
Abstract
Preconditioning brain with tumor necrosis factor alpha (TNF-α) can induce tolerance to experimental hypoxia and stroke and ceramide is a downstream messenger in the TNF-α signaling pathway. A hypoxic-ischemic (HI) insult in the immature rat injures brain primarily through apoptosis. Apoptosis is regulated by Bcl-2 family proteins. The authors explored whether ceramide protects against HI in the immature rat, and whether Bcl-2 family protein expression is involved. Hypoxia-ischemia was produced in seven-day-old rats by ligating the right carotid artery, followed by 2 hours of 8% oxygen exposure. Thirty minutes after HI, C2-ceramide (150 μg/kg) was injected intraventricularly. Infarct volume was measured 5 days later. C2-ceramide reduced HI-induced brain damage by 45% to 65% compared with HI/dimethyl sulfoxide (DMSO) (vehicle control) or HI only groups. In separate experiments, brains of sham-operated control and HI only animals and animals subjected to HI plus C2-ceramide or DMSO infusion were sampled 6 hours, 24 hours, and 5 days after treatments and analyzed for Bcl-2, Bcl-xl, and Bax expression (Western blotting), and apoptosis (TUNEL assay). Augmented Bcl-2 and Bcl-xl levels in the C2-ceramide treated group were associated with a significant decrease in TUNEL-positive cells. The results support a protective role for ceramide in neonatal HI.
Although the brain of a newborn exhibits a higher tolerance to hypoxia and ischemia than that of an adult, morbidity from perinatal asphyxia remains high (Volpe, 1995). Recently, increasing attention has been focused on the induction of tolerance to hypoxic-ischemic insults (HI). A period of cerebral hypoxia or ischemia insufficient to produce brain damage can provide protection against a subsequently more severe insult (Vannucci et al., 1998). The mechanism for this phenomenon is not completely understood. In a previous in vivo study, the authors found that TNF-α plays an important role in induction of tolerance to ischemia (Nawashiro et al., 1997). The authors' in vitro experiments identified ceramide as a downstream signaling step in the preconditioning pathway (Ginis et al., 1999; Liu et al., 2000). Hypoxia-ischemia modeled in a neonate is different from a stroke modeled in an adult. Cell death in the former is mainly because of apoptosis, but in the latter necrosis is predominant (Ferrer et al., 1994). The underlying mechanisms for the neuroprotection induced by hypoxic or ischemic preconditioning in the immature rat have not been fully elucidated, but probably involve expression of genes and synthesis of proteins such as Bcl-2 family proteins (Bossenmeyer-Pourie and Daval, 1998). Members of the Bcl-2 family are protooncogenes. Some of the members, such as Bcl-2 and Bcl-xl, have antiapoptotic functions and other members, such as Bax, are proapoptotic (Oltvai et al., 1993). In the current study, the authors hypothesized that C2-ceramide plays a role in induction of tolerance to HI that could involve Bcl-2 proteins.
MATERIALS AND METHODS
Animal model
The protocol was approved by the Animal Care and Use Committee (ACUC) of the National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (NIH). All surgical procedures were performed on postnatal day 7 in unsexed Sprague–Dawley rats (Charles River Laboratories, Wilmington, MA, U.S.A.). The animals were subjected to a well-characterized neonatal HI model (Rice et al., 1981). Briefly, the pups were deeply anesthetized by methoxyflurane inhalation and placed on a warm water pad. The right common carotid artery of each pup was exposed, isolated from the vagus nerve and jugular vein, and ligated with 4–0 surgical silk. The incision then was sutured and the animal was allowed to recover in a warm environment. The whole surgical procedure was completed in less than 5 minutes. After a recovery of 1 hour, the pups were placed in glass chambers, which were submerged in a water bath (37°C), and exposed to a mixture of 8% O2 and 92% N2 for 2 hours. The animals were divided into four groups. In group 1 (HI/ceramide), 30 minutes after HI, the pups were deeply anesthetized again and 150 μg/kg C2-ceramide (Sigma, St. Louis, MO, U.S.A.) in 1 μL dimethyl sulfoxide (DMSO) was infused into the lateral ventricle by stereotaxic injection within 1 minute. Preliminary studies determined this C2-ceramide dose to be optimal. The lamda suture was used as a surface anatomic landmark, and the injection site was at anterior-posterior (AP) 1.5 mm, lateral (L) −1.5 mm, depth (D) −4 mm. After 1 hour of recovery, the pups were returned to their dam. In group 2 (HI/DMSO), 1 μL DMSO was infused into the lateral ventricle as in group 1. In group 3, animals were subjected to HI without C2-ceramide or DMSO infusion (HI only). Group 4 was composed of sham-operated animals that underwent the same operation except that the exposed carotid artery was not ligated and there was no exposure to hypoxia (sham-operated). Group 3 and group 4 received no subsequent intraventricular injections.
