
Abstract
As one of the key determinants of ischemic injury, cerebrovascular endothelial cell (EC) degeneration may be dependent upon the generation of the free radical nitric oxide (NO) and the subsequent induction of programed cell death (PCD). Although the mechanisms that can prevent EC injury are most likely multifactorial in origin, the metabotropic glutamate receptor (mGluR) system may represent a novel therapeutic approach for ECs given the ability of the mGluR system to reverse neuronal cell injury. This study examined the modulation of individual subtypes of mGluRs during anoxia and NO toxicity in primary rat cerebrovascular ECs. Cell injury was determined through trypan blue dye exclusion, intracellular lactate dehydrogenase release, DNA fragmentation, membrane phosphatidylserine (PS) exposure, and cysteine protease activity. Anoxia, through the generation of NO, and exposure to exogenous NO were directly toxic to ECs. Exposure to NO rapidly decreased EC viability from 98% ± 2% to 40% ± 9%, increased DNA fragmentation from 2% ± 2% to 61% ± 9%, and increased membrane PS exposure from 3% ± 3% to 66% ± 6% over a 24-hour period. Activation of the mGluR system significantly increased EC survival through the prevention of NO-induced DNA fragmentation and cellular membrane PS residue exposure. In contrast, antagonism of the mGluR system failed to prevent PCD. Cytoprotection by the mGluR system was dependent, at least in part, upon the direct inhibition of NO-generated caspase 1- and caspase 3-like activities. Further investigation into the ability of the mGluR system to prevent PCD in ECs may open new therapeutic avenues for the treatment of cerebrovascular injury.
Keywords
Vascular endothelial cells (ECs) form an antithrombotic and antiinflammatory barrier throughout the cardiovascular system (Staub, 1981; Sage et al., 1982). As a result, they play an important role in regulating vascular tone (Palmer et al., 1988; Moncada et al., 1991), immune surveillance (Palmer et al., 1988; Moncada et al., 1991), and vascular permeability (Gudgeon and Martin, 1989). Intact endothelium acts as a sensor and transducer to regulate physiologic functions of the vascular wall. Yet, dysfunction or injury of vascular ECs can become a critical step in the development of several disease states such as atherosclerosis, intravascular coagulation, and ischemic injury (Kurose et al., 1994; Grammas et al., 1997).
In the central nervous system, the free radical nitric oxide (NO) plays an important role in neurotransmission, synaptic plasticity, and the regulation of gene expression. In the cardiovascular system, NO also emerges as an important regulator of a variety of physiologic responses including inhibition of platelet aggregation, modulation of inflammatory cell adhesion, and prevention of smooth muscle proliferation (Suematsu et al., 1994; Li and Forstermann, 2000).
Nitric oxide also has a significant role as a mediator of cardiovascular and central nervous system damage during ischemic injury, reperfusion insults, and inflammation (Moncada et al., 1991; Bhagat and Vallance, 1999). Endothelial cell injury can be initiated by several different stimuli that may ultimately lead to apoptosis or programed cell death (PCD). Programed cell death is composed of two independent pathways that consist of nuclear DNA degradation and the loss of membrane asymmetry with exposure of membrane phosphatidylserine (PS) residues (Vincent and Maiese, 1999a; Maiese and Vincent, 2000b). In contrast to genomic DNA fragmentation, exposure of membrane PS residues is considered to be an early marker for PCD and allows cells to be phagocytized for subsequent destruction (Rimon et al., 1997; Savill, 1997). Although PCD in ECs can serve to maintain the cellular population in the vascular wall, excessive cellular DNA degradation and the exposure of PS residues in ECs without effective modulation may lead to disturbed endothelial function and precipitate atherogenesis (Dimmeler et al., 1997; Galle et al., 1999).
