
Abstract
The proto-oncogenes bcl-2 and bcl-x-long have been shown to suppress apoptotic cell death in a variety of in vitro systems and cell lines, including neurons. An alternatively spliced form of bcl-x, bcl-x-short, is a promoter of apoptotic death. Whether these genes are induced after ischemia or play any role in determining the fate of ischemic neurons is unknown. To begin to address this issue, we studied the expression of bcl-2, and bcl-x mRNA and protein after global ischemia in the rat. Ischemia was induced in isoflurane-anesthetized rats by the four-vessel occlusion method. mRNA expression was studied by Northern blot analysis at 24 h after ischemia and by in situ hybridization at 2, 4, 8, 24, and 72 h after 15 min of global ischemia. Protein expression was studied using both immunocytochemistry at 4, 8, 16, 24, and 72 h after ischemia and Western blot analysis from tissue harvested at 16, 24, and 72 h after ischemia. Western blots showed that bcl-x-long is the predominant form of bcl-x protein expressed in both normal and ischemic brain. Both bcl-2 and bcl-x-long mRNA were expressed in CA1, CA3, and the molecular layer of the dentate after ischemia. However, bcl-2 and bcl-x protein were expressed only in CA3 and dentate. Thus, while bcl-2 and bcl-x-long mRNA were expressed in both surviving and dying neurons, their proteins were expressed in neurons destined to survive. These results support potential roles for these two apoptosis suppressor proteins in promoting survival after cerebral ischemia.
Apoptosis, a form of programmed cell death, plays a critical role in the regulation of development and maintenance of many adult tissues, including those of the nervous system (Saunders, 1966). Apoptosis is associated with the activation of a genetic program in which apoptosis effector genes promote cell death, while repressor genes enhance cell survival. Among the apoptosis repressor genes studied in mammalian cells, the proto-oncogene bcl-2 has attracted the most attention. Bcl-2 protein has been found to enhance the survival of many, but not all, cell types exposed to a variety of adverse stimuli (Vaux et al. 1988; Hockenbery et al., 1990; Sentman et al., 1991; Strasser et al, 1991). Moreover, several studies have implicated bcl-2 as a potential regulator of neuronal survival. For example, overexpression of bcl-2 has prevented primary neurons and neuronal cell lines from trophic factor withdraw or glutathione depletion-induced cell death (Garcia et al., 1992; Allsopp et al., 1993; Batistatou et al., 1993; Kane et al., 1993).
In the central nervous system, bcl-2 is expressed in a large population of neurons during embryonic development (LeBrun et al., 1993; Novack and Korsmeyer, 1994), but levels are greatly reduced or undetectable in the majority of postmitotic central nervous system (CNS) neurons of adult brains (Merry et al., 1994). However, bcl-2-deficient mice exhibited normal development and maintenance of the CNS (Veis et al., 1993). These findings suggest that other genes may also be important in controlling cell death in the CNS. One of the candidates is bcl-x, a newly isolated gene with significant homology to bcl-2. This alternative splicing of the gene produces at least three proteins: bcl-x-long (bcl-x-l), bcl-x-β, and bcl-x-short (bcl-x-s). The bcl-x-l form promotes cell survival. Bcl-x-β shares the anti-apoptotic effects of bcl-x-l and is identical to bcl-x-l, although the hydrophobic tail region of bcl-x-β is missing (Gonzalez-Garcia et al., 1995). The bcl-x-s form blocks bcl-2 function and promotes cell death. Only bcl-x-l and bcl-x-β have been readily detected in adult CNS. Both of these proteins suppress apoptotic death in neurons (Boise et al., 1993; Gottschalk et al., 1994; Gonzalez-Garcia et al., 1995). Moreover, bcl-x-deficient mice exhibited extensive apoptotic cell death during the development of CNS and other systems (Motoyama et al., 1995).
Our group and others have recently shown that bcl-2 immunoreactivity was induced in sublethally injured neurons following cerebral ischemia (Shimazaki et al., 1994; Chen et al., 1995a; Krajewski et al., 1995), suggesting that bcl-2 may be an inducible endogenous neuroprotectant in the adult brain. To further address this hypothesis, we studied the transcriptional and translational expression of bcl-2 and bcl-x genes in a rat model of transient global ischemia, in which delayed neuronal cell death occurred in the hippocampal CA1 region. It has been suggested that apoptosis is involved in this process (MacManus et al., 1993; Kihara et al., 1994; Nitatori et al., 1995).
METHODS
Animal model
Global ischemia was induced in isoflurane-anesthetized rats using the four-vessel method described by Pulsinelli (Pulsinelli et al., 1982) with modifications. Male Sprague–Dawley rats (300–350 g) were ventilated with 1.5% isoflurane in a mixture of 25% O2 and 73.5% N2O (vol/vol). The left femoral artery was cannulated for blood pressure monitoring or blood gas and glucose sampling. Rectal temperature was continuously monitored and kept at 37–37.5°C with a heating pad and lamp. Brain temperature was monitored by a 29–Ga thermocouple implanted in the left striatum and allowed to decline spontaneously from 36.2 ± 0.1 to 34.2 ± 0.5°C during ischemia. Within 5–8 min of reperfusion, brain temperature returned to preischemia range in all animals. Animals were placed in a stereotaxic frame, and both vertebral arteries were coagulated and transected at the level of the junction of first and second cervical vertebrae. The bilateral common carotid arteries (CCAs) were then exposed. Bilateral external carotid arteries were ligated and the anesthesia was discontinued. Then, 3 min later, CCAs were occluded with microvascular clips for 15 min under continuous monitoring of the electroencephalograph, (EEG). The EEG became isoelectric in all rats studied within 10 s following CCAs occlusion. At variable time points following completion of ischemia, animals were killed according to the experimental protocols.
