
Abstract
Considerable interest has focused on the possibility of using viral vectors to deliver genes to the central nervous system for the purpose of decreasing necrotic neuronal injury. To that end, we have previously shown that a herpes simplex virus (HSV) vector expressing Bcl-2 could protect neurons from ischemia. In that study, vector was delivered before the ischemia. However, for such gene therapy to be of clinical use, vectors must be protective even if delivered after the onset of the insult. In the present study, we show that an HSV vector expressing Bcl-2 protects striatal neurons when delivered after focal ischemia. Rats were exposed to middle cerebral artery occlusion for 1 hour, followed by reperfusion, and damage was assessed 48 hours later. Delivery of the Bcl-2 vector 30 minutes after reperfusion (i.e., 1.5 hours after ischemia onset) prevented any significant loss of virally-targeted neurons in the striatum. In contrast, in rats microinfused with a vector only expressing a reporter gene, a highly significant loss of neurons occurred. By 4 hours into the reperfusion period (5 hours after ischemia onset), delivery of the Bcl-2 vector was no longer protective. These data show the efficacy of postinsult gene therapy strategies for the brain, underline the finite length of this temporal therapeutic window, and support the growing evidence attesting to the neuroprotective potential of Bcl-2.
Neurons are highly sensitive to hypoxia-ischemia. Given this susceptibility and their postmitotic nature, the development of effective protective therapeutic strategies is essential. One promising strategy to emerge has been the use of viral vectors such as herpes simplex virus (HSV) that allow the overexpression of exogenous genes in postmitotic cells. Herpes simplex virus vectors have the advantage of being both neurotropic and capable of packaging large amounts of DNA for delivery to neurons (Glorioso et al., 1992). We have generated an HSV vector, designated vα22βgalα4Bcl-2, bearing the human proto-oncogene bcl-2 gene and the Escherichia coli lacZ marker gene. Bcl-2 protects against both apoptotic and necrotic death in many cell types, although the precise mechanism(s) remains controversial (reviewed in Bredesen, 1995). We have previously shown that infection of cultured hippocampal neurons with vα22βgalα4Bcl-2 can protect against oxidative insults, hypoglycemia, and glutamate toxicity. Moreover, when delivered directly to the brain, vα22βgalα4Bcl-2 can protect striatal neurons against focal ischemia (Lawrence et al., 1996); similarly, a monocistronic HSV vector expressing Bcl-2 has also been reported to protect cortical neurons from focal ischemic injury (Linnick et al., 1995).
In those two previous reports, vector was delivered to cultures or the brain before the insult. If such gene-transfer strategies are to be useful clinically, they must be able to protect even if delivered after the insult (i.e., it is not possible to predict a neurological crisis in advance). In this report, we have explored the protective effect achieved by the delivery of the vα22βgalα4Bcl-2 to the striatum after, rather than in anticipation of middle cerebral artery occlusion (MCAO).
MATERIALS AND METHODS
Herpes simplex virus vectors were generated as previously described (Lawrence et al., 1995). Briefly, the amplicon plasmid pα22βgalα4Bcl-2 contained the human bcl-2 gene and the Escherichia coli lacZ gene under the control of the HSV α4 and α22 promoters, respectively. The HSV oriS and the “a” sequence were also included to provide the necessary cis-signals for replication and packaging of the amplicon DNA. pα22βgalα4Bcl-2 was transfected into cell line E5 (DeLuca et al., 1985) which was stably transfected with the HSV immediate early gene a4. Superinfection with the HSV α4 deletion mutant dl20 yielded vector, designated vα22βgalα4Bcl-2 which contained concatemers of the plasmid, and replication incompetent helper virus particles at a ratio of 1:2 (Lawrence et al., 1996). A control vector, designated vα22βgalα4bst, was generated bearing the bcl-2 gene with the insertion of a stop codon so as to produce a truncated dysfunctional gene product (Lawrence et al., 1996). Viral stocks were concentrated 5- to 10-fold in microcon 100 kd microfiltration devices (Amicon). Vector titer before concentration was 1 × 106 infectious particles/mL, and helper virus titer was 2 × 106 plaque forming units/mL.
