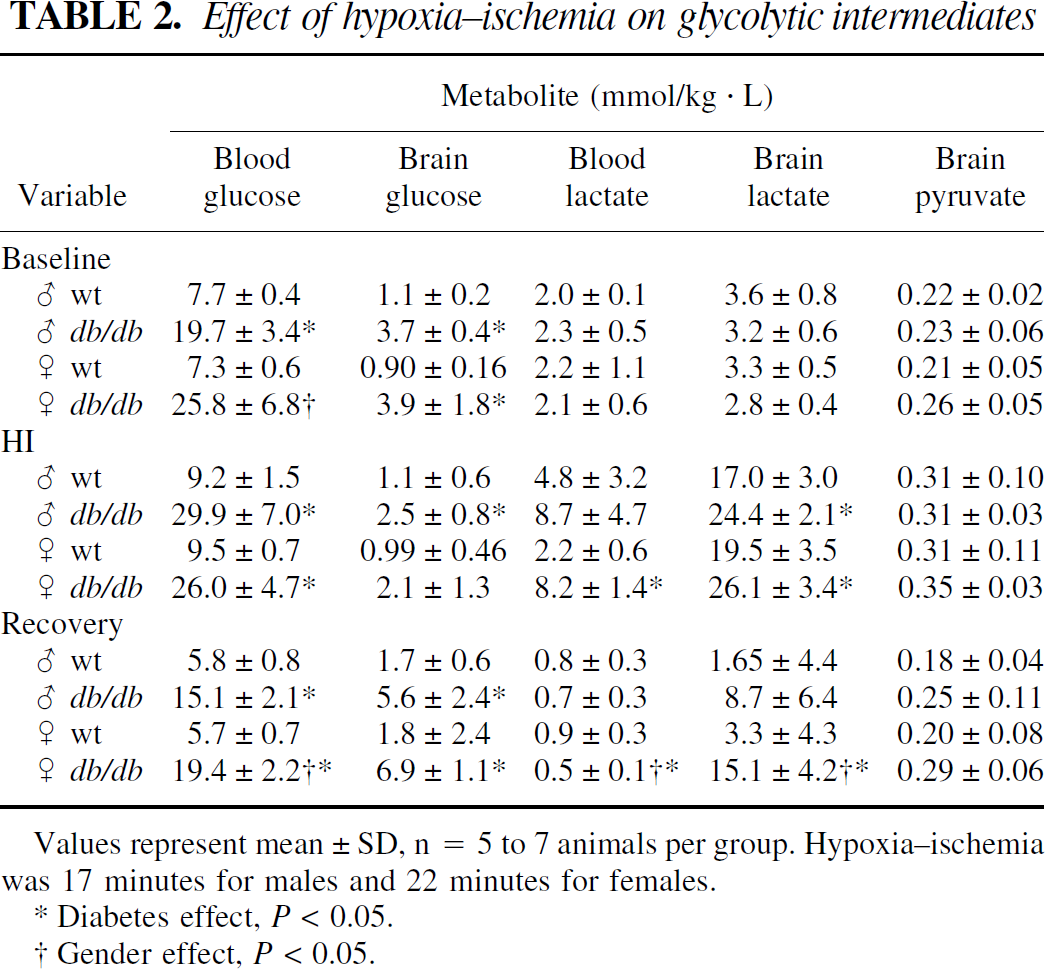
Values represent mean±SD, n = 5 to 7 animals per group. Hypoxia–ischemia was 17 minutes for males and 22 minutes for females.
Diabetes effect, P < 0.05.
Gender effect, P < 0.05.
Cerebral high energy phosphate reserves were also similar in all groups, with the exception of slightly elevated phosphocreatine (P~Cr) in both diabetic males and females (Fig. 4). By the end of HI, ATP and P~Cr decreased to comparable levels in control and diabetic animals, without apparent gender differences. ATP and P~Cr levels were restored in control mice at 4 hours of recovery but were below baseline values in the diabetic mice. To gain an estimation of the corresponding intracellular pH independent of lactate, brain creatine and ADP were measured in these brains; baseline, end-ischemic, and 4 hours of recovery values for intracellular pH were calculated based on the creatine kinase equilibrium (Fig. 4, bottom panel). There was no difference among groups of mice in baseline or end-ischemic pH, which was significantly reduced in all groups. However, at 4 hours of recovery, all male mice (control and diabetic) and control female mice demonstrated a somewhat elevated pH indicative of a postischemic alkaline shift. This was significantly retarded in the female diabetic mice. Whereas end-ischemic lactate was elevated to 8 to 10 mmol/L · kg in all animals, there were no changes in any other metabolites in the contralateral hemisphere (data not shown).
