
Abstract
Five minutes of bilateral common carotid artery occlusion in the Mongolian gerbil results in a selective, delayed death of CA1 pyramidal neurons. Although Bcl-2 appears to protect a variety of cells from cell death, the precise role of this apoptosis-regulating protein is complicated. We used immunoblots to estimate levels of Bcl-2, Bcl-xl, and Bax at various times after carotid occlusion. Rather than Bcl-2, Bcl-xl appears to be the predominant neuroprotective form of this family of proto-oncogenes in the gerbil hippocampus. After transient ischemia, Bcl-2 and Bcl-xl protein levels remained the same. However, Bax levels were dramatically increased at 6 hours after ischemia, compared with sham-operated animals, and were still elevated at 72 hours after ischemia. To monitor dimerization interactions among theses apoptosis-regulating molecules, immunoprecipitation studies were conducted. These studies demonstrated that Bcl-xl association with Bax increases after ischemia. Therefore, Bax may disrupt the more favorable Bcl-xl (Bcl-2) interactions necessary for normal neuronal functioning and thus promote transient ischemic death.
Transient global ischemia results in selective neuronal damage of hippocampal CA1 neurons. Although there is a delay of approximately 72 hours in the appearance of neuronal death (Fig. 1), markers related to the mechanism of ischemic death become apparent well before neurons die. Changes in the levels of the Bcl-2 family of proto-oncogenes have been reported as early as 6 to 8 hours after ischemia (Krajewski et al., 1995b; Chen et al., 1996; Antonawich et al., 1996). The precise role of these apoptosis-regulating molecules is complicated.
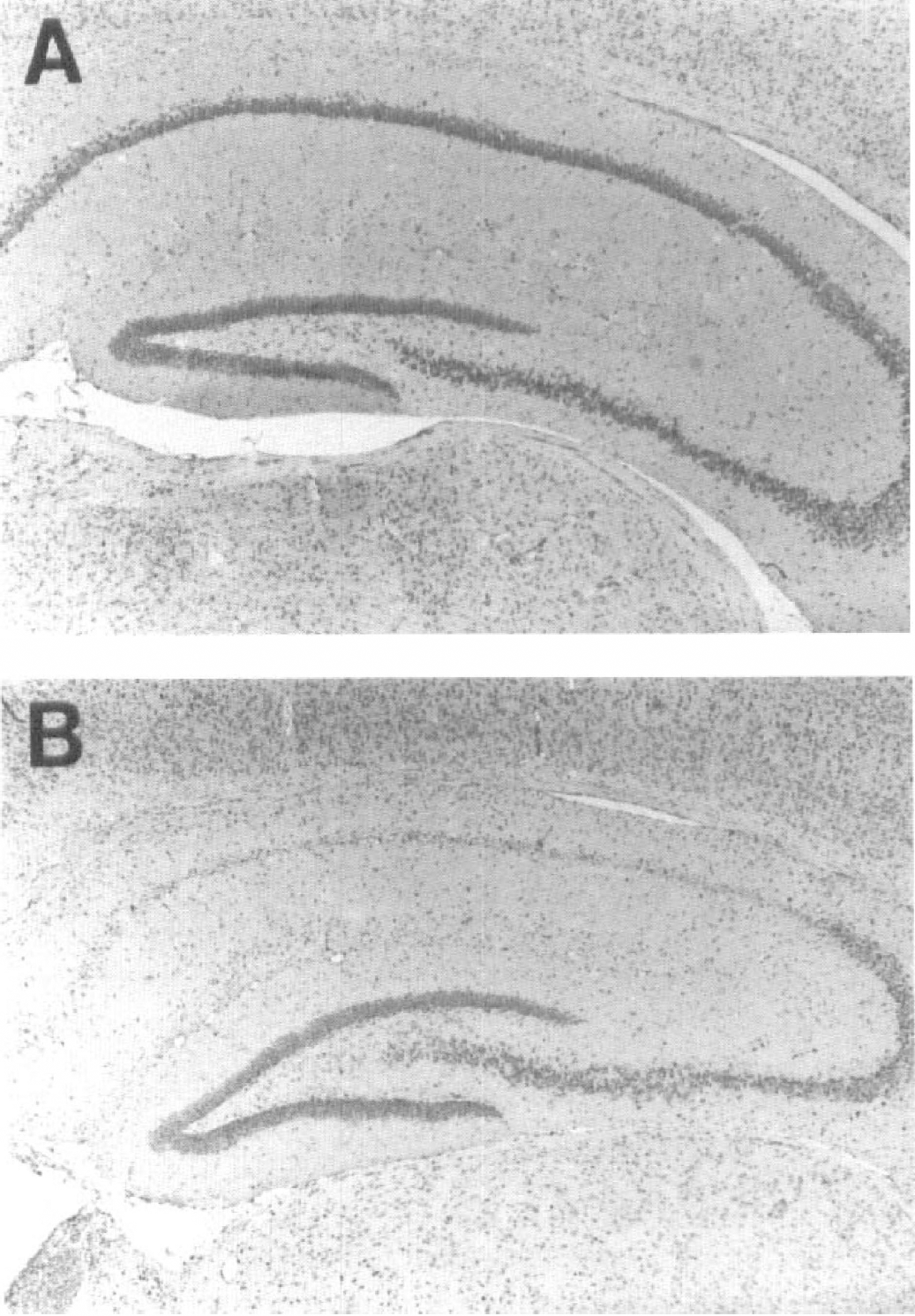
Hippocampal damage after transient ischemia.