In regards to the prevention of PCD, current interest has focused on the role of the metabotropic glutamate receptor (mGluR) system. At least eight genes encode for the mGluR subtypes and have been classified into three groups on the basis of sequence homology, signal transduction mechanisms, and agonist selectivity (Pin and Duvoisin, 1995; Bordi and Ugolini, 1999). These receptors are located throughout the central nervous system and are vital for both neuronal development and physiologic function. Activation of specific subtypes of mGluRs have been demonstrated to be neuroprotective against anoxia (Maiese et al., 1996), glutamate toxicity (Bruno et al., 1996), and free radical NO exposure (Vincent et al., 1999; Maiese et al., 2000).
Recently, ECs have been shown to express mGluRs (Krizbai et al., 1998; Gill et al., 1999). Yet, the function of the mGluR system in ECs remains to be defined. Because the cytotoxic actions of ischemic NO generation are relevant to the pathogenesis of vascular and neuronal diseases, research into the mechanisms underlying EC degeneration and EC protection during NO toxicity may have effective therapeutic implications for cerebrovascular and neurodegenerative disorders. In the current study, the authors examined the role of specific mGluR subtypes in rat cerebrovascular ECs during anoxia and subsequent NO toxicity. The current work illustrates that ischemic NO generation leads to both DNA degradation and membrane PS exposure in ECs. Cytoprotection by the mGluR system is broad in nature and effectively prevents the induction of the two independent pathways of PCD in ECs. The ability of the mGluR system to prevent PCD in ECs is mediated through the direct modulation of caspase 1- and caspase 3-like activities and may offer protection against both direct EC degeneration and subsequent thrombosis development.
MATERIALS AND METHODS
Endothelial cell culture
Vascular ECs were isolated from Sprague-Dawley adult rat brains by using a modified collagenase/dispase-based digestion protocol (Rupnick et al., 1988). The cerebrum was trimmed into blocks suspended in dissociation medium containing 1 mg/mL collagenase/dispase (Roche, Mannheim, Germany) in M199E with 0.5% antibiotic-antimycotic solution for 2 hours at 37°C. The slurry was homogenized with three complete strokes of a pestle tissue grinder. The resultant pellet was resuspended with 15% dextran (Sigma, Louis, MO, U.S.A.) in HEPES buffer and centrifuged at 4,000 g for 20 minutes at 4°C. The vascular pellet was resuspended in a colloidal silica gradient solution of 45% Percoll (Sigma in Dulbecco's phosphate saline and centrifuged at 20,000 g for 20 minutes at 10°C. The upper band, consisting primarily of single cells, was carefully removed and washed three times by centrifugation. The pellet then was resuspended in culture media consisting of M199E with 20% heat-inactivated fetal bovine serum, 2 mmol/L L-glutamine, 90 μg/mL heparin, and 20 μg/mL EC growth supplement (ICN Biomedicals, Aurora, OH, U.S.A.). Cultures were washed 1 hour after plating to remove debris and nonadherent cells and maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% room air. Experiments were performed with cells from the third passage. Cells were identified as endothelial by a cobblestone appearance with phase contrast microscopy, were positive with direct immunocytochemistry for factor VIII-related antigen, and were negative for glial fibrillary acidic protein immunocytochemistry.
Experimental treatments
Anoxia
To induce anoxia, EC cultures were deprived of oxygen by placing them into an anoxic chamber system (Sheldon Manufacturing, Cornelius, OR, U.S.A.). During experimental paradigms, the cultures were maintained in an anaerobic environment (95% N2, 5% CO2) at 37°C per the experimental paradigm. To determine whether NO mediates EC injury during anoxia, inhibition of the production of NO in an oxygen-free environment was performed with the nitric oxide synthase (NOS) inhibitors for inducible NOS (iNOS), N-(3-(Aminomethyl)benzyl)acetamidine-2HCl (1400W) (Biomol, Plymouth Meeting, PA, U.S.A.), and endothelial NOS (eNOS), N
Nitric oxide application
Nitric oxide administration was performed by replacing the culture media with media containing 6-(2-hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1-hexanamine (NOC-9) (Calbiochem) or sodium nitroprusside (SNP) (Sigma, St. Louis, MO, U.S.A.). More than one NO generator was used to ensure that the biologic effects observed were a result of NO generation rather than by-products of NO generations. After treatment with the NO donors, the cultures were placed in a normoxic, humidified incubator at 37°C with 5% CO2 for periods determined by the specific experimental paradigm.