Northern blot analysis
Specificity of the bcl-2 and bcl-x-l oligonucleotide probes was examined by Northern blot analysis. Hippocampal and cortical total RNA were extracted from normal brains and from brains subjected to 15 min ischemia followed by 24 h of reperfusion (n = 3 rats per sample). Then, 20 μg RNA/lane was electrophoresed through a 1% agarose-2.2 M formaldehyde gel for Northern blot analysis. Subsequently, mRNA was transferred to nylon filters and 40 mer antisense oligodeoxynucleotides (Biosynthesis Inc., Dallas, TX, U.S.A.) were constructed to be complementary to the regions of each gene shown in Fig. 1. A BLAST search of the Genbank database revealed no other homologous genes. The sequences used for bcl-2 was 5′-CACTGAATGCTCTCCGGTACCGCAGTTCAAACTCATCGCC-3′ (bcl-2 oligo) (Fig. 1) and for bcl-x-long was 5′-GGTGGTCATTCAGGTAGGTGGCCATCCAACTTGCAATCCG-3′ (bcl-x oligo). Oligodeoxynucleotides were 3′ end-labeled with 40 μCi [32P]dATP (specific activity > 106 Ci/mol) using terminal deoxynucleotidyltransferase (GIBCO BRL, Grand Island, NY, U.S.A.). Unincorporated nucleotides were separated by P-6-sephorose columns. The labeled probe was denatured by heat and hybridized to the filters at 42°C overnight. The washing procedure was performed under high stringency conditions in 2 × sodium/sodium citrate (SSC) buffer/0.1% sodium diodecyl sulfate (SDS) (45 min for three times, 52°C). Membranes were then exposed to Kodak X-OMAT AR films (Eastman Kodak Company, Rochester, NY, U.S.A.) using intensifier screens at −80°C for 72 h. Autoradiogram signals were quantified by a gel densitometric scanning program using an MCID image analysis system (St. Catharine's, Ontario, Canada). To control for variation in the amount of RNA in different samples, all blots were rehybridized with an oligonucleotide probe (5′-ACGGTATCTGATCGTCTTCGAACC-3′) corresponding to 18 S RNA. The original probe was stripped off in a solution containing 0.1 × SSC and 0.5% SDS at 100°C for 15 min. All densitometric values for bcl-2 and bcl-x-l were normalized to values for 18 S RNA obtained on the same lane.
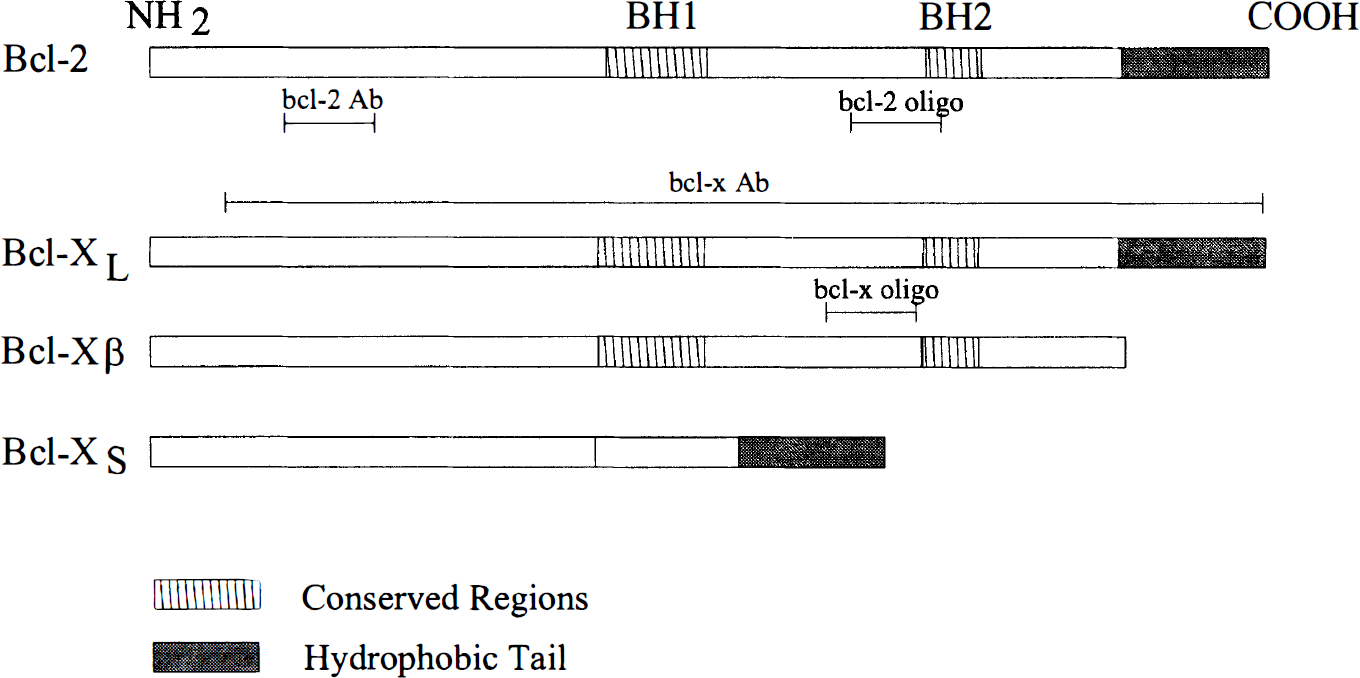
Schematic diagram of bcl-2 and bcl-x sequences and splice variants showing positions of the conserved homodimer binding regions (BH1 and BH2) and the hydrophobic tail. Bcl-2 Ab, region of protein sequence used to raise antibody against bcl-2; bcl-x Ab, region of protein sequence used to raise antibody against bcl-x; bcl-2 oligo, region of cDNA sequence used to construct antisense oligonucleotides for bcl-2; bcl-x oligo, region of cDNA sequence used to construct antisense oligonucleotides for bcl-2.
