
Abstract
This study examines the effects of middle cerebral artery (MCA) occlusion in the rat on blood to brain glutamine transport, a potential marker of early endothelial cell dysfunction. It also examines whether the effects of ischemia on glutamine transport are exacerbated by hyperglycemia. In pentobarbital-anesthetized rats, 4 hours of MCA occlusion resulted in a marked decline in the influx rate constant for [14C]L-glutamine from 16.1 ± 1.2 μL·g−1·min−1 in the contralateral hemisphere to 7.3 ± 2.5 μL·g−1·min−1 in the ischemic core (P < 0.001). This reduction was even greater in xylazine-ketamine-anesthetized rats in which the influx decreased to 2.6 ± 1.1 μL·g−1·min−1. This greater reduction appears related to the hyperglycemia induced by xylazine-ketamine anesthesia. Glucose injection in pentobarbital-anesthetized rats also resulted in a greater decline in [14C]L-glutamine influx in the ischemic core but had no effect on the contralateral tissue. The effects of hyperglycemia on glutamine transport in the ischemic tissue were associated with a decline in plasma volume, which may reflect either endothelial cell swelling or plugging of the microvasculature. The reduction in glutamine transport during ischemia was progressive, but even as early as 1 hour, there was a 60% and 40% decline in influx in hyperglycemic and normoglycemic rats, respectively. The fall in [14C]L-glutamine influx may reflect a dissipation of the endothelial cell [Na+] gradient. A decline in this gradient would affect many blood-brain barrier transporters with potentially deleterious effects on the ischemic brain.
Keywords
Recently, we have described a decrease in blood to brain taurine transport during focal cerebral ischemia in the rat (Stummer et al., 1995). This change might be related to the role of taurine in brain volume regulation (Thurston et al., 1980). However, it might also reflect a disruption in the Na+ gradient of the endothelial cells that form the blood-brain barrier (BBB) as blood to brain taurine transport is Na+ dependent (Benrabh et al., 1995). To evaluate this latter possibility, we have examined whether blood to brain glutamine transport is also decreased by cerebral ischemia, as this process is also Na+ dependent (Keep et al., 1997a). An understanding of glutamine transport during cerebral ischemia is important because glutamine is the most prevalent amino acid in blood and CSF and it is the major precursor of the excitatory amino acid glutamate. An alteration in Na+-dependent transport at the BBB during ischemia would be important as a diverse group of transporters rely on the endothelial Na+ gradient (e.g., Betz and Goldstein, 1978; Kollros et al., 1990; Pardridge et al., 1994; Tayarani et al., 1987).
In addition, this study examines whether the changes in blood to brain glutamine transport during focal cerebral ischemia are exacerbated by hyperglycemia. Many studies have found that hyperglycemia increases brain injury after ischemia with reperfusion. Data suggest that cerebrovascular dysfunction may contribute to this adverse effect because hyperglycemia leads to reduced CBF and greater BBB breakdown during reperfusion (Dietrich et al., 1993; Ginsberg et al., 1980; Venables et al., 1985). In contrast, the cerebrovascular effects of hyperglycemia during ischemia are uncertain. Measurements of CBF during ischemia have not shown a clear difference between hyperglycemic and normoglycemic animals (Venables et al., 1985), but we have recently found a marked (50%) reduction in cerebral plasma volume after middle cerebral artery (MCA) occlusion in hyperglycemic rats (Kawai et al., 1997c). The latter suggests that hyperglycemia does alter the cerebrovasculature during ischemia, a possibility further examined by the present measurements on blood to brain glutamine transport.
Abstracts on part of this work have previously been published (Kawai et al., 1997a, 1997b; Keep et al., 1997b).
MATERIALS AND METHODS
Animal preparations and experimental protocols
All procedures were approved by the University Committee on Use and Care of Animals at the University of Michigan. Adult male Sprague-Dawley (Charles River, Portage, MI, U.S.A.) rats weighing between 250 and 300 g were used for all experiments. Rats were allowed free access to food and water before the experiments. In a first group of experiments, the effect of MCA occlusion was examined in rats anesthetized with sodium pentobarbital (50 mg/kg) or with xylazine (10 mg/kg) and ketamine hydrochloride (50 mg/kg). The MCA was occluded using the suture method based on the surgical insertion of a nylon monofilament (3–0) through the external carotid artery (Longa et al., 1990). These rats were used to determine either cerebral plasma, red blood cell volume, BBB glutamine transport, or CBF at either 1, 2, or 4 hours after occlusion. Cerebral blood flow was determined using 4-[N-methyl-14C]-iodoantipyrine.
In the second group of experiments, rats were anesthetized with sodium pentobarbital (50 mg/kg), and acute hyperglycemia was induced by intraperitoneal administration of 2 mL of 50% D-glucose solution 20 minutes before MCA occlusion. Because of a small increase in plasma osmolality (approximately 20 mOsm/kg) after glucose injection, normoglycemic controls received a similar osmotic load of 18% D-mannitol solution. The effects of blood glucose were also examined by administering insulin (3 U/kg intraperitoneally) 60 minutes before xylazine-ketamine anesthesia to prevent the hyperglycemia normally associated with this anesthesia.
In all animals, body temperature in anesthetized rats was maintained at 37°C using a rectal thermometer and a feedback-controlled heating pad. Cannulas (PE-50) were placed in both femoral arteries for blood sampling and for measurement of blood pressure, and into a femoral vein for isotope injection.