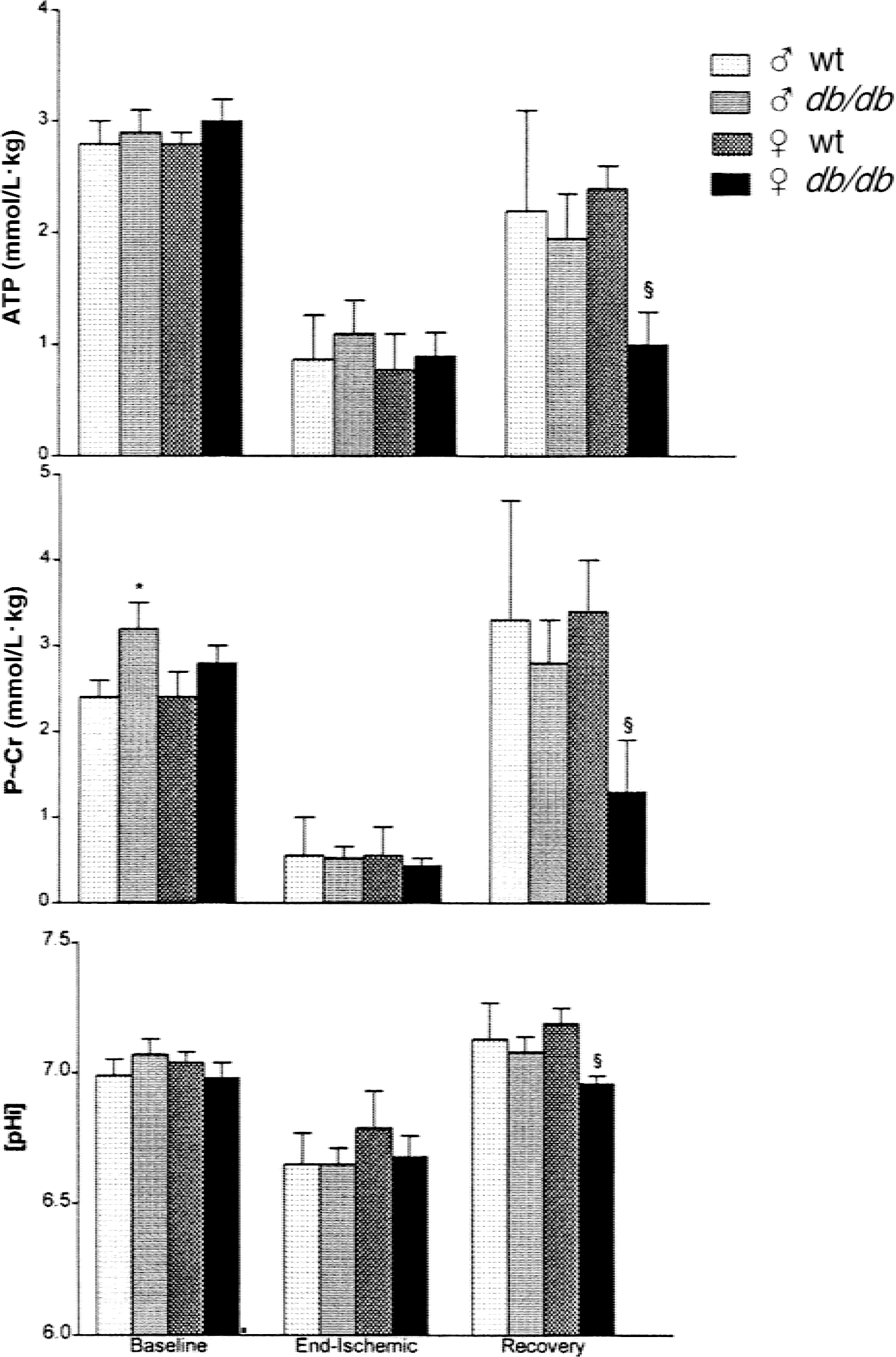
High energy reserves and intracellular pH [pHi] in male and female, control and db/db, mice. Effect of hypoxia–ischemia and early recovery. Male mice were subjected to 17 minutes of hypoxia–ischemia and female mice were subjected to 22 minutes of hypoxia-ischemia. At preischemic (baseline), 0 (end-ischemic), and 4 hours of recovery (recovery) animals were anesthetized and then decapitated. Heads were placed in liquid N2. Frozen brains were dissected from contralateral and ipsilateral hemispheres and extracted in perchloric acid. High energy metabolites were measured as described in Methods. Concentrations for ATP and P~Cr in comparison with calculated values for [pHi] calculated from the creatine kinase equilibrium (see text) are shown.