This family of proteins consists of both inhibitors and promoters of programmed cell death. Bcl-2 is expressed in high levels in neuronal tissues during embryonic development, but is downregulated in the adult CNS. It appears to protect a variety of cells from cell death in vitro (Garcia et al., 1992; Allsopp et al., 1993; Kane et al., 1993; Hockenberry et al., 1993; Reed, 1994). Bcl-2 can also provide protection from naturally occurring developmental cell death and focal ischemic damage in overexpressing transgenic mice (Martinou et al., 1994). One particularly interesting Bcl-2-related protein is Bcl-x. Whereas neuronal Bcl-2 levels decrease with age, Bcl-x expression is retained in the adult system (Gonzalez-Garcia et al., 1995). Bcl-x is 74% homologous to Bcl-2 and it also promotes cell survival. Bcl-x encodes two proteins, Bcl-xl and Bcl-xs, with opposing functions. The “L” form promotes survival whereas the “S” form is an inhibitor of Bcl-2 or Bcl-xl function (Krajewski et al., 1994b). Of particular interest is the fact that whereas Bcl-xs is expressed at high levels in cells undergoing a rapid rate of turnover, Bcl-xl is found in long-lived post-mitotic cells, such as the adult brain (Boise et al., 1993; Mituguchi et al., 1996). Bax is a protein homologous to Bcl-2 that forms a dimer with it and prevents the actions of Bcl-2 (Korsmeyer et al., 1993). Overexpression of Bax counters the death repressor effects of Bcl-2 in growth factor-deprived apoptotic death (Oltvai et al., 1993). The exact mechanism by which Bcl-2, Bcl-xl, and Bax influence cell survival is not known (Reed, 1997). Overexpression of Bax leads to the activation of specific proteases involved in apoptosis termed caspases (Jurgensmeier et al., 1997; Lin et al., 1996; Vekellis et al., 1997) and eventually cell destruction. This balance between the neuroprotective and neurodegenerative members of the Bcl-2 family of proto-oncogenes may explain the susceptibility of CA1 hippocampal neurons to global ischemic insult (Krajewski et al., 1995b).
This work focused on the role of Bcl-2, Bcl-xl, and Bax in ischemic damage. In the present study, immunoblotting methods were used to estimate the levels of Bcl-2, Bax, and Bcl-xl after carotid artery occlusion, and immunoprecipitation studies to monitor dimerization among Bcl-2 family proteins.
METHODS
Carotid occlusion
Adult male Mongolian gerbils were placed in an anesthetic container containing 3% halothane, 70% nitrous oxide, and 27% oxygen for 2 to 3 minutes. During the surgical procedure the anesthetic plane was maintained using 1.5% halothane anesthesia administered through a nose cone. The carotid arteries were exposed, isolated, and occluded for 5 minutes using microaneurysm clips. Throughout the occlusion period halothane was reduced to 0%. Body temperature was monitored using both a rectal and a temporalis muscle probe and maintained between 37.0° and 38.0°C. At the completion of the occlusion period, the clips were removed and reperfusion visually confirmed before the neck was closed with surgical wound clips. Sham surgeries were conducted in a similar manner except that the carotid arteries were not occluded.