One hour of ischemia was induced by the insertion of an intraluminal 3-0 nylon suture through the common carotid artery as previously described (Longa et al., 1989; Memezawa et al., 1992; Yoon and Steinberg, 1994; Lawrence et al., 1996). Thirty minutes after removal of the suture 6 μL of either vα22βgalα4Bcl-2 or vα22βgalα4bst was delivered bilaterally to the striatum of 250 to 300 g Sprague-Dawley rats (from bregma: AP = 0, ML = 3.5, with 2 injection sites of 3 μL each at DV = 4.5 and 3.5 mm). Another group of animals in a separate experiment received the vector 4 hours after removal of the suture. Cytopathogenicity of such vector/helper virus stocks was not detected when delivered to the striatum at the titers used (Ho et al., 1995). Infection with vα22βgalα4Bcl-2 resulted in consistent in vivo coexpression of βgal and Bcl-2, as assessed by immunostaining (data not shown).
Animals were killed 48 hours later and brain sections were prepared and analyzed as previously described (Lawrence et al., 1995). Briefly, X-gal/cresyl violet-stained sections were prepared and individual βgal-positive neurons were counted over successive sections by an individual blinded as to treatment. The efficacy of any intervention was expressed as the difference between the total number of βgal-positive neurons in the occluded hemisphere and in the unoccluded control hemispheres. Infarct damage was assessed by assigning a degree of severity on a scale of 0 to 5, in which 0 represented no visible damage in the occluded hemisphere and 5 represented complete ablation of the striatum and surrounding cortex (Lawrence et al., 1995). This allowed the exclusion of animals exhibiting 0 damage and a quantitative comparison of damage between the remaining animals in the Bcl-2 and control groups by Chi-square analysis. Five animals in the 30 minute postischemic Bcl-2 group, 4 animals in the 30 minute postischemic control group, 4 animals in the 4 hours postischemic Bcl-2 group, and 3 animals in the 4 hours postischemic control group were discarded by this criterion, leaving 13, 15, 9, and 11 animals, respectively in each treatment group.
A parallel study of time-course of expression was performed. Rats were occluded unilaterally and vα22βgalα4Bcl-2 was injected bilaterally 30 minutes into the reperfusion period. Animals were then killed at indicated times and numbers of βgal-positive neurons quantified.
A significant protection of neurons through introduction of a vector was detected by comparing the number of βgal-positive neurons in the ischemic and nonischemic hemispheres by paired Student's t-test. The magnitude of any such protection was compared between groups by unpaired Student's t-tests.
RESULTS
Microinjection of vα22βgalα4Bcl-2 caused very localized expression in the striatum, with the majority of βgal-positive cells within 0.5 mm of the injection site (Fig. 1). In nonischemic control striatal tissue, expression was shown by 4 to 6 hours after injection, with a dramatic increase by 8 to 9 hours (Table 1).
Number of βgal-positive neurons as a function of time after vα22βgalα4Bcl-2 injection and tissue status

Data indicate the number of βgal-positive neurons in the striatum within the single coronal section containing the tip of the microinfusion syringe. Vector was injected 30 minutes after the onset of ischemia. Two-way analysis of variance indicated a significant increase in expression over time in both non-ischemic and ischemic tissue (P < 0.01, 0.02, respectively), with expression being greater in nonischemic tissue at 8 to 9 hour and 12 hours (P < 0.01, 0.05, respectively, by Newman-Keuls post-hoc). n = 14, 3, and 4 for the 3 respective timepoints.