Blood-brain barrier glutamine transport and cerebral plasma volume measurement
The permeability of the BBB to [14C]L-glutamine was assessed by a modification of the method proposed by Ohno et al. (1978) for a single time point analysis as previously described (Stummer et al., 1995). A bolus of 0.2 mL saline containing 5 μCi [14C]L-glutamine was administered 5 minutes before decapitation of rats. After radioisotope injection, an arterial blood sample was continuously withdrawn at a constant rate to determine the integral of plasma radioactivity. For the simultaneous determination of plasma volume, 20 μCi of [3H]inulin was then injected into the femoral vein and allowed to circulate for 2 minutes. At the end of experiment, a terminal plasma sample was taken, and the rat was killed by decapitation. Brains were rapidly removed and separated into cortices and basal ganglia. Cortices were flattened on a piece of Parafilm and punches were made of ipsilateral and contralateral cortices to obtain tissue samples from the core of the MCA distribution, a ring of tissue surrounding these, hereafter called the intermediate zone, and the remainder of the cortex, hereafter called the outer zone, as described previously (Menzies et al., 1993). Samples were immediately weighed and digested in methyl-benzethonium hydroxide. Scintillation fluid was added to the brain and plasma samples and radioactivity was counted using a Beckman 3801 liquid scintillation counter (Fullerton, CA, U.S.A.).
The permeability of [14C]L-glutamine was assessed using a two-compartment model for blood to brain transfer (Ohno et al., 1978). The rate constant for unidirectional uptake (Ki) relating the brain concentration of tracer over time to the arterial plasma concentration can be defined as
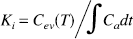
where Cev(T) is the terminal (time = T) concentration of extravascular tracer in either the brain or CSF and ∫Cadt is the integral of the arterial tracer concentration from time t = 0 to T. Cev(T) was calculated from total tracer counts Ctot(T) in the brain samples, final tracer plasma concentration Cpl(T), and plasma volume PV as
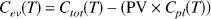
There are several points that should be addressed before trying to measure an influx rate constant for a compound such as [14C]L-glutamine. In particular, is [14C]L-glutamine the compound being transported, is the uptake of tracer unidirectional, and is plasma the source of isotope being taken up into brain or do erythrocytes also act as a source. Several pieces of evidence indicate that [14C]L-glutamine is the transported compound (although once into brain it is converted into several different compounds). We have examined plasma from rats injected with [14C]L-glutamine with paper chromatography using n-butanol:water:acetic acid (12:5:3) as the solvent. Using this method it appears that 92% and 88% of the isotope in plasma is still in the form of [14C]L-glutamine at 1 and 5 minutes, respectively. There is conversion of L-glutamine into L-glutamate, but even by 5 minutes this conversion is less than 10% (Dierks-Ventling et al., 1971), and in our experiments about 75% of isotope entry occurs within the first minute. Conversion to the slowly transported L-glutamate will, therefore, not apparently be enough to seriously affect the reported results. L-Glutamine can be converted into glucose intracellularly, but the rate of appearance of 14C-glucose in rat plasma after [14C]L-glutamine injection is very low (Aikawa et al., 1973). The values obtained for Kiin vivo are similar to those obtained using in situ perfusion (Keep et al., 1997a) in which the perfusate is directly infused into the cerebrovasculature and is not subject to metabolism by intestine, kidney, liver, and skeletal muscle.
In preliminary experiments, the unidirectionality of [14C]L-glutamine uptake was examined in rats undergoing no MCA occlusion. There was no significant difference in the Ki determined by a 1- and 5-minute isotope circulation time (18 ± 1 and 18 ± 2 μL·g−1·min−1, respectively, n = 4). This indicates that there was no significant efflux from the brain during these time points and, hence, unidirectional uptake was measured. In ischemic tissue, the Ki for [14C]L-glutamine is reduced, and therefore, efflux of isotope from the brain within 5 minutes is likely to be even less than in nonischemic tissue. The fact that the Ki for isotope entry did not vary from 1 to 5 minutes is also evidence that the presence of metabolites of [14C]L-glutamine in plasma are not making a significant difference to the measured Ki because an increase in uptake with time would be expected if [14C]L-glutamine was being converted into a more permeable substrate, whereas decreases in uptake with time would be expected if the [14C]L-glutamine was being converted to a less permeable compound. In these preliminary experiments, the uptake of [14C]L-glutamine into red blood cells was also examined. At the end of a 5-minute circulation, less than 0.5% of the isotope found in brain was present in red blood cells, an insignificant correction.
In one set of experiments the influx rate constants for [14C]L-glutamine and [3H]taurine were determined in the same animals. That experiment was the same as for [14C]L-glutamine alone except the animals received a single injection of isotope (5 μCi [14C]L-glutamine and 15 μCi [3H]taurine) 5 minutes before decapitation and the plasma volume correction was determined in another group of rats.
Cerebral red blood cell volume measurement
The effect of MCA occlusion on cerebral red blood cell volume in controls and hyperglycemic rats was determined with red blood cells tagged with chromium 51 using a method described in detail previously (Keep et al., 1995). In brief, 1.5 mL of blood from a donor rat was washed and centrifuged, and the packed red blood cells were incubated with 100 μCi of 51Cr for 1 hour at 37°C. The labeled red cells were then washed and kept on ice. Just before use, a 200-μL aliquot of cells was resuspended with 100 μL phosphate-buffered saline; this solution was injected into the femoral vein and allowed to circulate for 2 minutes. At the end of experiment, a terminal arterial blood sample was withdrawn and the animal was decapitated. Brain sampling and preparation were as for the cerebral plasma volume experiment. Whole blood samples were bleached with 30% H2O2 before counting. The cerebral red blood cell volume for each sample was calculated as (dpm/g brain) × (systemic hematocrit)/(dpm/mL whole blood).
Cerebral blood flow measurement
Cerebral blood flow was measured by the indicator fractionation technique (Van Uitert and Levy, 1978) using 10 μCi of 4-[N-methyl-14C]-iodoantipyrine as a blood flow marker. This method has been described in detail previously (Kawai et al., 1997c) and used a bolus injection of isotope and a 10-second circulation time.