In situ hybridization
Rats used for in situ hybridization were anesthetized with 4% chloral hydrate and decapitated 2, 4, 8, 24, and 72 h following 15 min of global ischemia (n = 3 per group). Brains were rapidly removed, frozen in 2-methylbutane at −30°C and stored at −80°C. Sections (20 μm) were cut on a cryostat at −20°C, collected on Probe-On Slides (Fisher Biotech, Pittsburgh, PA, U.S.A.), and processed for in situ hybridization using the method previously described (Chen et al., 1995b). Briefly, oligodeoxynucleotide probes were labeled with [135S]-dATP using terminal deoxynucleotidyltransferase and hybridized (1 × 106 cpm/ml) at 42°C for 18 h with the slides. After hybridization, sections were rinsed in 1 × SSC (150 mM sodium chloride and 15 mM sodium citrate, pH 7.4) at 55°C for 60 min with several changes of 1 × SSC, dehydrated, and exposed to a Kodak SB-5 film for 3 weeks. Control and ischemic brain slides were hybridized together and developed on the same film. Relative changes in mRNA expression were then quantified by determining the ratio of the optical density of the specified regions in ischemic brains versus controls using the MCID system. Cellular localization was evaluated by coating slides with Kodak NTB-2 emulsion. Sections were exposed at 4°C for 5 weeks, developed in D-19, and counterstained with cresyl violet.
Western blot analysis
Western blot analysis was performed to verify the specificity and efficiency of the monoclonal bcl-2 (Dako Co., Carpenteria, CA, U.S.A.) and bcl-x (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) antibodies used in these studies. The peptides used to produce these antibodies are shown in Fig. 1. Rats were killed at 16, 24, and 72 h following 15 min ischemia (n = 3 per group). Hippocampi were dissected, homogenized, and lysed. Then, 40 μg protein samples from each brain were loaded onto a 12% SDS-polyacrylamide gel. Western blots were performed as described previously (Sambrook et al., 1989), using the chemiluminescent detection system (Clontech, Palo Alto, CA, U.S.A.). The transferred polyvinylidene difluride (PVDF) membrane was incubated in bcl-2 or bcl-x antibody at a dilution of 1:200 at 4°C overnight, followed by washing procedures and secondary antibody reactions. Chemiluminescent substrate, 25 mM (Clontech Laboratories, Palo Alto, CA, U.S.A.) was applied to the side of the membrane containing the blotted proteins. The blot was wrapped in plastic wrap and exposed to a Kodak X-OMAT film.
Immunocytochemistry
Rats were given 100 U/kg heparin i.p. and anesthetized with 4% chloral hydrate. Five min later, they were perfused with 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS) (pH 7.4) at 4, 8, 16, 24, or 72 h following 15 min ischemia (n = 3 per group). Brains were removed, postfixed for 4 h, sectioned at 50 μm on a vibratome, and processed for immunocytochemistry staining for bcl-2 and bcl-x (1:100 dilution), using the avidin-biotin-horseradish peroxidase technique previously described (Chen et al., 1995a). As a control, alternate sections were incubated in the absence of primary antibodies.
RESULTS
Northern blot analysis
Bcl-2 and bcl-x-l oligonucleotide probes were hybridized in a single band at −2.5 and 2.7 kb, respectively, with both control and ischemic brain samples (Fig. 2). Both bcl-2 and bcl-x-l mRNAs were present at low levels in control brain but were increased in ischemic samples. Bcl-2 expression was increased 1.89-fold in the cortex and 2.49-fold in the hippocampus, compared to nonischemic brain. Bcl-x expression was also increased 1.95-fold in the cortex and 2.97-fold in the hippocampus, compared to nonischemic brain.