DISCUSSION
The current study contains three major findings. First, as expected, mice with genetic, Type II diabetes sustained increased mortality, histologic injury, and hemispheric edema after cerebral HI, relative to age-and sex-matched wild type mice. Second, both prolonged HI (30 minutes) and a shorter HI duration titrated to assure 75% survival resulted in larger infarctions in male versus female db/db mice. This difference was not explained by preservation of end-ischemic CBF or by diminished hemispheric swelling. Third, pHi and brain lactate recovery were markedly delayed in female versus male db/db mice. This finding was not linked to differences in intraischemic high energy phosphate depletion, suggesting that the initial biochemical insult was comparable among all animal groups. These results indicate that outcome from experimental stroke is gender- dependent in animals with Type II diabetes, with the female diabetic demonstrating a greater degree of protective adaptation to diabetic hyperglycemia than the male. The female db/db brain sustained increased hyperglycemia and prolonged lactacidosis relative to the male, yet remained resistant to infarction. Whereas this observation may seem predictable given the growing amount of evidence supporting enhanced ischemic tolerance in female animals (Hurn and Macrae, 2000; Roof and Hall, 2000), this is the first report of ischemic bioenergetics and pathology in the female diabetic brain. The extent to which reproductive steroids are important in this observation remains to be demonstrated.
The current focal cerebral ischemic model, originally developed in the authors' laboratory for use in the immature rat (Rice et al., 1981; Vannucci et al., 1996b; Levine, 1960), combines unilateral carotid artery ligation with a period of systemic hypoxia and results in unilateral infarction of defined striatal, hippocampal, and cortical brain regions. Initial experiments using a severe insult of 30 minutes duration demonstrated a significant diabetic and gender effect on mortality such that male db/db > male wild type and female db/db, but also that female db/db > wild type, nondiabetic female. This gradation was further reflected in the extent of gross damage in surviving animals. Because survivorship per se could have influenced these results, we examined a less severe insult and again determined that histologic injury was surprisingly low in the female diabetic. End-ischemic CBF was reduced within each brain region to 50% to 60% of contralateral structure, and neither the presence of diabetes nor female gender altered the level of CBF reduction. Blood flow reduction, combined with hypoxemia, resulted in tissue necrosis ipsilateral to carotid ligation in all animals, albeit less in females.
One important factor in the enhanced cell death associated with diabetic stroke is the presence of hyperglycemia and enhanced tissue acidosis consequent to excessive lactate accumulation (Myers, 1979; Plum, 1983; Siesjo et al., 1990, 1993). In the current study, rapid increases in lactate were measured in all animals, but the rise was approximately twice as great in the diabetics, such that there was little further increase by the end of the ischemic episode. By the end of the ischemic period, all mice had lactate values >16 mmol/L/kg, which is likely greater than the postulated threshold necessary for neuronal damage (Plum, 1983; Siesjo et al., 1990, Li et al., 1994). Furthermore, end-ischemic pHi, as estimated by the creatine kinase equilibrium, was not different among experimental groups. Thus, it is apparent from the results of both Table 2 and Fig. 4 that the end-ischemic metabolic state was not different among the four groups of mice.
Significant gender- and diabetes-related differences were present at 4 hours of recovery. The early postischemic restoration of the energy state in the control mice was significantly retarded in the diabetic mice, and this was more pronounced in the female brains. Whether secondary energy failure occurred was not addressed in this study. Furthermore, it should be emphasized that metabolite recovery levels represent averaged hemispheric values and include healthy and damaged/dying cells. Brain lactate normalized in nondiabetic mice with substantial restoration of high energy reserves, but not in db/db mice, particularly females. This was associated with elevated levels of brain glucose, indicating that lactate was serving as the preferential fuel, sparing glucose utilization. This phenomenon has been reported previously for both immature (Vannucci et al., 1994) and adult (Katsura et al., 1996) postischemic brain and could explain early reports of postischemic depression of cerebral glucose utilization in hyperglycemic animals (Kozuka et al., 1989).