Metabotropic glutamate receptor modulation
Ligands for the metabotropic glutamate receptor subtypes (S)-3,5-dihydroxyphenylglycine (DHPG), 2R,4R-4-aminopyrrolidine-2,4-dicarboxylate (APDC), L(+)-2-amino-4-phosphonobutyric acid (L-AP4), (RS)-1-aminoindan-1,5-dicarboxylic acid (AIDA), and methylphenylethynylpyridne (MPEP) were obtained from Tocris Cookson (St. Louis, MO, U.S.A.). All mGluR ligands were applied directly to the cultures as previously described per the experimental protocol (Vincent et al., 1997; Vincent and Maiese, 1999b; Maiese et al., 2000).
Assessment of cell survival and injury
Endothelial cell injury was determined by bright field microscopy using a 0.4% trypan blue dye exclusion assay after treatment with the NO donors. For trypan blue dye exclusion, the mean survival was determined by counting 8 randomly selected nonoverlapping fields with each containing approximately 10 to 20 cells (viable + nonviable) in each well in a 24-well Petri dish. In addition, the leakage of intracellular lactic dehydrogenase (LDH) into culture medium was determined to examine membrane integrity. Intracellular lactic dehydrogenase activity was measured by using Sigma Diagnostics Lactate Dehydrogenase (St. Louis, MO, U.S.A.) agents.
Assessment of DNA fragmentation
Genomic DNA fragmentation was determined by the terminal deoxynucleotidyl transferase nick end labeling (TUNEL) assay. Briefly, cells were fixed in 4% paraformaldehyde/0.2% picric acid/0.05% glutaraldehyde in phosphate-buffered saline solution (PBS: 10 mmol/L KH2PO4, 37 mmol/L Na2HPO4, 87 mmol/L NaCl, 53 mmol/L KCl, pH 7.4). Cells were permeabilized using 0.1% Triton X-100, then endogenous peroxidase was blocked using 0.3% H2O2 in methanol. The 3‘-hydroxy ends of cut DNA were labeled with biotinylated dUTP (Promega, Madison, WI, U.S.A.) using the enzyme terminal deoxytransferase (Promega). Incorporation of the label was detected using streptavidin-peroxidase (Sigma) and visualized with 3,3‘-diaminobenzidine (Vector Laboratories, Burlingame, CA, U.S.A.).
Reversible membrane phosphatidylserine residue assay
Per the authors' prior protocols (Vincent and Maiese, 1999a; Lin et al., 2000; Maiese and Vincent, 2000b), annexin V conjugated to phycoerythrin (PE) was purchased from R&D Systems (Minneapolis, MN, U.S.A.). A 30 μg/mL stock solution was diluted directly before use to 3 μg/mL in warmed (37°C) binding buffer (10 mmol/L HEPES, pH 7.5, 150 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1.8 mmol/L CaCl2). The growth medium was removed from culture plates and then 300 μL of diluted annexin V conjugate was applied to the EC layer. Plates were incubated at 37°C in a humidified atmosphere in the dark for 10 minutes and then rinsed twice using fresh binding buffer. The ECs were examined using a Leitz DMIRB microscope (Leica, McHenry, IL, U.S.A.) and Oncor Image 2.0 imaging software (Oncor, Gaithersburg, MD, U.S.A.). Images were acquired using both transmitted light, as well as fluorescent single excitation light at 490 nm and detected emission at 585 nm.
After examination, the annexin V label was detached by washing three times in a calcium-free dissociation buffer (10 mmol/L HEPES, pH 7.5, 150 mmol/L NaCl, 5 mmol/L KCl, 2.8 mmol/L MgCl2) that contained magnesium as a replacement cation. The use of a calcium-free buffer does not alter EC survival or EC programed cell death (Vincent and Maiese, 1999a; Lin et al., 2000; Maiese and Vincent, 2000b). Plates then are reexamined to confirm that the annexin V was completely removed and then returned to the incubator for a further specified period. Each randomly selected field of cells uses “blinded” assessment and is continuously imaged through a cooled CCD system with further identification through precise x-y labeled coordinates. The quantitation of PS residue exposure is obtained through the direct counting of annexin V labeled ECs.