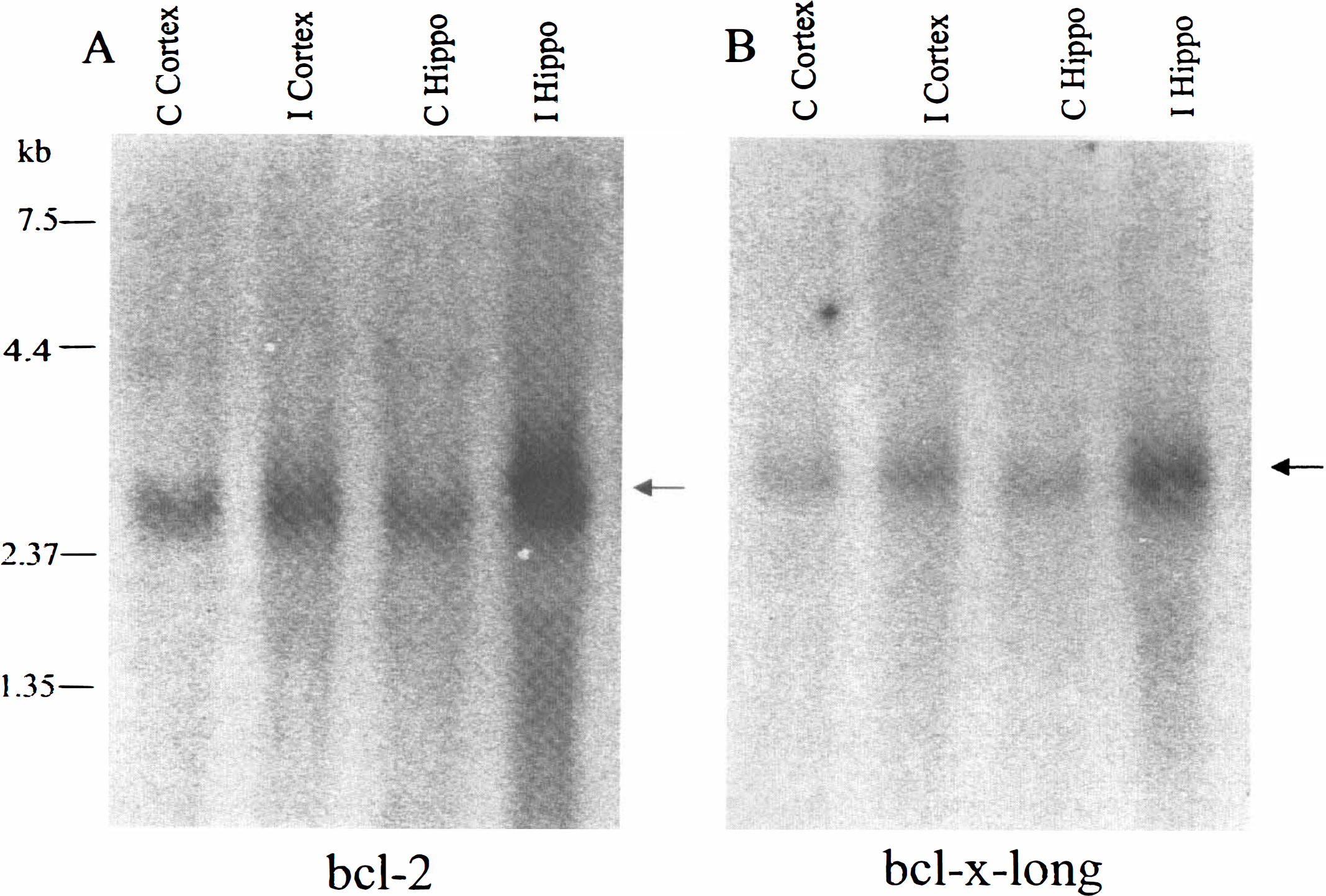
Northern blot analysis of bcl-2 (
In situ hybridization
In control rats, in situ hybridization revealed low basal expression of both bcl-2 and bcl-x-l mRNAs. Sections hybridized with sense oligonucleotide probes, as a negative control, revealed a low level of nonspecific background signal (data not shown).
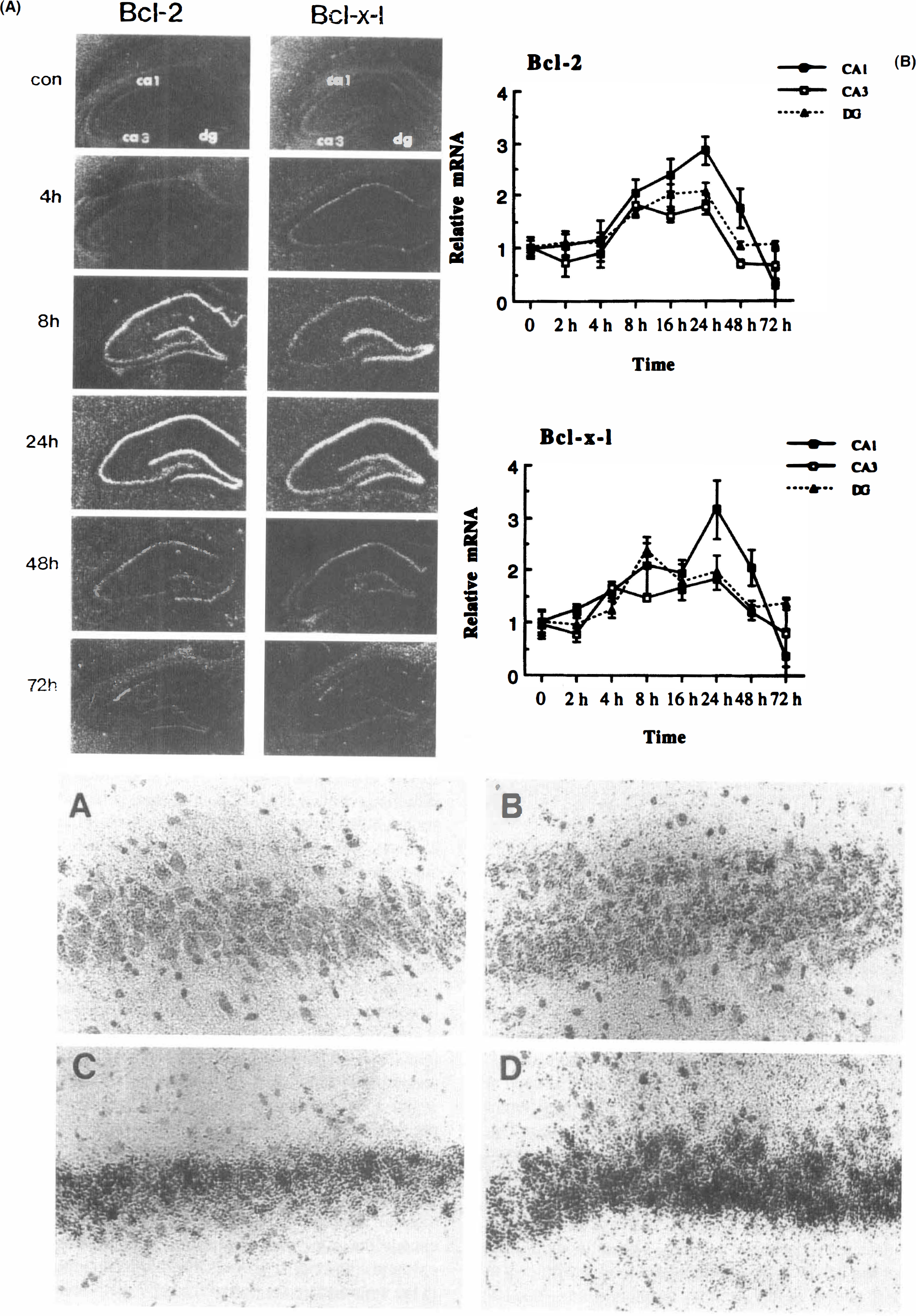
The effect of transient global ischemia on bcl-2 and bcl-x-l mRNA expression in representative in situ hybridizations is shown in Fig. 3A; the relative changes of mRNA measured in the hippocampus are summarized in Fig. 3B. Four hours following ischemia, a slight elevation of bcl-x-l mRNA was detectable in CA1 and CA3 pyramidal cell layers. This mRNA level was further increased at 8 h and peaked at 24 h in CA1 and CA3 regions and the dentate gyrus (DG). An elevation of bcl-2 mRNA level was detectable at 8 h in CA1-3 pyramidal cell layers and the granular layer of the DG, and remained high at 24 h in these regions. Both bcl-2 and bcl-x-l mRNA levels remained elevated in the CA1 at 48 h, but the signal decreased below basal