Statistical analysis
Comparisons between ipsilateral and contralateral tissues were by paired t test. Influx rate constants, blood volumes, or CBF in normoglycemic and hyperglycemic rats were compared by unpaired t test. Values at different time points were compared by analysis of variance (ANOVA) with Newman-Keuls multiple comparison test. Differences were considered to be significant at the P < 0.05 level. All values are presented as means ± SD.
RESULTS
Physiologic parameters
Physiologic parameters from the rats are given in Table 1. Most physiologic parameters were similar between pentobarbital- and xylazine-ketamine-anesthetized rats. One exception was that the xylazine-ketamine-anesthetized rats were moderately hyperglycemic compared with pentobarbital-anesthetized control rats. Glucose injection produced severe hyperglycemia at the time of MCA occlusion. However, the level of glycemia returned to normal values within an hour. Both glucose- and mannitol-injected rats displayed increased hematocrit and plasma osmolality.
Physiologic parameters
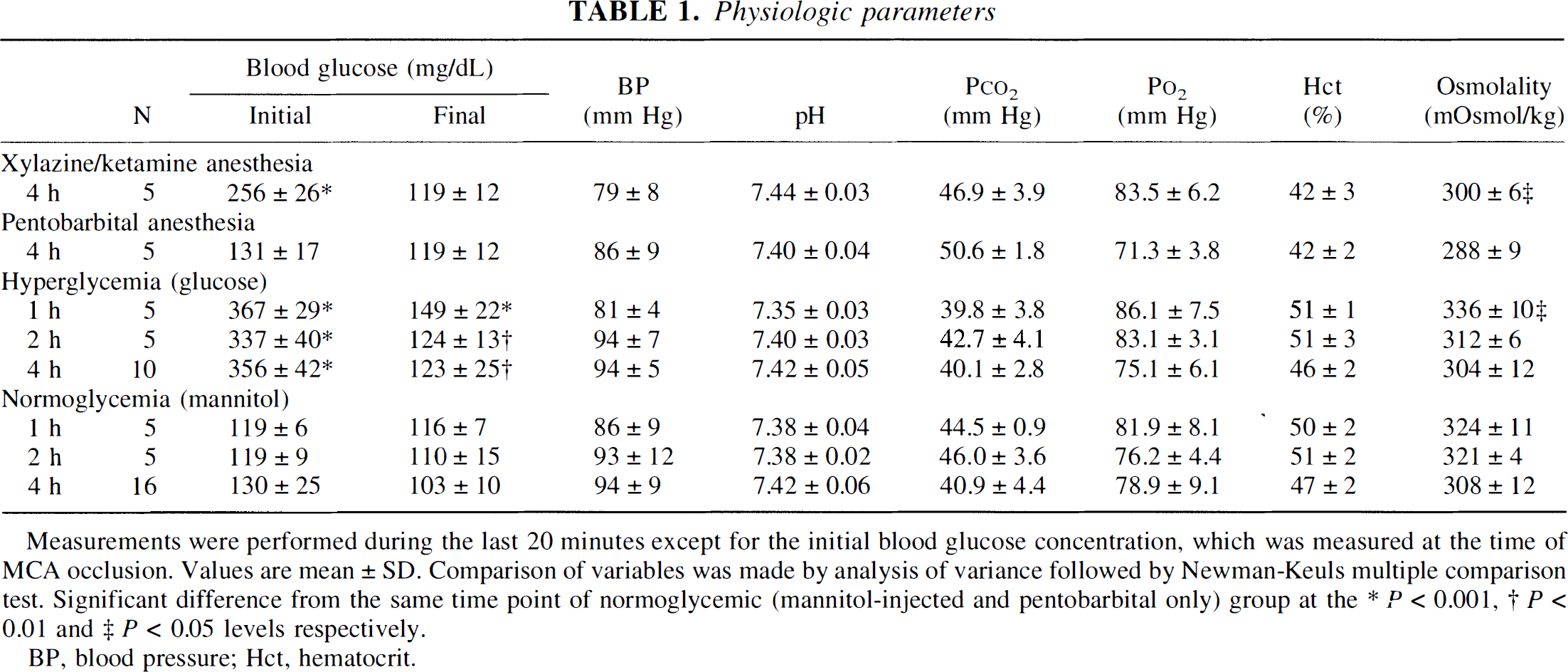
Measurements were performed during the last 20 minutes except for the initial blood glucose concentration, which was measured at the time of MCA occlusion. Values are mean ± SD. Comparison of variables was made by analysis of variance followed by Newman-Keuls multiple comparison test. Significant difference from the same time point of normoglycemic (mannitol-injected and pentobarbital only) group at the ∗ P < 0.001, † P < 0.01 and ‡ P < 0.05 levels respectively.
BP, blood pressure; Het, hematocrit.
Blood-brain barrier glutamine transport
Four hours after MCA occlusion, the Ki for glutamine in the contralateral cortex did not vary with the anesthetic agent used for the occlusion (Fig. 1). With both anesthetics, the ipsilateral cortex showed a marked reduction in glutamine Ki (Fig. 1), with the degree of reduction being greatest in the core of the MCA territory. The reduction in the core, however, was greater with xylazine-ketamine compared with pentobarbital anesthesia (2.6 ± 1.1 and 7.3 ± 2.5 μL·g−1·min−1, respectively; P < 0.01).