Antisera
We tested various antibodies in an attempt to find one capable of detecting gerbil Bax (Table 1). Antibodies, both commercial sources and produced (Krajewski et al., 1994a, 1994b; 1995b), corresponded to amino acid fragments located throughout the sequences of mouse and human Bax. Interestingly, the most effective proved to be an anti-human Bax antibody. It was raised against amino acids in close proximity to the N-terminus (Santa Cruz Biotechnology N-20, Santa Cruz, CA, U.S.A.). This antisera was also effective in detecting mouse Bax. For the detection of Bcl-2 and Bcl-x, recombinant rabbit anti-mouse antisera was used (Krajewski et al., 1995).
Bax antibodies
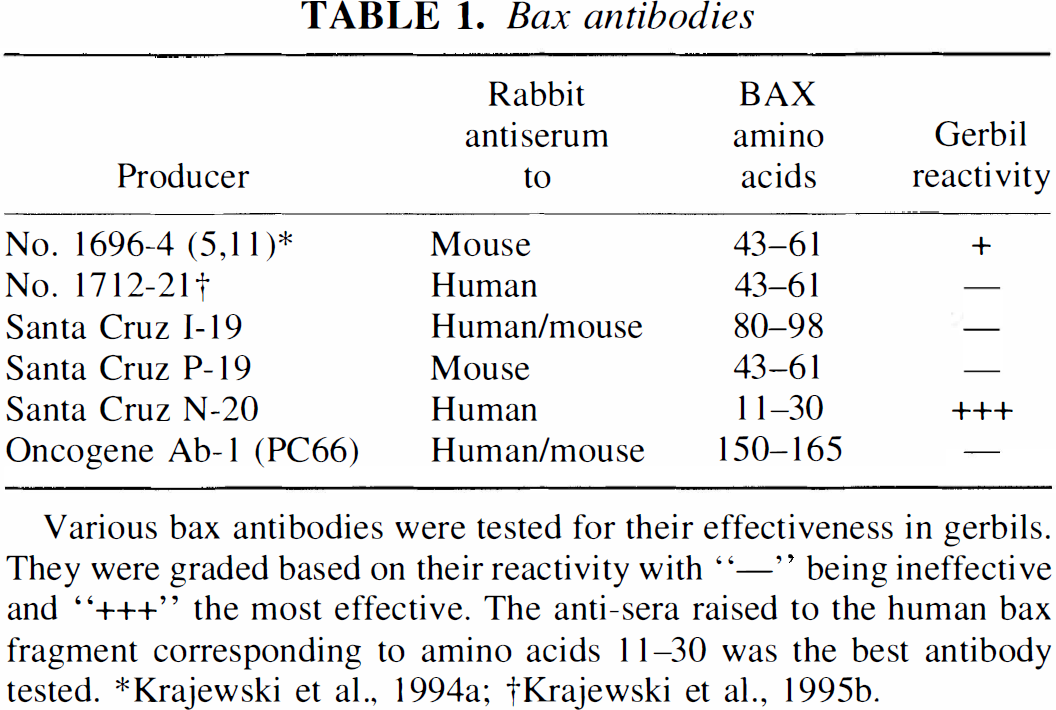
Various bax antibodies were tested for their effectiveness in gerbils. They were graded based on their reactivity with “—” being ineffective and “+++” the most effective. The anti-sera raised to the human bax fragment corresponding to amino acids 11–30 was the best antibody tested.
Krajewski et al., 1994a
Krajewsk,i et al., 1995b.
Immunoblotting
Immediately after decapitation, mouse and gerbil whole hippocampal brain tissues (c57BL/6 Taconic, Germantown, NY) were dissected and homogenized in RIPA buffer containing protease inhibitors. After sonication on ice and centrifugation (16,000g for 5 to 10 minutes), the serum was collected. The protein levels were quantified using a BCA protein assay (Pierce Inc., Rockford, IL, U.S.A.), and 50-μg protein samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)/immunoblotting (12.5% gels), using either rabbit anti-mouse Bcl-2, Bcl-x (1:1,000 and 1:3,000, respectively) (Krajewski et al., 1995a) or rabbit antihuman Bax (1:1,000, N-20; Santa Cruz Biotechnology Inc.) specific preabsorbed antisera. This was followed by 0.15 μg/mL biotinylated goat anti-rabbit IgG for 1 hour at room temperature and horseradish peroxidase avidin-biotin complex reagent (Elite; Vector Labs Burlingame, CA, U.S.A.) for 1 hour, then visualized with 10 minutes exposure to diaminobenzidine.