control level at 72 h, consistent with the histologic cell death in this region. Emulsion-coated slides revealed that silver grains occurred predominantly in neuronal elements (Fig. 3C).
Western blot analysis
As shown in the representative Western blot in Fig. 4A, anti-human bcl-2 antibody detected a band at ∼26 kDa, the predicted size for the bcl-2 protein. Bcl-2 protein was present at a very low level in control samples, but the protein level was elevated in ischemic samples. The bcl-2 antibody also recognized an additional band at 52 kDa, although the level was not significantly different between control and ischemia samples. As shown in Fig. 4B, bcl-x immunoreactivity was evident on a single band at 26 kDa, the predicted size for bcl-x-l protein. Bcl-x-l protein was present at a relatively high level in control samples, and a two- to threefold increase in the signal was detected in ischemic samples. The antibody did not detect the bands at the appropriate site of the bcl-x-s or bcl-x-β protein in any samples tested.
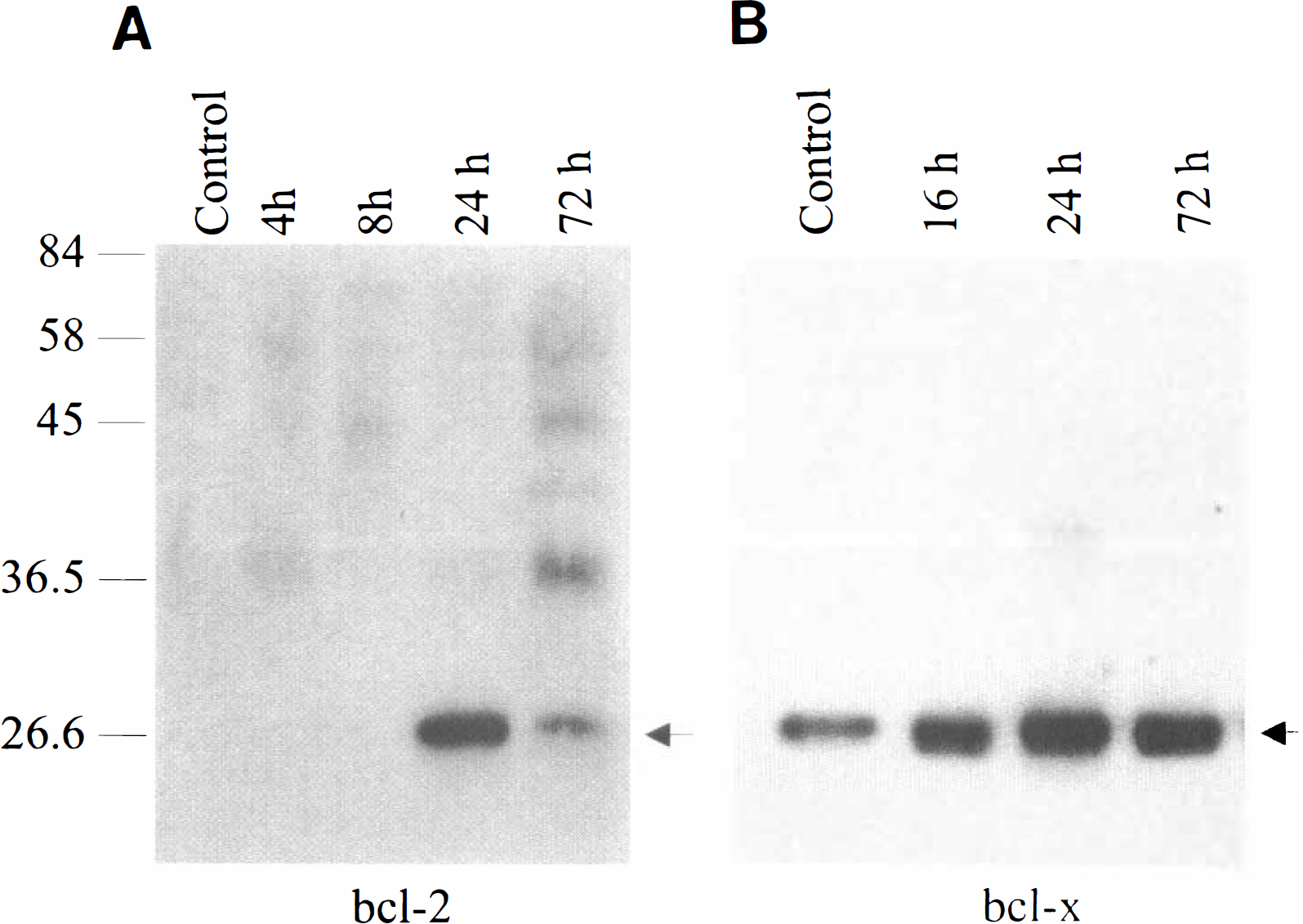
Western blot analysis of bcl-2 (