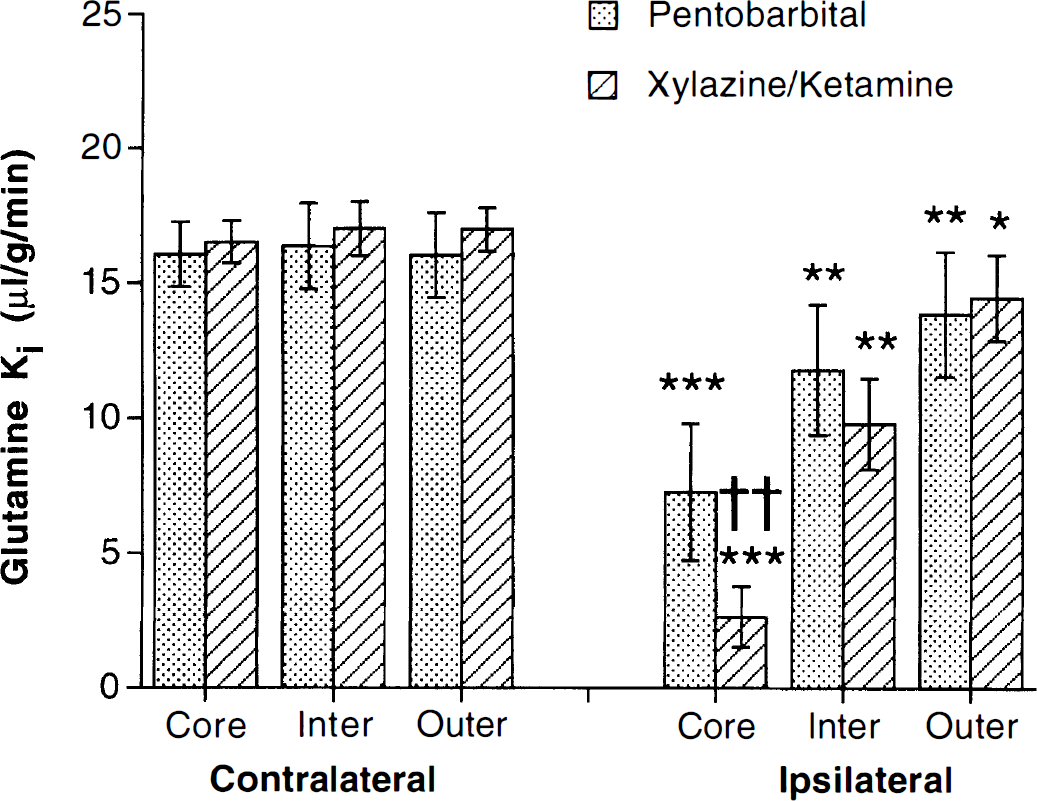
The influx rate constant (Ki) for [14C]L-glutamine in tissues contralateral and ipsilateral to the site of middle cerebral artery (MCA) occlusion. Occlusions were performed under pentobarbital or xylazine-ketamine anesthesia. Measurements were made at 4 hours of occlusion. The contralateral and ipsilateral cortices were divided into core, intermediate (inter), and outer zones. The values are means ± SD; n = 5. *, **, and *** indicate a significant reduction in the ischemic tissue compared with contralateral at the P < 0.005, P < 0.01, and P < 0.001 levels; †† indicates a significant difference between the results from the two anesthetics at the P < 0.01 level.
The greater decrease in glutamine Ki in the ischemic core of the xylazine-ketamine-anesthetized rats appears to be related to the hyperglycemia induced by this anesthesia because pretreatment of rats with insulin prevented the increase in blood glucose (blood glucose at occlusion, 78 ± 13 mg/dL) and resulted in an ischemic glutamine Ki of 11.0 ± 5.1 μL·g−1·min−1 in the core of the MCA territory, which was significantly greater (P < 0.001) than that found without insulin (2.6 ± 1.1 μL·g−1·min−1). Insulin did not affect the contralateral glutamine Ki.
Whether hyperglycemia results in a greater reduction in glutamine Ki during ischemia was further examined in glucose- and mannitol-injected rats (Fig. 2). Again, after 4 hours of ischemia, the ischemic tissue Ki was reduced in both sets of animals compared with the contralateral tissue (P < 0.001), but the reduction in glutamine Ki was significantly greater in the core of the MCA territory in the glucose-injected rats (P < 0.01). The Ki for glutamine in the contralateral cortex did not differ between the two sets of animals (Fig. 2).
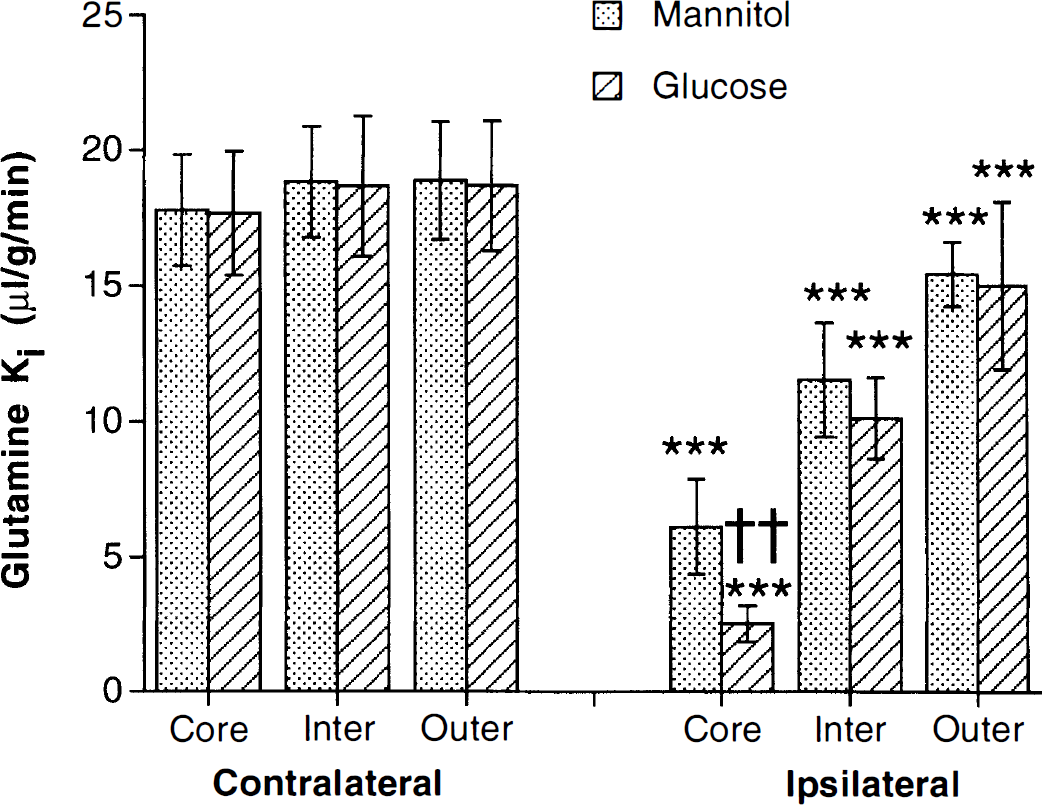
The influx rate constant (Ki) for [14C]L-glutamine contralateral and ipsilateral to the site of MCA occlusion after 4 hours. Experiments were in pentobarbital-anesthetized rats that received either a glucose or a mannitol injection 20 minutes before occlusion. The values are means ± SD; n = 5 to 8. *** indicates a significant reduction in the ischemic tissue compared with contralateral at the P < 0.001 level; †† indicates a significant difference between glucose- and mannitol-injected rats at the P < 0.01 level.