The relation between preischemic hyperglycemia, whether acute or chronic as in the diabetic, and the generation of lactate, reduction in tissue pH, energy depletion, and ultimate damage is far from clear. Infarct size because of hyperglycemia during transient middle cerebral artery occlusion in cats correlated better with the degree of tissue acidosis than with lactate levels or ATP depletion (Wagner et al., 1992). Further differences in ischemic outcome have been reported for acutely hyperglycemic versus chronically hyperglycemic (diabetic) rats, with better outcome observed in the diabetics, despite comparable levels of preischemic hyperglycemia, lactate accumulation, and energy depletion (Wagner and Lanier, 1994). These studies, along with other studies, have led to a general hypothesis that the diabetic brain has adapted such that it can protect itself from the deleterious effects of lactic acidosis caused by cerebral ischemia; however, the mechanism or mechanisms underlying this adaptation have yet to be determined. All of these earlier studies were conducted in male animals. The current results suggest that the female db/db mouse has an even greater capacity to withstand prolonged lactacidosis and slower restoration of energy phosphates. The basis for such an adaptation could relate to enhanced astrocytic resistance to acidosis or altered metabolic efficiency with use of lactate as a neuronal fuel. For example, estrogen increases activity of several key glycolytic enzymes (Kostanyan and Nazaryan, 1992), modulates glucose transporter function within the blood–brain barrier (Shi et al., 1997), and, in humans, can improve carbohydrate metabolism and decrease insulin requirements (Bush and Miller, 1987; Brussaard et al., 1997).
Gender-linked differences in ischemic tolerance have been well characterized, although the mechanisms accounting for protection of the female brain are not clear (Hurn and Macrae, 2000; Roof and Hall, 2000). Only one very recent study has examined this in the context of diabetes. Female biobreeding type I (BB) rats, a genetically insulin-dependent diabetic animal model, sustain smaller infarcts after middle cerebral artery occlusion than their male counterparts (Toung et al., 2000). Furthermore, chronic estrogen treatment reduces stroke damage in the BB male to an even larger degree than in nondiabetic male rats (Toung et al., 2000). The current data indicate that a similar situation might exist for Type II diabetes—that is, that although diabetes exacerbates stroke injury, females enjoy a level of protection relative to their male counterparts. These experiments do not establish the basis for an apparent interaction among diabetes, ischemia, and gender. However, we hypothesize that there is an overlap between mechanisms of acidotic brain injury and the actions of reproductive steroids. For example, cerebral acidosis leads to increased generation of free radicals (Lundgren et al., 1992; Morimoto et al., 1996; Wei et al., 1997; Wei and Quast, 1998) and iron-catalyzed oxygen radical damage (Siesjo et al., 1993; Rehncrona et al., 1989; Hurn et al., 1995). The antioxidant defense systems in the brains of male db/db mice have been shown to be partially compromised (Makar et al., 1995), suggesting vulnerability to radical-induced lipid peroxidation. However, both estrogen (Hall et al., 1991; Culmsee et al., 1999; Kume-Kick and Rice, 1998; Vedder et al., 1999) and progesterone (Roof and Hall, 2000) have antioxidant and anti-lipid peroxidation actions in brain, respectively. Female gerbils are less vulnerable to cerebral ischemia than their male littermates and this has been attributed to estrogeninduced inhibitionof iron-catalyzed lipid peroxidation in brain (Hall et al., 1991). Further experiments are needed to determine whether the relative protection enjoyed by female diabetic animals, Type I or II, is mediated through hormonal scavenging of oxygen radicals.
In conclusion, this study demonstrates that diabetic hyperglycemia in a genetic model of NIDDM, the db/db mouse, exacerbates HI injury and that this effect is gender-dependent. Whether estrogen is directly involved in amelioration of diabetic complications in stroke rather than providing generalized neuroprotection in all forms of ischemic injury remains to be shown. However, this model allows for an experimentally controlled gradation of insult so that it will be possible to dissect the cellular and molecular events set into motion after cerebral ischemia and design therapeutic interventions.
Footnotes
Acknowledgment
The authors thank Dr. Robert C. Vannucci for helpful comments during the preparation of the manuscript.