Immunoprecipitation
After sacrificing, whole hippocampi were homogenized in NP-40 isotonic lysis buffer containing a protease inhibitor cocktail (Sedlak et al., 1995). Because NP-40 lysis buffer lacks the detergent sodium dodecyl sulfate (SDS), included in the immunoblot RIPA lysis buffer, Bcl-xl-Bax interactions remain intact. Protein levels were determined as described above, and 1 mg of protein was incubated for 24 hours with Bax antisera (4 μL/mg protein). Immune complexes were collected using Protein-A/G Sepharose (10 μL/μg antibody), electrophoresed using 12.5% SDS-polyacrylamide gels, then detected by immunoblotting as described above.
Semiquantitative and statistical analysis
The JAVA image analysis system (Jandel Scientific, Corte Madera, CA, U.S.A.) was used to measure protein band intensities. The ischemic intensities were adjusted for trial difference by standardizing them to the sham ischemic intensities. A non-parametric univariate analysis and Student's t tests were used to compare the various ischemic time periods and sham-operated control animals. Statistical analyses were carried out using the Statistical Analysis System (SAS Institute, Cary, NC, U.S.A.).
RESULTS
Bcl-xl appears to be the predominant neuroprotective form of this family of apoptosis-regulatory genes in the gerbil hippocampus. Although abundant Bcl-2 levels are seen in both the mouse and gerbil control thymus tissue, hippocampal protein levels are quite low (Fig. 2A). Bcl-xl levels are quite easily detected in the hippocampus. Bcl-xl appears as a prominent band at 29 kDa in each of the mouse homogenates, with an additional slightly higher molecular weight faint band in the thymus and two additional higher bands in both the hippocampal and cortical homogenates. Unlike the Bcl-x1 protein bands obtained with mouse, gerbil hippocampal Bcl-x1 demonstrates a triplet of bands of approximately 29, 27.5, and 26 kDa mass (Fig. 2B). A band corresponding to the short form of Bcl-x was not observed in our gerbil hippocampal tissue.