Assessment of hypoxic–ischemic injury
At P12 days, 5 days after injury, the brains were removed, frozen on dry ice, and sectioned (20 μm). Sections were postfixed over paraformaldehyde vapors and stained with cresyl violet (Barks et al., 1995). A computerized image analysis system (NIH Image 1.62) was used to measure cross-sectional areas of the striatum, anterior hippocampus, and cortex bilaterally in every eighth 20-μm coronal section (≥13 sections per brain). The volume of intact tissue was measured. Regional volumes were calculated by summation of cross-sectional areas and integration (Swanson et al., 1990).
Western blot
Pups were killed at 6 hours, 24 hours, and 5 days after HI. Each hemisphere was homogenized in 0.5 mL lysis buffer, containing 10% glycerol, 0.05 mol/L tris-buffered saline (TBS), 1 mmol/L phenylmethylsulfonyl fluoride, 1 μmol/Lcalpain inhibitor I, 1 μg/mL leupeptin, and 1 μg/mL aprotinin. Lysates were centrifuged at 18,500x g (4°C) for 5 minutes and the supernatant was aspirated and stored at −70°C until used. Tissue protein was measured by Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, U.S.A.) and adjusted for each blot. One hundred microgram protein samples were loaded on to 14% sodium dodecyl sulfate-polyacrylamide gels after denaturing in sodium dodecyl sulfate gel-loading buffer in boiled water for 4 minutes. After electrophoresis, proteins were electrotransferred to the polyvinylidene difluoride membrane at a constant voltage of 25 V for 90 minutes. After transfer, the membrane was washed twice with TBS plus 0.1% Tween-20 (TBST, pH 7.4) and then preincubated with a blocking buffer (5% nonfat dry milk in TBST) at 4°C overnight. The polyvinylidene difluoride membrane then was incubated with mouse monoclonal Bcl-2, Bcl-xl, or Bax primary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) 1:1000, 1:500, and 1:1000 dilutions, respectively, in TBST at room temperature for 2 hours. After washing, blots were incubated with secondary anti-mouse antibody conjugated with horseradish peroixdase 1:4000 dilution at room temperature for 1 hour. Immunoblots were visualized by means of Amersham enhanced chemiluminescence (ECL)(Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.) and Kodak X-Omat films (Kodak, Rochester, NY, U.S.A.). In positive control experiments, the positive control standards purchased from Santa Cruz Biotechnology were used to identify the signals of Bcl-2, Bcl-xl, and Bax.
Semiquantification of the Western blots
The intensity of the corresponding western blot band was measured by using NIH Image 1.62. Data are calculated as the ratio of the signal intensity in the ischemic hemisphere compared with that in the contralateral hemisphere.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling staining (TUNEL)
Pups were killed at 6 hours, 24 hours, and 5 days after HI and the brains were immediately removed and frozen on dry ice. Coronal (20 μm) sections were cut on a cryostat and stored at −70°C until used. The authors used the in situ cell death detection kit (Boehringer Mannheim, Indianapolis, IN, U.S.A.) according to the manufacturer's instructions for TUNEL staining. Four sections were taken from each brain at bregma +0.20 mm and −2.8 mm (Paxinos and Watson, 1986), two sections in each level. Samples were analyzed with a Zeiss Axiovert 100 light microscope (20x objective). Digitized images of 10 microscopic fields per hemisphere in each of the 4 sections were generated using a digital CCD Camera C4742–95–12 (Hamamatsu) and Zeiss Axio Version 2 software. The same microscope and camera settings were used for all samples. The number of TUNEL-positive cells within each image was determined by means of the Scion Image (NIH Image for PC) computer program.
Statistics
Means and SD were calculated and differences were tested with analysis of variance followed by a Fisher PLSD at a significance level of P < 0.05, for the two-tailed test.
RESULTS
Measurement of infarct volume
In the HI only or HI/DMSO group, the volume of the uninjured cortex, striatum, and anterior hippocampus in the ischemic hemisphere was about half of the contralateral side (Fig. 1A to 1C). No differences between ipsilateral and contralateral hemispheres in the sham-operated group (data not shown) existed. After injection of 150 μg/kg C2-ceramide in the HI/ceramide group, the uninjured volume of ischemic cortex, striatum, and hippocampus was augmented by 67%, 63%, and 45%, respectively, at 5 days after injury.