Immunocytochemistry and histopathology
Bcl-2 immunoreactivity was not significantly detectable in control brains (Fig. 5) or in sections incubated without primary antibody (data not shown). At 24 and 72 h after ischemia, bcl-2 immunoreactivity was widespread in CA3 pyramidal neurons and, to a lesser extent, in DG granule cells; glial induction of bcl-2 was also seen in the hippocampus at 72 h (Fig. 5). However, few, if any, bcl-2 positive neurons were present in CA1 regions at any time points studied.
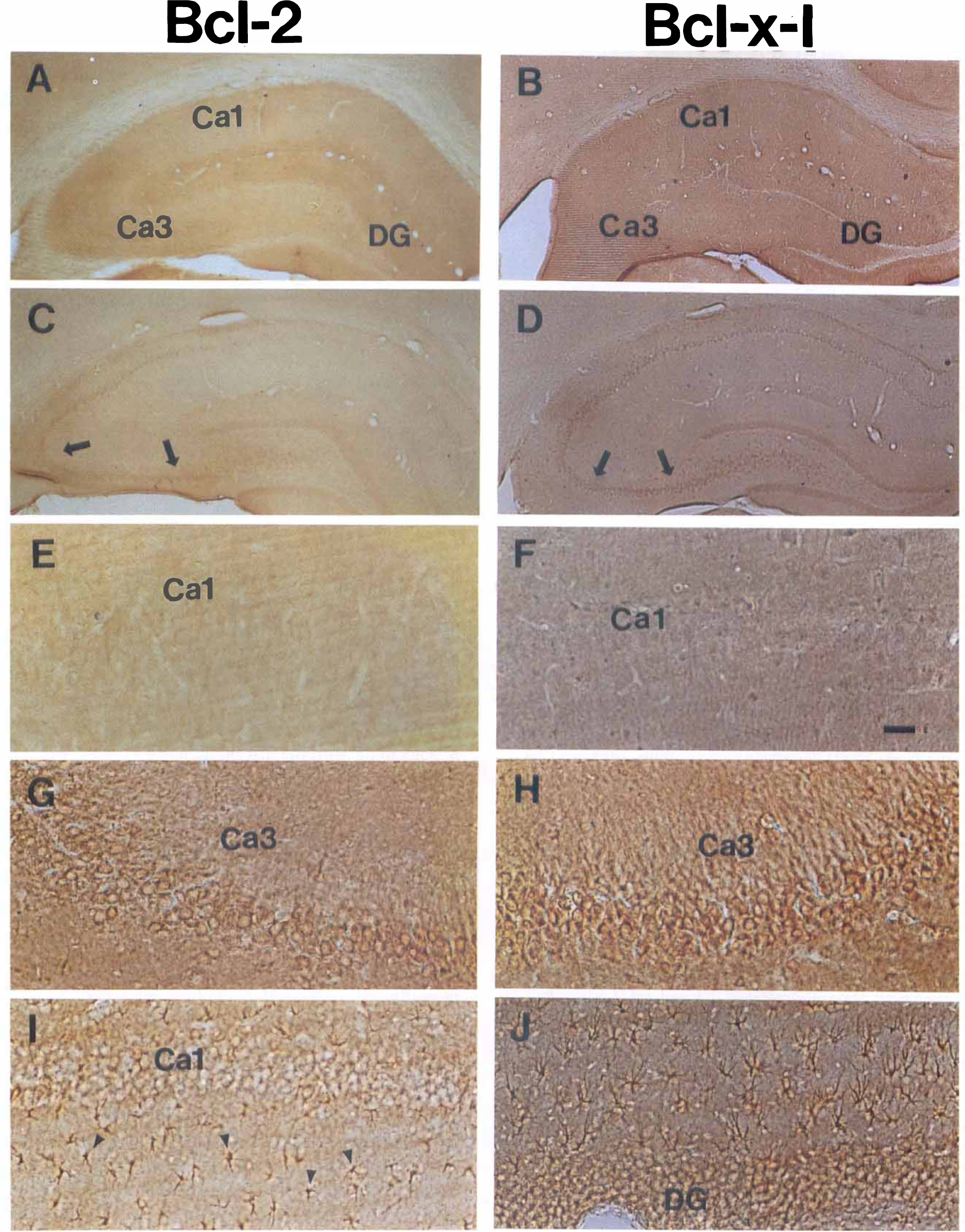
Representive bcl-2 and bcl-x immunostaining in the hippocampus of control brains (
Basal bcl-x immunoreactivity was present in control brains. A faint staining for bcl-x was detected in neurons in layers 2, 3, and 5 of the neocortex and in some neurons in the hippocampal CA1 and CA3 regions. At 16, 24, and 72 h after ischemia, an elevation of bcl-x-l immunoreactivity was present in CA3 pyramidal neurons (Fig. 5), and in some DG granule cells and neurons in layers 3 and 5 of the cortex (data not shown). Bcl-x-l immunoreactivity was not increased in CA1 regions at any time point.