The time course of the decline in glutamine Ki in the core of the MCA territory was examined in the glucose- and mannitol-injected rats. By 1 hour of occlusion, there was a reduction in the ischemic glutamine Ki in both groups of animals (Fig. 3), but the reduction was again greater in the glucose- compared with the mannitol-injected rats (61% ± 11% versus 42% ± 8%). The decrease in glutamine Ki in the ischemic core was progressive, with the reduction in ischemic Ki being 86% ± 4% and 65% ± 9% by 4 hours in the glucose- and mannitol-injected rats.
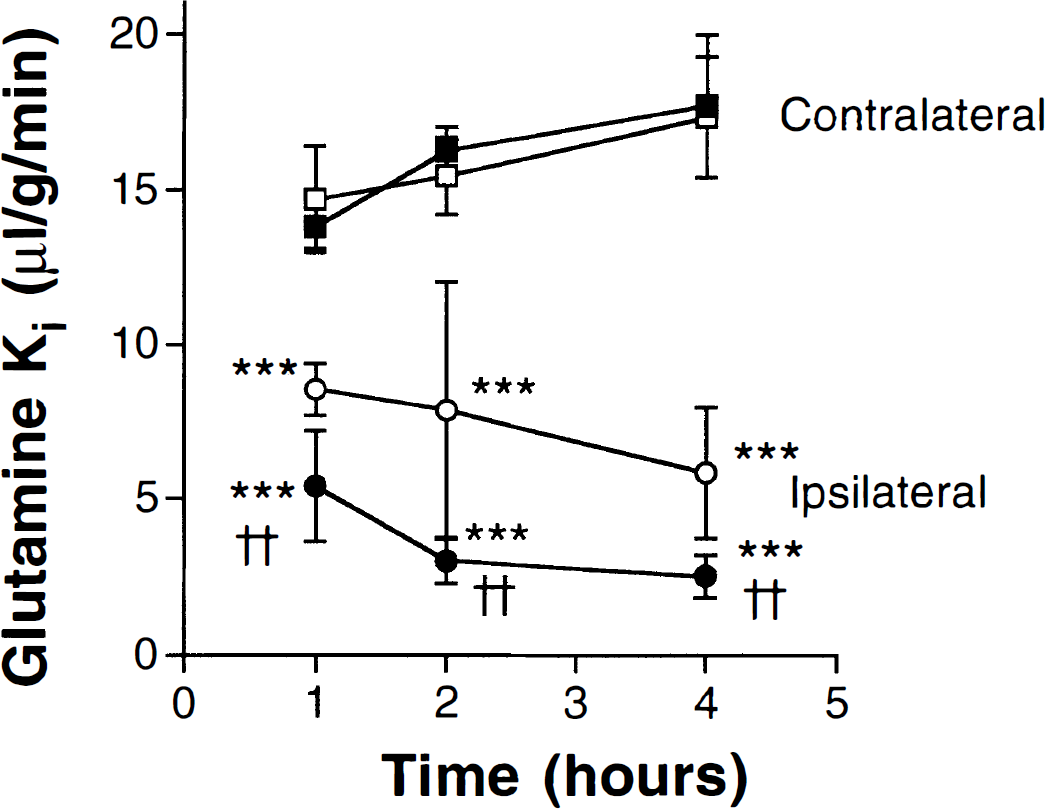
The influx rate constant (Ki) for [14C]L-glutamine in the cortex ipsilateral and contralateral to the MCA occlusion. Measurements were made in rats injected with glucose (filled symbols) and mannitol (open symbols) 1, 2, and 4 hours after MCA occlusion. Values are means ± SD; n = 5 to 8; †† indicates a significant difference at the P < 0.01 level between glucose- and mannitol-injected rats; *** indicates a significant difference (P < 0.001 level) between ipsilateral and contralateral tissue.
Blood-brain barrier glutamine and taurine transport
To better examine whether the ischemic changes in glutamine transport are related to previously reported changes in blood to brain taurine transport, the influx rate constants for glutamine and taurine were measured simultaneously in xylazine-ketamine-anesthetized rats after 4 hours of MCA occlusion (Fig. 4). These results from the core, intermediate, and outer zones demonstrate an apparently linear relation between glutamine and taurine Ki.