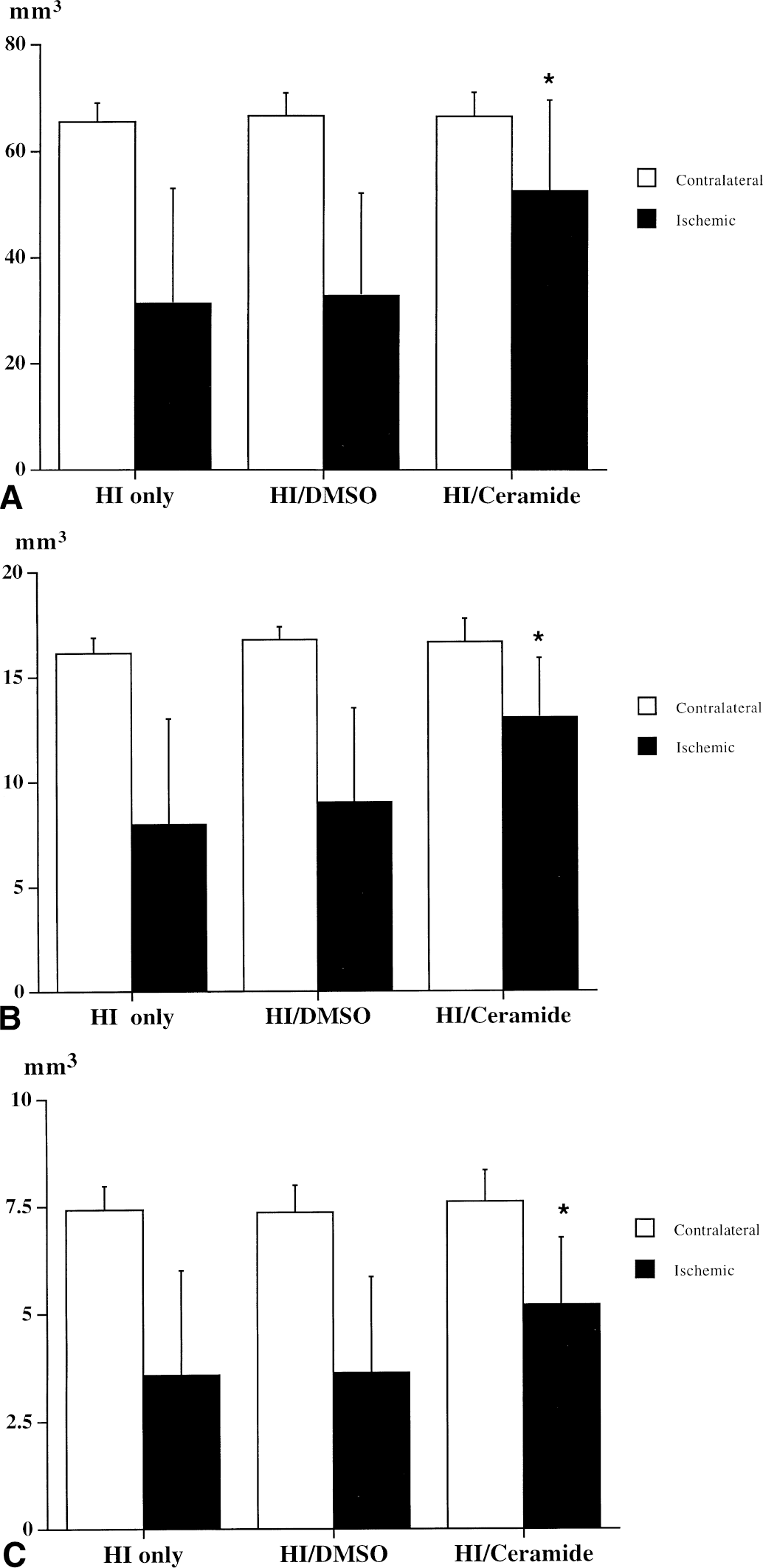
Effect of intraventricular C2-ceramide 150 μg/kg injected at 30 minutes after HI on the volume of cortex
Western blot
In control pups, there was a background expression of Bcl-2 proteins in both hemispheres. Hypoxia–ischemia itself did not produce any changes in expression of Bcl-2 proteins in this model. However, 6 hours after HI in the HI/ceramide group, Bcl-2 and Bcl-xl expression in the ischemic hemisphere was approximately twice as high as in the contralateral hemisphere, indicating that C2-ceramide up-regulates expression of these antiapoptotic proteins (Fig. 2A to 2B, Fig. 3). There was no difference in Bax expression (Figs. 2C and 3). Twenty-four hours after HI in the same group, Bcl-2, in the ischemic hemisphere of ceramide-injected pups, remained 90% increased, but Bcl-xl had decreased to approximately 40% greater than baseline (Fig. 2A to 2B). Five days later, no differences in Bcl-2 and Bcl-xl between two hemispheres were observed in any of the groups (Fig. 2A to 2C).