Histological studies revealed that few morphological changes occurred in the hippocampus at 24 h after ischemia, but the majority (>95%) of neurons in the CA1 disappeared or showed pyknotic changes at 72 h.
DISCUSSION
Because energy failure is a common characteristic of necrotic cell death, one would assume that new gene expression and apoptosis do not occur and would not be an important mechanism of cell death in ischemia. However, a number of studies have suggested that new gene expression is involved in the delayed neuronal death of CA1 hippocampal neurons after global ischemia. Energy stores are rapidly restored after global ischemia (Shimizu, et al., 1993). Protein synthesis inhibitors prevent delayed death of CA1 neurons (Goto et al., 1990; Shigeno et al., 1990; Papas et al., 1993). DNA fragmentation has been detected; this suggests that endonucleases are activated (MacMannus et al., 1993; Kihara et al., 1994; Nitatori et al., 1995). Some of the histological attributes of apoptosis are present (Nitatori et al., 1995), although this was not supported by some other studies (Deshpande et al., 1992). Thus, since cell death after ischemia may require new gene expression and have some of the attributes of apoptosis, it is not unrealistic to hypothesize that genes in the apoptotic family could play a role in determining the fate of ischemic neurons.
Bcl-2 and bcl-x are two well-characterized genes that regulate apoptotic cell death in mammals. Basal expression of bcl-2 and bcl-x-l mRNA was detectable, although at low levels, in control nonischemic brains. Several different bcl-2 transcripts have been described in rodents; these all appear to have the same open reading frame and encode the same protein (Castren et al., 1994). Differences in transcript length attributable to polymorphisms in the nontranslated region of the bcl-2 mRNAs appear to be tissue, species and strain specific. The bcl-2 mRNA detected by Northern analysis in this study was similar in size to a bcl-2 transcript fond in mouse brain and Wistar rat thymus (Negrini et al., 1987; Castren et al., 1994). These results also confirm the observation that a short mRNA transcript is the predominant bcl-2 mRNA expressed in Sprague Dawley extragonadal tissues, including brain (Tilly et al., 1995), although multiple transcripts have been described in the Wistar rat (Castren et al., 1994).
In contrast to mRNA, bcl-2 and bcl-x-l protein were differentially expressed in control brains. While bcl-2 immunoreactivity was very low in the Western blot analysis and hardly detected in immunocytochemical sections, bcl-x-l was present at a much higher level in the Western blot and was readily detectable with immunocytochemistry in many cortical and some hippocampal neurons in normal controls. These observations are consistent with previous findings that bcl-x-l, but not bcl-2, protein is expressed in a significant amount in the normal adult rodent brain (Boise et al., 1993; Merry et al., 1994). Furthermore, bands at the appropriate locations for bcl-x-s and bcl-x-β were not detectable in Western blots, suggesting that neither bcl-x-β nor bcl-x-s is a major product of the bcl-x genome in either control or ischemic rat brain. These results are in keeping with the observation that bcl-x-l mRNA is the major transcript of bcl-x expressed in the adult mouse brain (Gonzalez-Garcia et al., 1995).
Results show that bcl-2 and bcl-x-l mRNA were induced mainly in the hippocampus with similar expression patterns following global ischemia. The CA1 pyramidal cell layer exhibited the highest expression signals for both mRNAs, followed by the granular cell layers of the DG and the CA3 pyramidal layer. Induction of both mRNAs in CA1 neurons was more persistent (at least 48 h) than was their induction in CA3 neurons and the DG (24 h). Finally, induction for both genes in CA1 neurons peaked at 24 h and then decreased below basal level at 72 h, coinciding with the histologic death of these neurons.
Immunocytochemistry studies revealed a somewhat different pattern for bcl-2 and bcl-x-l protein accumulation compared to mRNA expression in ischemic brains. Both bcl-2 and bcl-x-l immunoreactivity were detected at a high level in CA3 pyramidal neurons and in many DG granule cells at 24 or 72 h following ischemia. But while expression of both bcl-2 and bcl-x-l mRNA was induced in CA1 neither bcl-2 nor bcl-x protein was expressed there. These results are in accordance with the observations that lethal ischemia inhibits bcl-2 expression in CA1 in gerbil global ischemia (Shimazaki et al., 1994) and that neither bcl-2 protein nor bcl-x protein is expressed in CA1 after cardiac arrest in rats (Krajewski et al., 1995). The discrepancy between in situ data and immunocytochemistry results suggests that translation of bcl-2 and bcl-x mRNA is blocked in CA1 after ischemia. This result is not surprising, since inhibition of protein synthesis is a well-known consequence of ischemia (Vass et al., 1988; Kiessling et al., 1993). Thus, bcl-2 and bcl-x protein are not expressed in CA1, and apoptotic death may be unopposed.