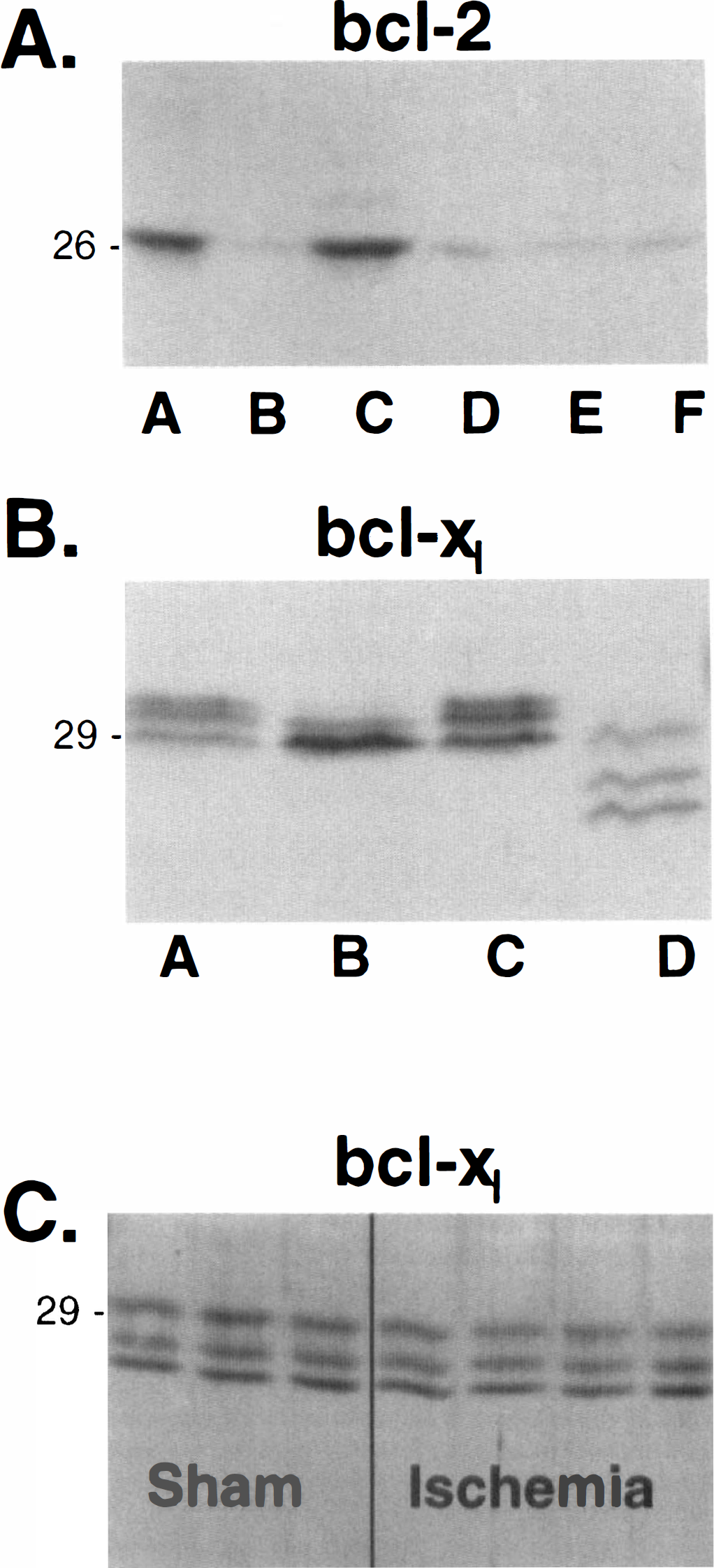
Immunoblot analysis of neuroprotective Bcl-2 family members.
Gerbil Bax protein proved more difficult to detect. Antisera directed to recombinant mouse Bax was not effective in the gerbil. Various antibodies were tested, but the best was based on an N-terminal fragment of human Bax (Table 1). Fig. 3 shows an example of an immunoblot analysis obtained by use of this antibody on various ischemic and control tissue samples. Ischemic hippocampal Bax levels were significantly increased compared with sham-operated animals (P < 0.01) at 6, 24, and 72 hours after ischemia (n = 10 per group, Fig. 4; Table 2). These Bax levels were approximately 60% higher in the ischemic compared with the sham-operated animals. However, there were no statistically significant Bax level differences between the three postischemic time points. Hippocampal Bcl-xl and Bcl-2 levels show little or no change at any of the three postischemic time points (n = 8 and 6 per group, respectively, Table 2).
Protein band intensities after transient ischemia

Bcl-2, bcl-x1, and bax protein band intensities were measured at various times, after 5 minutes of carotid occlusion, using the JAVA image analysis system. Each of the intensity values from the various trials were standardized by expressing them as a ratio, relative to sham control values. Statistical analysis was accomplished using the Statistical Analysis System (SAS Institute, Cary, NC, U.S.A.). Only bax demonstrated a statistically significant effect after ischemia (∗P < 0.01). Bax levels were elevated approximately 60% within 6 hours of ischemia.