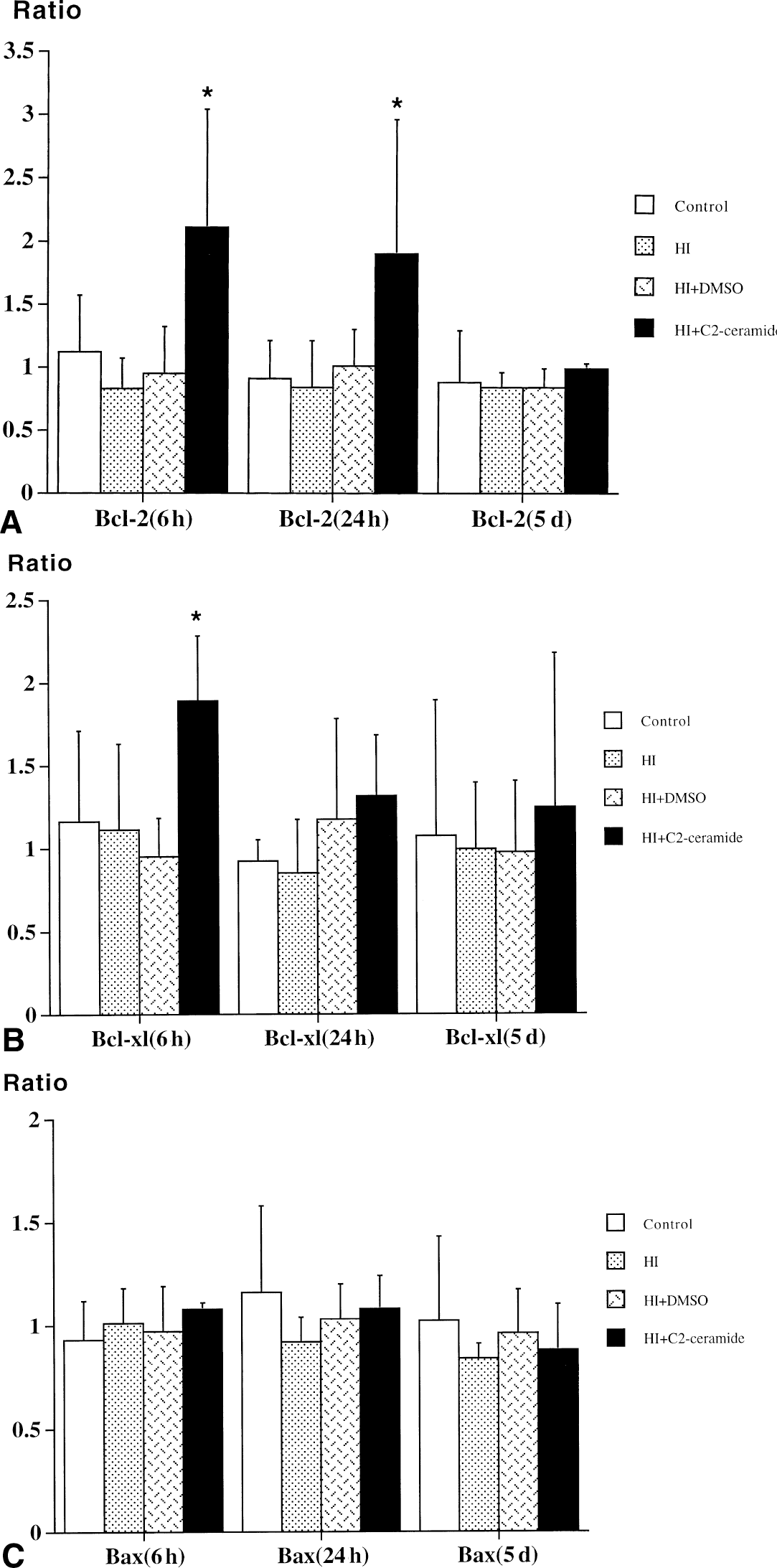
Effect of intraventricular C2-ceramide 150 μg/kg injected 30 minutes after hypoxic–ischemic injury (HI) on the expression of Bcl-2
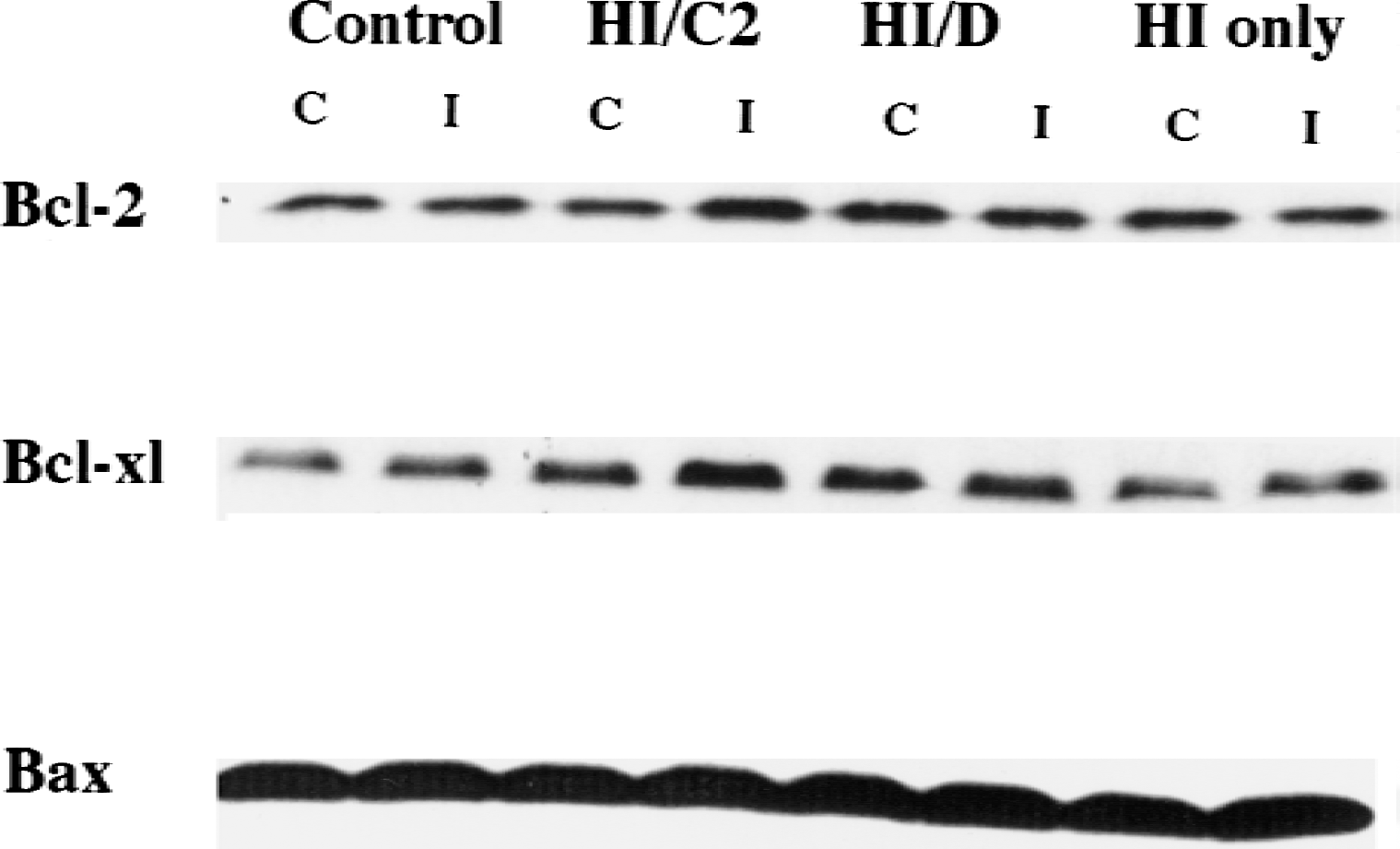
Expression of Bcl-2, Bcl-xl, and Bax proteins in the ischemic and contralateral brain hemispheres at 6 hours after hypoxic–ischemic injury (HI). The band intensity of Bcl-2 and Bcl-xl from the ischemic side is greater than that from the contralateral side in the HI/ceramide group. Control, sham-operated control; HI/C2, HI/ceramide; HI/D, HI/DMSO; C, contralateral side; I, ischemic or ipsilateral side.