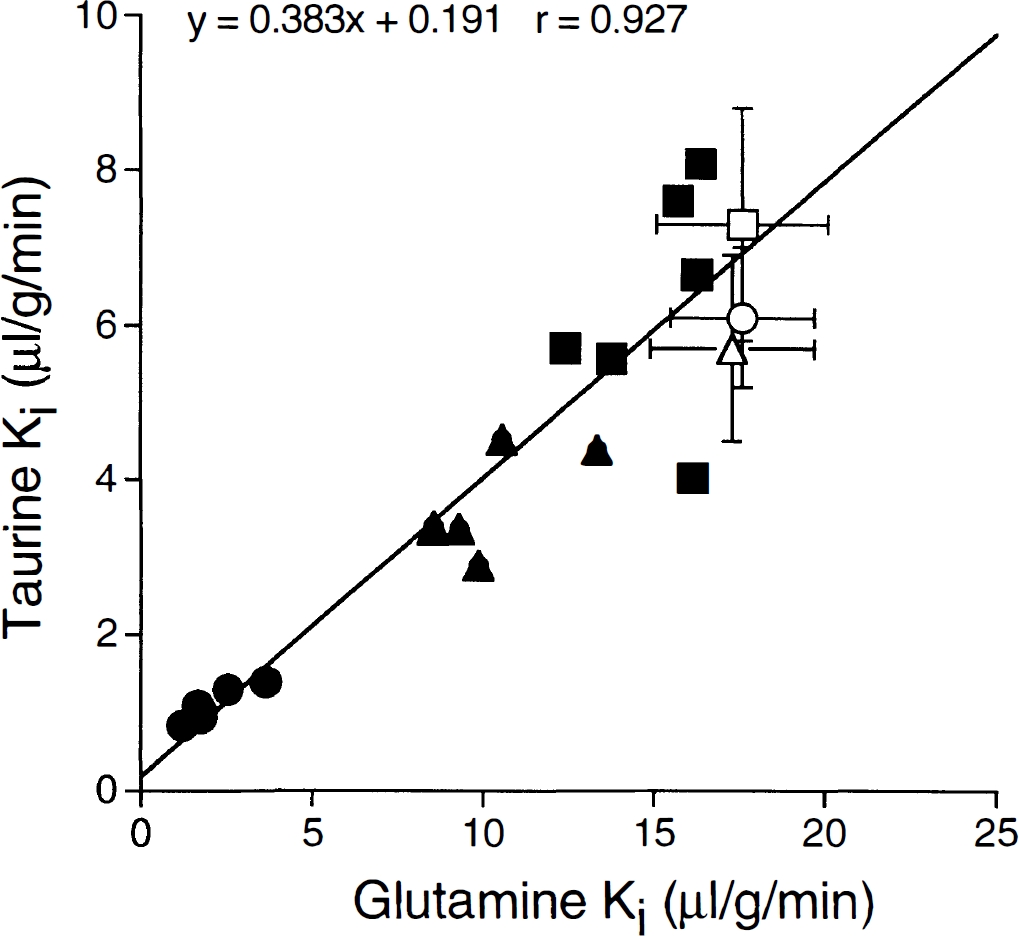
Relation between the influx rate constants for [3H]taurine and [14C]L-glutamine in rats undergoing 4 hours of MCA occlusion (xylazine-ketamine anesthesia). The results shown are from 6 rats. The squares, triangles, and circles are values from the outer, intermediate, and core zones, respectively. The filled symbols are ipsilateral to the occlusion and the open symbols (mean ± SD) are contralateral. The regression line is through the ipsilateral values only.
Cerebral blood volume
The changes in glutamine transport during ischemia in mannitol-injected rats were not associated with a change in cerebral plasma volume. After 4 hours of ischemia none of the ipsilateral tissue samples differed significantly in plasma volume from the contralateral cortex (Fig. 5). However, in glucose-injected hyperglycemic rats, there was a marked reduction in plasma volume in the ischemic core after 4 hours (Fig. 5). As with glutamine transport, this effect of ischemia on plasma volume was progressive, with there being an 11% ± 11% reduction by 1 hour, but 27% ± 20% and 45% ± 13% reductions at 2 and 4 hours (the latter reductions being significant at P < 0.05 and P < 0.01, respectively).
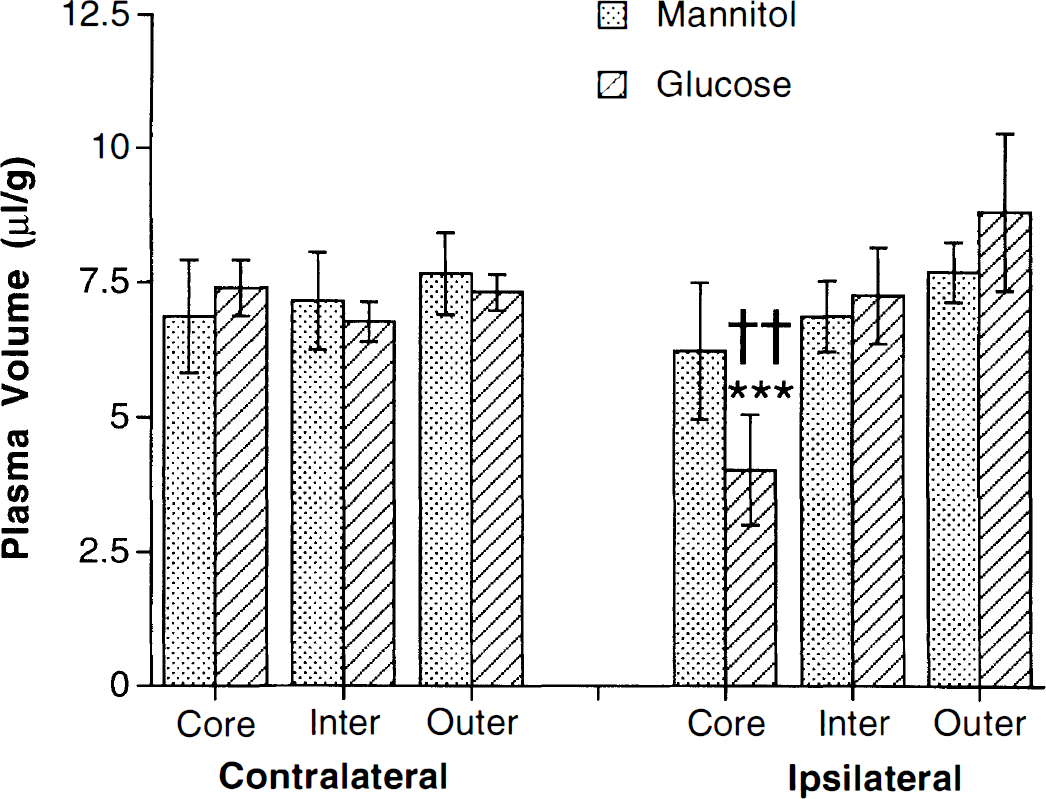
Cerebral plasma volume ([3H]inulin space) contralateral and ipsilateral to the site of MCA occlusion after 4 hours. Experiments were in pentobarbital-anesthetized rats that received either a glucose or a mannitol injection 20 minutes before occlusion. The values are means ± SD; n = 5 to 10. *** indicates a significant reduction in the ischemic tissue compared with contralateral at the P < 0.001 level; †† indicates a significant difference between glucose- and mannitol-injected rats at the P < 0.01 level.