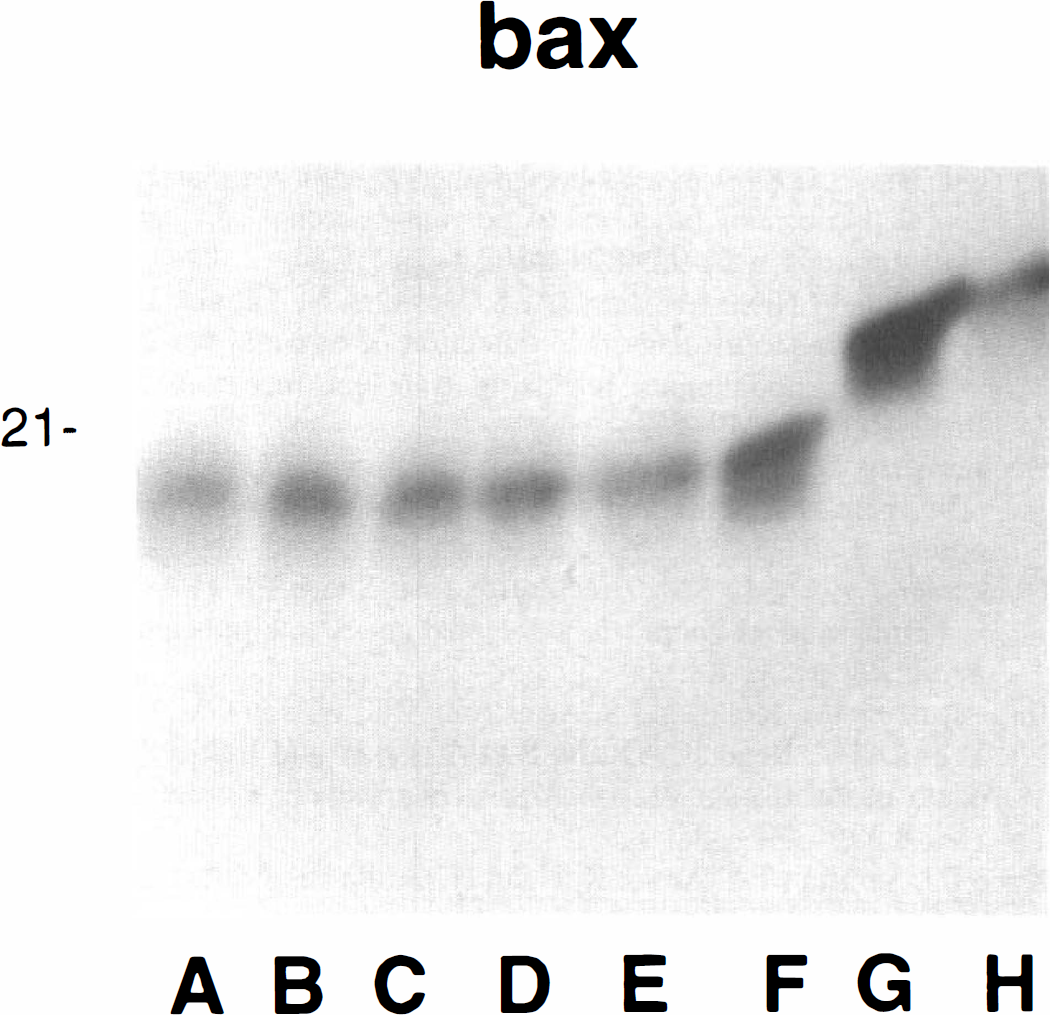
Immunoblot detection of Bax. Bax detected with rabbit antisera raised against an n-terminus fragment of human bax. A: 6-hour sham ischemia; B: 6-hour ischemia; C: 72-hour sham ischemia; D: 72 hour ischemia; E: control gerbil hippocampus; F: gerbil thymus; G: mouse thymus; H: mouse hippocampus. Gerbil Bax is not easily detected with anti-mouse antisera but does stain using antisera to a human N-terminal fragment. This is a representative immunoblot that included one animal from each of the listed experimental groups. Quantitative analysis was conducted using 10 animals per group.