TUNEL staining
TUNEL-positive cells were widely expressed in the ischemic hemisphere (ipsilateral to the carotid artery ligation) in the HI/ceramide, HI/DMSO, and HI only groups compared with sham-operated controls. However, there were fewer TUNEL-positive cells in the ischemic hemisphere of the HI/ceramide group than in HI/DMSO and HI only animals. There was no statistical difference in TUNEL-positive cell counts between ischemic and contralateral hemispheres in the HI/ceramide group at 6 hours after HI; however, after 6 hours, 24 hours, and 5 days in the HI/DMSO and HI only groups, the number of TUNEL-positive cells in the ischemic side was significantly increased compared with the contralateral side (Table 1). Comparison of the HI-induced apoptotic damage among different treatment groups also revealed a beneficial effect of C2-ceramide. At 6 hours and 5 days after HI, the number of TUNEL-positive cells in the injured hemisphere in the HI/ceramide group was significantly less than that in HI/DMSO or HI only groups (Table 1 and Fig. 4).
Effect of ceramide on the number of TUNEL-positive cells in each brain hemisphere at 6 hours, 24 hours, and 5 days after HI

Values represent the counts of TUNEL-positive cells (mean±SD).
Comparison of the TUNEL-positive cells in ischemic hemisphere with the positive cells in contralateral hemisphere in same group significant at P < 0.05.
Comparison of the number of TUNEL-positive cells in ischemic hemisphere in the group against the number of TUNEL-positive cells in ischemic hemisphere in the HI/ceramide group significant at P < 0.05 (n = 5 to 6). HI, hypoxic-ischemic injury; DMSO, dimethyl sulfoxide.
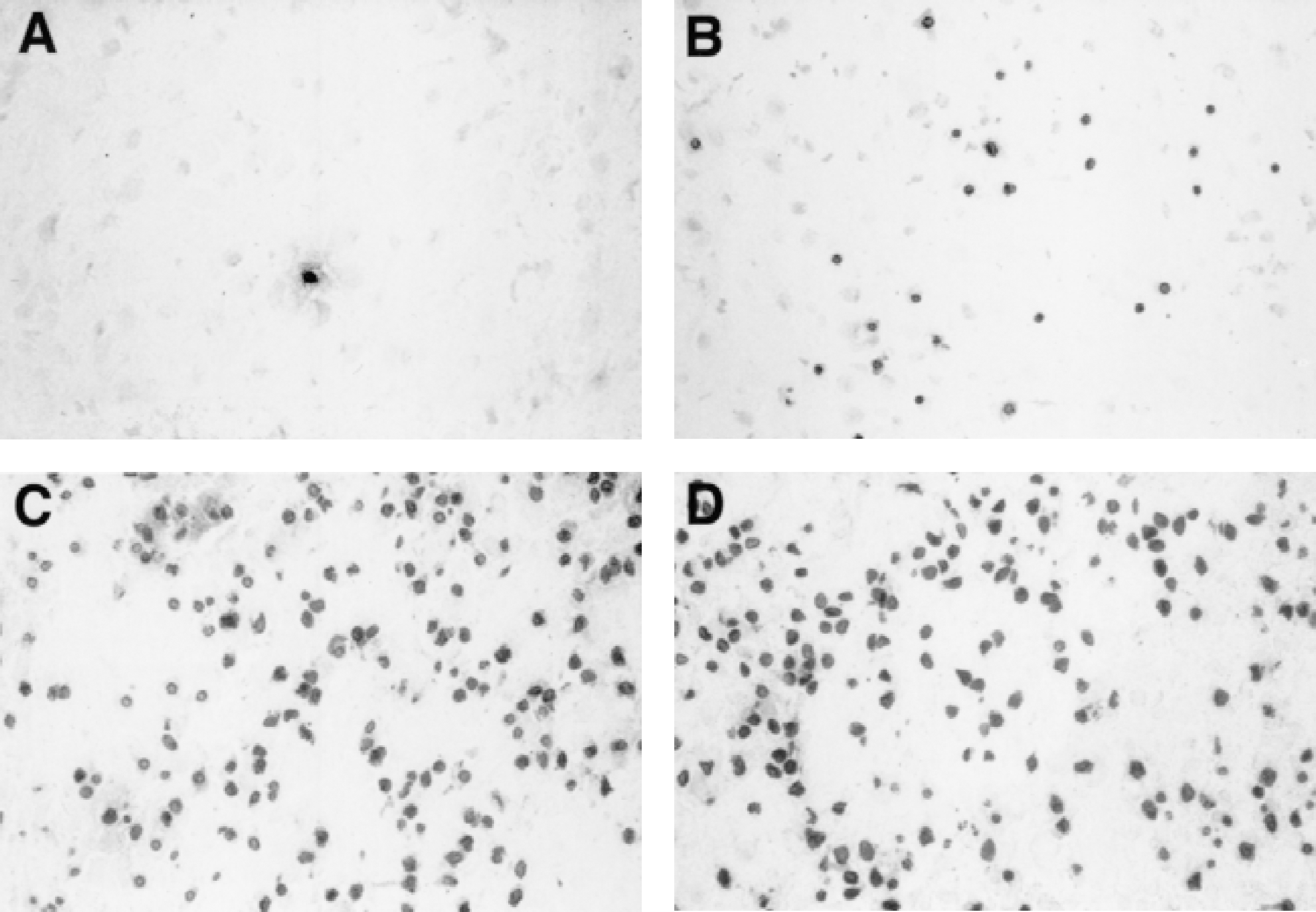
Photomicrographs of TUNEL-stained sections in the ischemic cortex (common carotid artery ligation side) at 5 days after hypoxic–ischemic injury (HI).
In all treatment groups, there was a trend toward a decrease of constitutive TUNEL-positive cells with age. The number of TUNEL-positive cells in an uninjured contralateral hemisphere was significantly less in 12-day-old pups (5 days after treatments) than in 8- to 9-day-old animals (6 and 24 hours after treatments) (Table 1).
DISCUSSION
Induced tolerance to ischemia is a phenomenon in which exposure of animals or cells to sublethal stress such as spreading depression, hypoxia, ischemia, seizures, or hyperthermia induces tolerance to subsequent, otherwise lethal ischemia. The mechanism of such ischemic tolerance in the brain is still unclear. Excitatory amino acids, adenosine receptors and ATP-dependent potassium channels, heat shock proteins, Bcl-2, superoxide dismutase, and activation of the extracellular signal-regulated protein kinase have