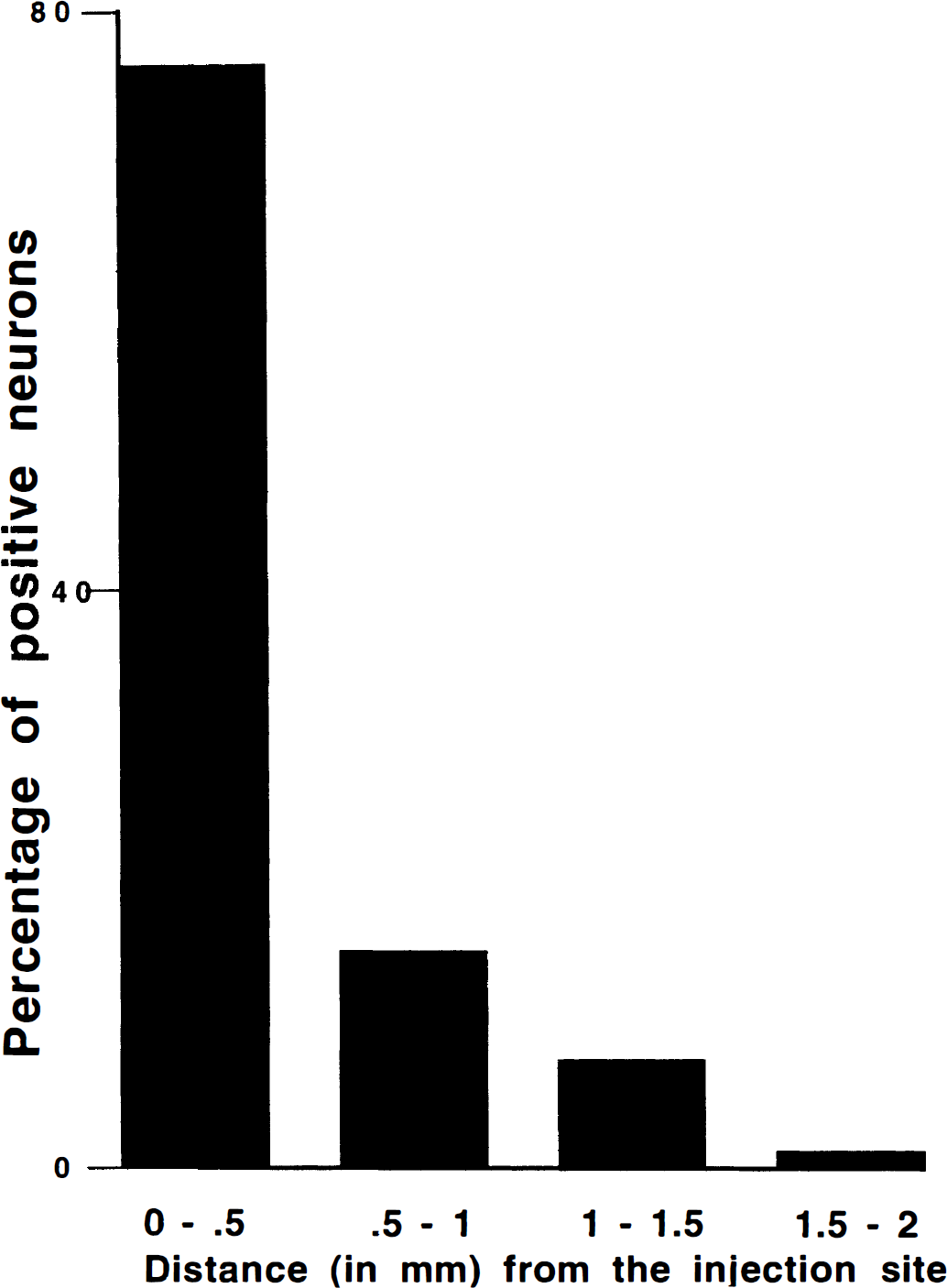
Histogram showing the distribution of βgal-positive neurons as a function of distance from the microinjection site (n = 8).
Occlusion of the middle cerebral artery for 1 hour caused a focal ischemic infarct within the medial and lateral striatum. The delivery of vα22βgalα4Bcl-2 and vα22βgalα4bst after the occlusion resulted in expression of βgal in striatal neurons, some of which lay within the lesioned cell field (Fig. 2). The extent of expression in this ischemic tissue was blunted and delayed when compared with expression non-ischemic control tissue (Table 1); this likely reflects the decrease in protein synthesis that is typical in ischemic tissue (Kleihues and Hossmann, 1971). Despite this, delivery of vα22βgalα4Bcl-2 was associated with decreased neuron loss. When vector was microinfused 30 minutes into the reperfusion period (i.e., 1.5 hours after ischemia onset), the number of surviving βgal-positive neurons in vα22βgalαBcl-2 rats did not differ significantly between the hemisphere undergoing MCAO and the unoccluded control hemisphere (Fig. 3: P > 0.05 by paired Student's t-test comparing numbers of βgal-positive neurons in the ischemic and nonischemic side), implying a protective effect of the vector. In contrast, in rats infused with vα22βgalα4bst, MCAO was associated with a significant loss of βgal-positive neurons, relative to the contralateral side (P < 0.001 by paired Student's t-test). As such, the extent of ischemia-induced loss of βgal-positive neurons was significantly greater in vα22βgalα4bst rats than in vα22βgalα4Bcl-2 rats (P < 0.05, unpaired Student's t-test). The two vector groups did not differ in the amount of total MCAO damage within the region of infusion (Bcl-2 damage = 3.54 ± 1.05; control damage = 3.27 ± 1.16; X2 = 1.98, not significant), indicating that the differential survivorship of vα22βgalα4Bcl-2- verses vα22βgalα4bst-infected neurons did not arise from the differing degrees of ischemia endured. By 4 hours into the reperfusion period (i.e., 5 hours after ischemia onset), there was a significant loss of βgal-positive neurons in both vα22βgalα4Bcl-2 and vα22βgalα4bst rats (P < 0.05 and 0.001, respectively, by paired Student's t-test comparing ischemic and control hemispheres); the magnitude of such loss did not differ between those two groups (not significant by paired Student's t-test). In animals receiving either the Bcl-2 or the control vector 4 hours after MCAO there was also no significant difference in the total MCAO damage within the region of infusion (Bcl-2 damage = 2.56 ± 1.23; control damage = 2.54 ± 1.44; X2 = 1.36, not significant).