The reduction in cerebral plasma volume in the ischemic core of glucose-injected rats was matched by a decline in cerebral red blood cell volume. In the contralateral core of the MCA territory, cerebral red blood cell volume was not significantly different between glucose- and mannitol-injected rats (1.9 ± 0.2 and 2.0 ± 0.2 μL/g) after 4 hours of MCA occlusion. However, in the ipsilateral ischemic core it was only 1.0 ± 0.4 μL/g in glucose-injected rats compared with 1.8 ± 0.2 μL/g in mannitol-injected animals (P < 0.01). The percent reduction in cerebral red blood cell volume in the ischemic core (50% ± 15%) in glucose-injected rats was similar in magnitude to that found in cerebral plasma volume (45% ± 13%).
Cerebral blood flow
Cerebral blood flow was examined in glucose- and mannitol-injected rats using 4-[N-methyl-14C]-iodoantipyrine. Contralateral hemisphere CBF 4 hours after MCA occlusion was not significantly different between the two glucose and mannitol groups (58 ± 10 and 58 ± 8 mL·100 g−1·min−1, respectively) and although the CBF in the ipsilateral tissue was about half in glucose-compared with mannitol-injected rats (7.5 ± 4.5 versus 14.7 ± 8.9 mL·100 g−1·min−1), this did not reach significance.
DISCUSSION
This study extends two previous observations on the effects of focal ischemia on the cerebrovasculature. We have found a reduction in the influx rate constant for glutamine at the BBB during MCA occlusion that is similar to that described earlier for taurine (Stummer et al., 1995). We have also found evidence that hyperglycemia exacerbates this reduction in glutamine transport indicating, as suggested by changes in cerebral plasma volume (Kawai et al., 1997c), that hyperglycemia has effects on the cerebrovasculature during ischemia as well as its known effects during reperfusion (Dietrich et al., 1993; Ginsberg et al., 1980; Kawai et al., 1997c; Venables et al., 1985).
Glutamine transport during ischemia
During MCA occlusion, we have previously reported a decline in blood to brain [3H]taurine transport (Stummer et al., 1995). This was in contrast to previous studies on another amino acid, leucine, which has been examined during ischemia (Brust, 1991; Sage et al., 1984) and anoxia (Betz et al., 1975). It was uncertain whether the reduction in taurine influx was caused by downregulation of the taurine transporter, dissipation of the endothelial cell Na+ gradient that drives BBB taurine transport, or loss of taurine from brain during ischemia causing an increase in plasma taurine concentration during passage through the ischemic brain and saturation of the taurine transporter. The reduction in influx did not appear to be related to a decline in the surface area of perfused capillaries as the decline in taurine uptake was much greater than a slight decline in the uptake of α-aminoisobutyric acid, an amino acid not transported from blood to brain (Stummer et al., 1995). We therefore examined whether there would be similar changes in the uptake of glutamine transport into brain because we have recently found that this uptake, like taurine, is Na+-dependent and apparently mediated by system N (Keep et al., 1997a). The present study demonstrates that glutamine transport, like that for taurine, is markedly reduced by MCA occlusion. After 4 hours of occlusion, there is no net loss of glutamine from the brain (ipsilateral and contralateral glutamine brain concentrations: 25 ± 3 and 25 ± 2 μmol/g dry weight; Stummer and Keep, unpublished observations). This suggests that changes in plasma glutamine concentration during the passage through the ischemic tissue is not the cause of the reduction in influx rate constant. In addition, when taurine and glutamine transport were measured simultaneously, there was a very close relationship between the decline in the influx rate constants for both amino acids during ischemia. Thus, unless both glutamine and taurine transport are regulated in exactly the same manner, it is unlikely that these changes in influx reflect downregulation of the respective transporters. In contrast, a dissipation of the endothelial [Na+] gradient might be expected to have a similar effect on both taurine and glutamine transport, and this may be the cause of the marked decline in the influx rate constants for these two amino acids in the ischemic tissue. Leucine transport, if this explanation is correct, would not be expected to decline with ischemia because that amino acid is transported by the Na+-independent system L (Smith, 1991).