been implicated as neuroprotective mechanisms in preconditioning (Chen and Simon, 1997; Singer, 1999). However, signaling pathways leading to activation of these genes or activation of signaling kinases are not well defined. In the authors' previous studies, TNF-α has been shown to play an important role in preconditioning for induction of ischemic tolerance and has been identified as a downstream mediator of preconditioning stimuli, such as hypoxia or lipopolysaccharide-pretreatment (Nawashiro et al., 1997; Tasaki et al., 1997). Ceramide, a sphingolipid, which can be produced in the cell through sphingomyelin (SM) hydrolysis or by de novo synthesis, is a lipid mediator in TNF-α signaling. Biologic effects of ceramide mimic those of TNF-α and include the regulation of cell proliferation and differentiation, inflammation, and apoptosis (Hannun, 1997). C2-ceramide is a short acyl chain (acetyl) analog of ceramide that is cell permeable. C2-ceramide has been widely used for elucidation of ceramide effects in cells. Thus, exogenous ceramide can protect hippocampal neurons against injury induced by glutamate (Goodman and Mattson, 1996). It can also protect against apoptosis induced by nerve growth factor withdrawal (Ito and Horigome, 1995) and against free radical–mediated forms of cell injury (Greenlund et al., 1995). Recently, the authors have shown that ceramide protects cortical neurons from hypoxic injury and mediates hypoxia tolerance induced by hypoxic preconditioning (Liu et al., 2000). To address whether C2-ceramide has effects in HI, the authors used C2-ceramide to examine whether it could confer cerebroprotection in a neonatal model of HI and studied expression of several proteins that were candidate molecules for any protective effect. The results of this investigation show that animals injected intracerebroventricularly with C2-ceramide have approximately 45% to 65% less brain damage than the control group. This protective effect of ceramide compares very favorably with other cytoprotectants used in this model, such as the adenosine deaminase inhibitor, deoxycoformycin (DCF; 2.5 mg/kg intraperitoneally) (Gidday et al., 1995), and caspase inhibitors (Cheng et al., 1998). Most importantly, ceramide was effective when injected 30 minutes after HI, which makes ceramide potentially advantageous for clinical application.
In contrast to the adult central nervous system in which ischemia causes most cells to die through necrosis, HI in the neonatal brain leads mainly to death through apoptosis (Ferrer et al., 1994). Apoptosis is a morphologically distinct form of cell death, which is characterized by nuclear condensation, membrane blebbing, and DNA fragmentation.