But, if protein synthesis is inhibited, how could apoptosis effector proteins be expressed? The answer appears to be that selective proteins are translated in ischemic neurons fated for delayed neuronal death. For example, the apoptosis effector gene p53 is expressed in ischemic neurons within an infarction after middle cerebral artery occlusion (Chopp et al., 1992). Bax, an apoptosis effector protein, is also expressed prior to death of CA1 after ischemia (Krajewski et al., 1995). We have recently shown that cyclooxygenase 2 protein, which could be an apoptosis effector via its generation of oxygen-free radicals, is expressed in CA1 neurons 24 h after global ischemia (Nakayama et al., 1995). The efficacy of protein synthesis inhibitors in preventing delayed neuronal death after global ischemia also suggests that translation of these selective proteins must occur in order to result in delayed neuronal death in the CA1 (Goto et al., 1990; Shigeno et al., 1990; Papas et al., 1993). Thus, severely injured neurons may “selectively” translate adverse proteins using their partially functional protein synthesis capacity. In such circumstances, the adverse gene products in these neurons may be dominant and mediate cell death. Therefore, further experiments designed to study expression of the functionally balanced genes such as bcl-2/Bax or bcl-x-l/bcl-x-s in the same neuronal populations may help us understand the role of these genes in ischemic injury.
Bcl-2 immunoreactivity was induced in glial cells in the hippocampal white matter 72 h after ischemia. Bcl-2 positive glial cells in the middle region of the white matter are astrocytes, since they reacted positively with the glial filament acidic protein antibody; however, the glial cells localized adjacent to the CA1 region were mainly microglia, since they reacted with the OX-6 antibody (data not shown). Thus, both microglia and astrocytes expressed bcl-2 protein. This was consistent with our previous findings that bcl-2 immunoreactivity was induced in glia in the border zone of infarction following focal ischemia (Chen et al., 1995a). The significance of this bcl-2 induction in glial cells is unclear. Merry reported that microglia comprise the major cell population expressing bcl-2 in the adult brain (Merry et al., 1994), suggesting that bcl-2 may be important for the maintenance of these cells after development. It has been shown that microglia are activated following cerebral ischemia (Morioka et al., 1993; Kato et al., 1994; Lehrmann et al., 1995) and generate oxygen-derived free radicals (Chao et al., 1992). The time for glial bcl-2 induction in this study is comparable to the reported time course for glial activation after global ischemia (Kato et al., 1994). Therefore, by reducing the intracellular generation of oxygen-free radicals, bcl-2 protein may protect activated microglia from their endogenous oxidants (Hockenbery et al., 1993; Kane et al., 1993; Veis et al., 1993). Further studies are required to address these issues.
The major finding of this study is that expression of both bcl-2 and bcl-x-l mRNA and protein is increased in CA3 after global ischemia, a region that is ischemic, yet survives. Thus, expression of bcl-2 and bcl-x may promote survival of these ischemic neurons. The ability of bcl-2 and bcl-x-l gene products to inhibit neuronal apoptosis in vitro is well documented, but the mechanism for this protection is unclear. Kane et al (1995) showed that bcl-2 expression may prevent necrotic neuronal death as well as apoptosis. This observation suggests that, instead of inhibiting a cellular death program directly, bcl-2 may modulate a generalized cellular process (such as generation of oxygen-derived free radicals) that may contribute to either apoptotic or necrotic death (Kane et al., 1995). Two observations support a role for bcl-2 as an endogenous neuroprotectant in vivo: overexpression of bcl-2 via an amplicon Herpes simplex vector decreased infarction, although marginally, after focal ischemia (Linnik et al., 1995); and a transgenic mouse that overexpresses bcl-2 had significantly smaller infarction after middle cerebral artery occlusion than did wild type controls (Martinou et al., 1994).
In summary, this study demonstrated a differential expression pattern for bcl-2 and bcl-x-l at their transcriptional and translation levels in the rat brain subjected to transient global ischemia. While bcl-2 and bcl-x-l mRNA were expressed in both surviving and dying neurons, their proteins were expressed primarily in neurons destined to survive. These results support a possible neuroprotective role for these two apoptosis suppressor genes in cerebral ischemic injuries. However, in order to confirm that expression of these proteins after cerebral ischemia promotes survival rather than being an epiphenomenon, further studies, in which synthesis of these proteins is specifically altered after ischemia, are required.
ADDENDUM
After submission of this article, we published the results of a study of the expression of the bcl-2 family protein Bax after global ischemia in rat (Chen et al., 1996). The proapoptotic protein Bax was primarily expressed in CA1 neurons which die after ischemia. We also colocalized the expression of Bax with the bcl-2 protein and found that bcl-2 was expressed in CA3 neurons which survive after ischemia, as was reported here.
Footnotes
Acknowledgment:
This work was supported, in part, by NIH grant NS 24728 (R.P.S.) and Department of Veterans Affairs Merit Review Program (S.H.G.). We thank Joyce Horner, Tim Lowry, and Michele Heubrin, and Kendra Basta for technical support, Amy Karolski for secretarial support, and Tanya Spewock for technical editing.