It is well known that neuronal and glial cell Na+ gradients dissipate during cerebral ischemia because of reduced ATP production, with subsequent failure of the cell membrane Na+/K+-ATPase, and the excitatory amino acid-mediated opening of nonselective cation channels. The effect of ischemia on the intracellular [Na+] of the cerebral endothelial cells has not been determined. The effect of MCA occlusion on the influx rate constant for sodium 22 has been examined, and those studies have generally indicated a small stimulation during ischemia (Betz et al., 1994b). However, the relation of this measure to intracellular [Na+] is uncertain because of the potential role of active, facilitative, and diffusional (channel or paracellular) transport in Na+ movement from blood to brain (Ennis et al., 1996).
The fall in blood to brain glutamine transport may have important consequences during ischemia. The plasma glutamine concentration in rat is about 500 μmol/L. Thus, with an influx rate constant of 16 μL·g−1·min−1, glutamine influx is about 2 μmol/g wet weight over 4 hours. During ischemia in normoglycemic rats, this number fell to about 1 μmol/g whereas in hyperglycemia rats it fell to about 0.3 μmol/g. Considering that total brain glutamine content is only about 4.5 μmol/g wet weight (Clarke et al., 1989), this alteration in influx would result in a 20% to 40% reduction in brain glutamine during ischemia if glutamine efflux from the brain and glutamine metabolism remained constant. The absence of any net loss of brain glutamine during MCA occlusion (see above) suggests that some of the net loss of brain glutamate that occurs in MCA occlusion (Keep et al., 1997b) may result from a net conversion to glutamine.
If there is a dissipation of the endothelial cell Na+ gradient, its effects would not be limited to systems N (glutamine) and β (taurine) amino acid transport. Sodium-dependent system A transport is present on the abluminal membrane of the BBB (Betz and Goldstein, 1978). It maintains low brain extracellular concentrations of several amino acids that are neurotransmitter precursors (Betz et al., 1994a). Thus, an alteration in the rate of system A transport may have adverse consequences for the brain and contribute to ischemic brain damage. Other non-amino acid transporters would also be changed. For example, cerebral endothelial cells have Na+/H+ antiport (Ennis et al., 1996) and Na+-dependent myo-inositol (Kollros et al., 1990), system ASC amino acid (Tayarani et al., 1987), and adenosine (Pardridge et al., 1994) transport, changes in which may adversely affect endothelial or parenchymal cell function.
Effect of hyperglycemia on glutamine transport during ischemia
Recently, we have noted a marked decrease (50%) in cerebral plasma volume during focal ischemia (MCA occlusion) in hyperglycemic but not normoglycemic rats (Kawai et al., 1997c). This appears to indicate that there are cerebrovascular changes during the ischemic event in hyperglycemic rats. This hypothesis is supported by the present finding that the reduction in the influx rate constant for glutamine during ischemia is further exacerbated in hyperglycemic animals. Yet to be determined is the extent to which cerebrovascular changes during ischemia participate in the known cerebrovascular dysfunction during reperfusion in hyperglycemic animals, including lower CBF, BBB disruption, and hemorrhagic conversion (de Courten-Myers et al., 1992; Dietrich et al., 1993; Ginsberg et al., 1980; Kawai et al., 1997c; Venables et al., 1985).
The present study compares glucose- with mannitol-injected rats. Because there is some evidence that mannitol may reduce ischemic brain damage (Little, 1978), it was possible that rather than glucose enhancing the reduction in glutamine transport during ischemia it was mannitol lessening the reduction. However, the reduction in glutamine transport obtained in mannitol-injected rats in the ischemic core at 4 hours (35% ± 9% of contralateral) was not significantly different from that obtained in pentobarbital-anesthetized rats receiving no mannitol (45% ± 14%). In addition, the reduction in glutamine transport observed in xylazine-ketamine-anesthetized rats, an anesthesia that induces hyperglycemia (Kawai et al., 1997c), could be ameliorated by injection of insulin, again indicating that hyperglycemia modulates BBB glutamine transport during ischemia.
Unlike the situation in normoglycemic rats, MCA occlusion in hyperglycemic rats is associated with a fall in the plasma volume of the ischemic tissue. After 4 hours of MCA occlusion in rats in which hyperglycemia was induced by glucose injection or xylazine-ketamine anesthesia, there was a 50% reduction in plasma volume (Kawai et al., 1997c). There may be a relationship between this decline in plasma volume and the additional reduction in glutamine transport that occurs during ischemia in hyperglycemic rats as both changes occur over a similar time course. A disruption of the endothelial cell Na+ gradient would result in endothelial cell swelling and, potentially, a reduction in capillary luminal diameter and thus a fall in plasma volume. In favor of this hypothesis, Paljärvi et al. (1983), examining the effect of acute hyperglycemia on endothelial cells after global ischemia and reperfusion, reported endothelial cell swelling and decreased luminal diameter. Alternately, the correlation between the decline in glutamine transport and that in cerebral plasma volume in hyperglycemic rats might reflect a partial failure of capillary perfusion. However, the extent to which plugging occurs in MCA occlusion is a matter of controversy (e.g., Buchweitz-Milton and Weiss, 1988; Theilen et al., 1994). It has been suggested that during ischemia some vessels may be partially occluded and allow plasma but not red blood cell flow. Our finding of an equal decrease in cerebral plasma and red blood cell volume suggests that any such partial occlusion is not a major factor in these volume changes.
Increased lactate production and decreased tissue pH have been proposed as the mechanisms by which hyperglycemia enhances parenchymal damage after cerebral ischemia. These factors may also play a role in the greater endothelial cell dysfunction described here in hyperglycemic rats.
In conclusion, this study demonstrates a major reduction in BBB glutamine transport during focal cerebral ischemia. Because major disruption of the BBB, as evinced by albumin entry, does not occur until after 6 hours of MCA occlusion (Menzies et al., 1993), these changes in transport are an early sign of BBB dysfunction during ischemia, a dysfunction that is enhanced by hyperglycemia.