Unlike necrosis, apoptosis is a programed suicide process requiring activation of several genes and proteins (Steller, 1995). Apoptosis is also a common response to neuronal injury, particularly during the neonatal period in mammals. Several families of molecules that regulate apoptosis have been reported (Reed, 1994). The Bcl-2 family proteins have been the focus of a great deal of research. Bcl-2 is an acronym for B-cell lymphoma/leukemia-2 (Reed, 1994). The Bcl-2 family includes a large number of intracellular proteins with opposing effects on the regulation of apoptosis. Some members, such as Bcl-2 and Bcl-xl (Parsadanian et al., 1998), function as apoptosis inhibitors, but Bax promotes apoptosis (Oltvai et al., 1993). The ratio of Bcl-2 to Bax determines survival or death after an apoptotic stimulus (Oltvai et al., 1993).
The intracellular locations of Bcl-2 proteins include the nuclear envelope, parts of the endoplasmic reticulum, and the mitochondrial membrane (Reed, 1994). It was first reported in 1988 that Bcl-2 can prolong cell survival by blocking programed cell death (Vaux et al., 1988) and is confirmed by the following studies (Wang et al., 1999; Chen et al., 2000). To block apoptosis, Bcl-2 acts upstream to prevent the activation of caspases, inhibit free radical formation, regulate calcium sequestration, block the proapoptotic actions of other members of the Bcl-2 family such as Bax and Bad (Merry and Korsmeyer, 1997), and block the release of cytochrome c from mitochondria (Yang et al., 1997). It has also been reported that Bcl-2 can act at a point downstream of ceramide (Zhang et al., 1996).
In the current study, ceramide increased the expression of Bcl-2 and Bcl-xl proteins as early as 6 hours after HI. Both Bcl-2 and Bcl-xl expression were significantly increased in the HI/ceramide group as compared with the HI/DMSO, HI-only, and sham-operated control groups. The level of Bax remained unchanged in all groups.
Up-regulation of Bcl-2 and Bcl-xl over a 24-hour period could explain the protective effect of C2-ceramide. In a model of permanent middle cerebral artery occlusion in Bcl-2 transgenic mice, the volume of brain infarction was reduced by 50% as compared with wild-type mice because of a reduction in apoptosis (Martinou et al., 1994).
After preconditioning, Bcl-2 is overexpressed and appears to be the most important gene that inhibits apoptosis (Maulik et al., 1999). In the authors' studies, overexpression of Bcl-2 and Bcl-xl coincides with an impressive neuroprotective effect of ceramide that was detectable 5 days after HI. Similarly, the protection induced by preconditioning is related to up-regulation of Bcl-2, which prevented apoptosis (Martinou et al., 1994). In Bcl-xl transgenic mice, hypoxia–ischemia produces approximately 40% to 60% less damage by reducing apoptosis, compared with wild type neonatal mice.
Overexpression of Bcl-xl prevented apoptosis of cortical and hippocampal neurons in a hypoxic–ischemic paradigm in neonatal mice (Parsadanian et al., 1998) and in adult mice (Wiessner et al., 1999). The current results show that at 6 hours and 5 days after HI, the number of TUNEL-positive cells determined by TUNEL assay had dramatically decreased in the lesioned hemisphere after ceramide application when compared with TUNEL-positive cell counts in HI/DMSO and HI only groups (Table 1). Also, the density of TUNEL-positive cells decreased with the increase of animals' age in the control group. TUNEL-positive cell counts were significantly less in 12-day-old pups than in 8-day-old pups. This observation is consistent with previous studies of rat brain development in which apoptosis has been shown to play a physiologic role (Romijn et al., 1991; Raff et al., 1993).
In conclusion, C2-ceramide, injected 30 minutes after HI, significantly protected neonatal brain against hypoxic–ischemic injury. In response to C2-ceramide, Bcl-2 and Bcl-xl levels increased and apoptosis was inhibited. These observations may have useful clinical implications. The neonatal hypoxic–ischemic brain injury model corresponds to perinatal asphyxia in humans, which leads to pathologic conditions such as cerebral palsy (Volpe, 1995). Ceramide analogs, such as C2-ceramide, that can augment expression of antiapoptotic genes may become useful therapeutic agents after neonatal HI.
Footnotes
Acknowledgment
The authors thank Mrs. Mary Crawford for excellent secretarial assistance.