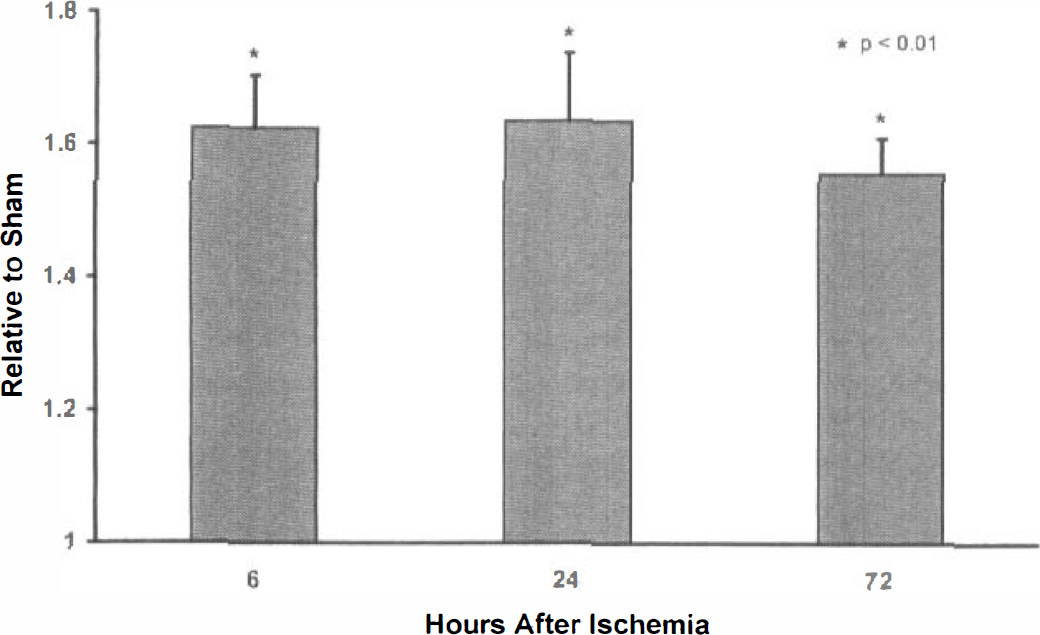
Hippocampal Bax protein levels. Five minutes of transient global ischemia significantly (n = 10 per group; P < 0.01) increases Bax levels compared with sham-operated control animals at 6, 24, and 72 hours after ischemia. These Bax levels are approximately 60% higher in the ischemic compared with the sham-operated animals. There were no statistically significant Bax level differences between the three postischemic time points.
From the immunoblot data, 6 and 72 hours were chosen to evaluate dimerization. Although it appears that most Bcl-xl is not bound to Bax in a nonischemic state, after ischemia there was an increase in Bax-associated Bcl-xl (Fig. 5). This increase in Bax-associated Bcl-xl was evident at both 6 and 72 hours after 5 minutes of carotid artery occlusion.
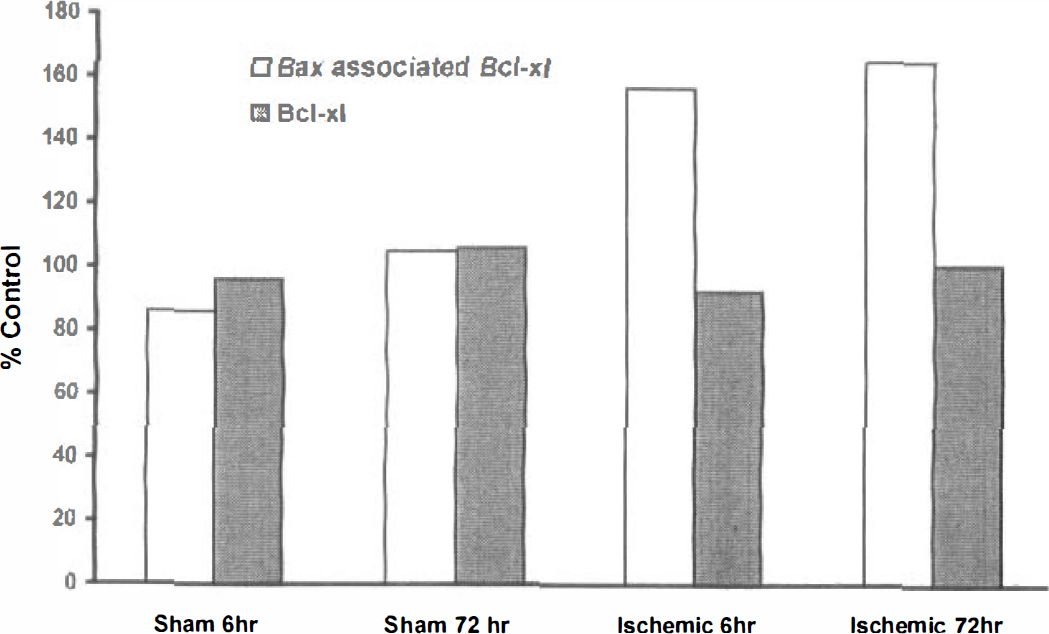
Dimer-associated Bcl-xl. Hippocampal dimer-associated Bcl-xl levels were increased after 5 minutes of carotid artery occlusion compared with control animals at 6 and 72 hours after ischemia. Dimerized Bcl-xl was detected by immunoprecipitation with Bax antisera using 1 mg of tissue. The Bcl-xl values included on the above graph are the protein levels from nondimerized pooled samples used merely for comparison. The “% control” is a ratio comparison with nonsurgically manipulated (shelf) control animal levels.