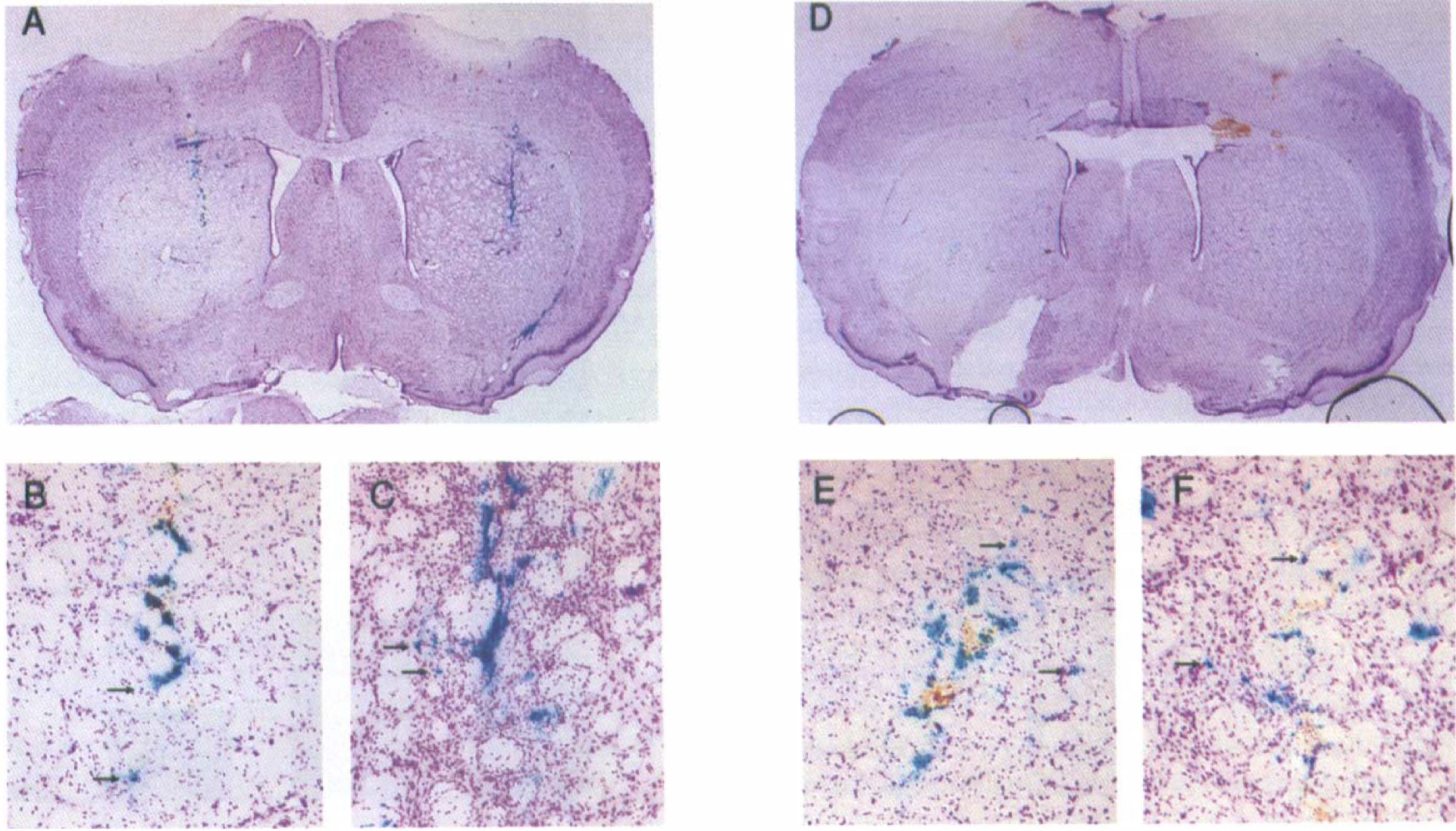
A representative striatal section from an animal that had been infected with vα22βgalα4Bcl-2 reveals the extent of middle cerebral artery occlusion damage in the left hemisphere
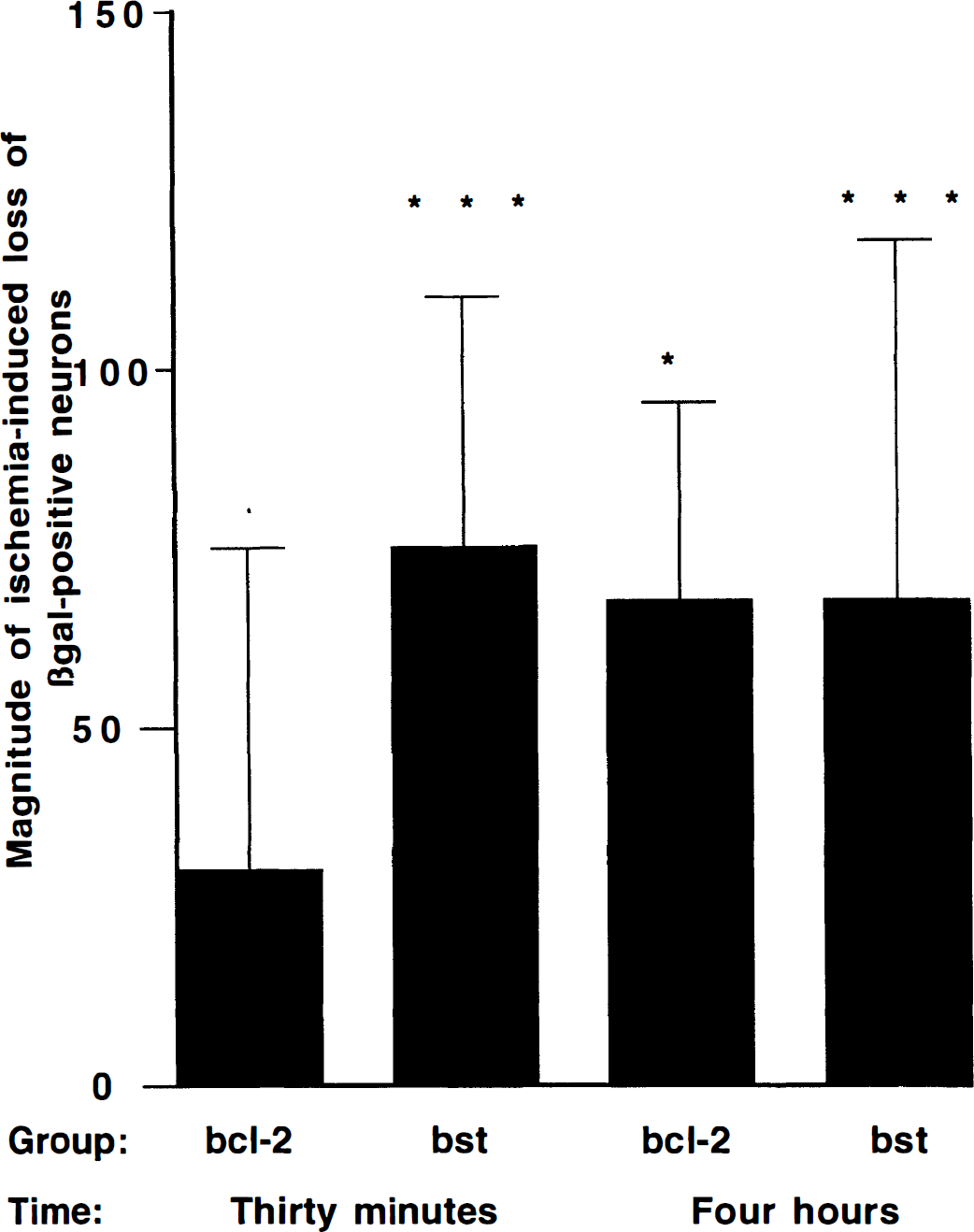
Magnitude of ischemia-induced loss of βgal-positive neurons as a function of vector treatment. X-axis indicates the difference in the number of βgal-positive neurons when comparing the ischemic and nonischemic hemispheres; a positive number indicates an ischemia-induced loss. Among animals injected with vector 30 minutes and 4 hours into the reperfusion period, nonischemic control hemispheres averaged 99 ± 57 and 131 ± 72 βgal-positive neurons, respectively. n = 13, 15, 9, and 11 for the four groups. *,***Ischemia-induced loss of βgal-positive neurons (when compared with nonischemic contralateral hemisphere) at the P < 0.05, and P < 0.001 levels of significance, respectively.