DISCUSSION
Although the exact mechanism by which Bcl-2 family proteins influence ischemic cell survival is not known, a favorable Bcl-2 to Bax ratio of expression may be essential for cell survival. After focal ischemic insults, both Bax and Bcl-2 mRNA levels are reported to increase in the peripheral region of a cerebral infarct. However, Bax protein levels appear to increase more than Bcl-2 (Simon et al., 1995; Chen et al., 1996). Recent global ischemia studies in rats and gerbils have revealed increases in Bax immunohistochemical staining after transient ischemia in the CA1 region (Krajewski et al., 1995b; Chen et al., 1996; Hara et al., 1996). Although an increase in Bcl-2 and Bcl-x mRNA levels in the CA1, CA3, and dentate hippocampal regions has been reported, this increase in message is not indicative of available tissue protein levels because of the decreased protein synthesis associated with ischemia. Vulnerable CA1 neurons do not demonstrate elevated protein levels whereas the other hippocampal regions do (Chen et al., 1997; Honkaniemi et al., 1996). The net effect is a shift in the balance of expression from a Bcl-2 (Bcl-xl) to a Bax dominance (Zhang et al., 1995), resulting presumably in caspase activation and apoptotic cell death. The balance between Bcl-2 and Bax may explain the relatively greater susceptibility of CA1 hippocampal neurons compared with CA3 neurons exposed to the same global ischemic insult (Krajewski et al., 1995b).
Our immunoblot data derived from a model of transient global ischemia confirm the current immunohistochemistry data suggesting an increase in Bax protein levels (Fig. 4) without substantial changes in Bcl-2 or Bcl-xl (Table 2). Consistent with previous reports (Mituguchi et al., 1996; Honkaniemi et al., 1996; Chen et al., 1997; Alonso et al., 1997), our data suggest that although hippocampal Bcl-2 levels are quite low, Bcl-x1, which is primarily found in postmitotic cells, serves as the major hippocampal neuroprotective member (Fig. 2). Furthermore, the protein bands obtained for Bcl-xl in the gerbil are somewhat different from those of the mouse. Because several alternatively spliced versions of Bcl-x mRNA have been described that produce two anti-apoptotic proteins, Bcl-x1 and Bcl-x1 ΔTM (transmembrane) (Fang et al., 1994), with similar molecular masses, this is most probably a result of alternate splicing in the gerbil. Using whole hippocampal homogenates, we did not detect an increase in postischemic Bcl-xl levels, even though an overall increased mRNA expression has been demonstrated (Chen et al., 1997; Honkaniemi et al., 1996).
Although the antisera used is capable of recognizing both forms of Bcl-x in human and mouse tissue (Krajewski et al., 1994b), it appears that Bcl-xs may not be expressed in gerbil hippocampal tissue. However, because the gerbil Bcl-x sequence in not yet cloned, we were unable to produce in vitro translated protein to confirm the specificity of this antibody. For the same reason, we do not know whether the lack of Bcl-xs can be explained by the dilutional effects of Bcl-xs-negative cells.
This is the first report to examine hippocampal Bcl-xl-Bax dimerization. Using immunoprecipitation, we demonstrated that Bcl-xl association with Bax increases after ischemia (Fig. 5). These studies suggest that initially there is an excess of Bcl-xl compared with Bax, and with the presence of more ischemia-induced Bax protein additional interactions become possible. This increased Bcl-xl-Bax interaction becomes apparent as early as 6 hours after ischemia, suggesting that it represents an early event associated with ischemia. This heterodimerization interaction is persistent and could be detected even at 72 hours after ischemia when significant cell death becomes apparent. These experiments suggest that Bax elevations may be involved in ischemic cell death by disrupting cytoprotective Bcl-xl (or Bcl-2) interactions with itself or other proteins, thus rendering hippocampal neurons susceptible to ischemic cell death.
Footnotes
Acknowledgements
The authors thank Mrs. Doris Boehle for her assistance in the preparation of this manuscript and Ms. Priscilla Wu for her expert technical assistance.