DISCUSSION
Previous studies have shown that HSV vectors expressing Bcl-2 can protect primary hippocampal cultures and the intact brain when delivered before necrotic insults (Linnick et al., 1995; Lawrence et al., 1996); moreover, overexpression of Bcl-2 in transgenic animals also protects against ischemia (Martinou et al., 1994). We now demonstrate that protection against a focal ischemic insult can be achieved by vector delivery within a limited window after the insult. Specifically, delivery of the Bcl-2 expressing vector 30 minutes, but not 4 hours into the reperfusion period was significantly protective. This was assessed by the survivorship of βgal-positive neurons. The assumption that positive X-gal staining in vα22βgalα4Bcl-2-infected neurons indicates Bcl-2 over-expression is supported by the consistent coexpression of the two gene products in striatal neurons using this bicistronic system (Lawrence et al., 1996) and the fact that Bcl-2 expression under the α4 promoter is more conservative than β-gal expression under the α22 promoter (Ho et al., 1995b). We have tested this assumption more explicitly with a vector expressing β-gal and HSP72 and using the same promoters as in the current study; we observe greater than 95% coexpression of the two up to 40 hours after infection (Fink et al., 1997).
This finding of protection by Bcl-2 at 1.5 hours, but not 5 hours after ischemia onset, gives some insight as to the time-course of degenerative events after ischemia. In nonischemic tissue, expression was shown by 4 to 6 hours, with a substantial increase by the 8- to 9-hour mark. This agrees with our previous work with the HSV α4 promoter, which also showed that expression is then sustained over a 48- to 72-hour period (reviewed in Ho et al., 1995b). Expression in ischemic striatal tissue was significantly decreased, probably reflecting the well-known inhibition of protein synthesis during ischemia (as first demonstrated by Kleihues and Hossmann, 1971). In such tissue, a substantial increase in expression did not occur until the 12-hour mark. These data suggest that the Bcl-2 is protective against events occurring within a time window of approximately 13 to 16 hours after reperfusion (assuming that Bcl-2 expression is delayed in the same manner as is β-gal expression, an assumption supported by the findings reviewed in the previous paragraph). This delayed protective time window agrees with findings showing the efficacy of postinsult delivery of NMDA receptor antagonists (Levy, 1990; Steinberg et al., 1995) and, more broadly, with the delayed nature of necrotic neuron death.
The precise mechanisms of Bcl-2 action remain quite controversial. A number of early investigators attributed its protective potential to its antioxidant properties (Behl et al., 1993; Kane et al., 1993; Zhong et al., 1993 a,1993 b). This was based on the ability of Bcl-2 to prevent oxidative death and the associated oxygen radical accumulation and lipid peroxidation (Hockenbery et al., 1993; Kane et al., 1993). In support of such an antioxidant function, infection with vα22βgalα4Bcl-2 reduced the amount of dichlorofluorescein fluorescence in hippocampal cultures after adriamycin exposure (Lawrence et al., 1996). Subsequent work has shown Bcl-2 protection against a variety of insults in extremely low oxygen environments (Jacobson and Raff, 1995), suggesting mechanisms independent of oxygen radical biology. Some of the most plausible routes involve the ability of the protein to dimerize and thereby inactivate proapoptotic proteins such as BAX (reviewed in Bredesen, 1995).
The finding in this article is of a similar theme as our previous studies showing an HSV vector expressing the glucose transporter gene is protective if delivered within a few hours after various necrotic insults (Lawrence et al., 1996b;Dash et al., 1996). These observations not only provide insight into the time course of neuronal injury, but support the potential therapeutic application of viral vector technologies. At present the restricted and local extent of expression (as shown in Fig. 1) represent serious limitations to such applications. Moreover, the decrease in protein synthesis that follows ischemia (resulting in decreased expression of potentially neuroprotective genes) show some specific challenges for carrying out gene therapy in the CNS after ischemic insults. Nevertheless, the demonstration of efficacy using postinsult intervention suggests that as these technical limitations are overcome, HSV vectors and other new viral vector strategies may offer a promising means of delivering neuroprotective genes to the central nervous system.