Assessment of cysteine protease activity
At specific times after NO exposure, caspase 1 and caspase 3-like activities were determined as previously described (Maiese and Vincent, 1999; Lin et al., 2000). Cells were harvested and resuspended in 100 μL lysis buffer (25 mmol/L HEPES, pH 7.5, 5 mmol/L MgCl2, 1 mmol/L ethyleneglycoltetracetic acid, 0.5% Triton X-100, 10 μg/mL phenylmethylsulfonyl fluoride (PMSF), 1 μg/mL aprotinin, 1 μg/mL leupeptin). Cell suspensions were homogenized using a sonic dismembrator (Model 550; Fisher Scientific, Pittsburgh, PA, U.S.A.), pulse mode (0.5 seconds on, 0.5 seconds off for 30 seconds), in an Eppendorf tube on ice. An aliquot of supernatant containing 30 μg protein was incubated with a 250 μmol/L colorimetric substrate for caspase 1 (Ac-YVAD) or for caspase 3 (Ac-DEVD) (Calbiochem, San Diego, CA, U.S.A.). The total reaction mixture volume was 100 μL in reaction buffer (50 mmol/L HEPES, pH 7.5, 10 mmol/L DTT, 1% sucrose, 0.1% CHAPS). Reaction mixtures were incubated at 37°C and the absorbance was measured at 405 nm for 1 hour. The mean rate of substrate cleavage in micromoles per minute per gram protein (μmol/ min/g) was calibrated with standard p-nitroaniline solutions.
Cysteine protease modulation
Modulation of cysteine protease activity in ECs was performed by using the irreversible and cell permeable caspase inhibitors Z-YVAD-FMK (YVAD) and Z-DEVD-FMK (DEVD) obtained from Pharmingen (Livermore, CA, U.S.A.). Inhibitors were added directly to the cultures at concentrations of 10 μmol/L, 50 μmol/L, and 85 μmol/L 1 hour before NO application.
Statistical analysis
For each experiment involving assessment of endothelial survival, DNA degradation, membrane PS exposure, and caspase activity, differences between groups were statistically analyzed by means of analysis of variance (ANOVA).
RESULTS
Nitric oxide production during anoxia is toxic to endothelial cells
Because NO is believed to be one of the modulators of cellular injury during a variety of insults, the authors initially examined whether endogenous NO production is a mediator of EC injury during an anoxic environment. The enzyme NOS in ECs was inhibited in an oxygen-free environment with the NOS inhibitors 1400W and L-NMMA. The agent 1400W is a selective inhibitor of inducible NOS (Garvey et al., 1997) and the agent L-NMMA is a competitive inhibitor of all three isoforms of NOS (Reif and McCreedy, 1995). The NOS inhibitors were added directly to the culture media 1 hour before anoxic exposure and cell survival was subsequently determined 24 hours later. To examine the effect of different time periods of anoxic exposure on EC cultures, EC survival was determined by trypan blue dye exclusion after a 6-, 12-, and 24-hour period of anoxia. As shown in Fig. 1, a significant decrease in cell survival was seen in EC cultures exposed to anoxia for the duration of 12 hours (48% ± 4%) and 24 hours (29% ± 7%), but not for a 6-hour (91% ± 3%) period when compared with untreated control (95% ± 4%). Administration of L-NMMA (1000 μmol/L) during anoxia significantly increased cell survival to 69% ± 8% and 60% ± 11% in cultures exposed to 12- and 24-hour periods of anoxia, respectively. Treatment of ECs with 1400W (100 μmol/L) also significantly increased EC survival to 73% ± 8% and 65% ± 5% after a 12- and 24-hour period of anoxic exposure. There was no significant difference in EC viability between the administration of the two different types of NOS inhibitors for each time period of anoxic exposure.
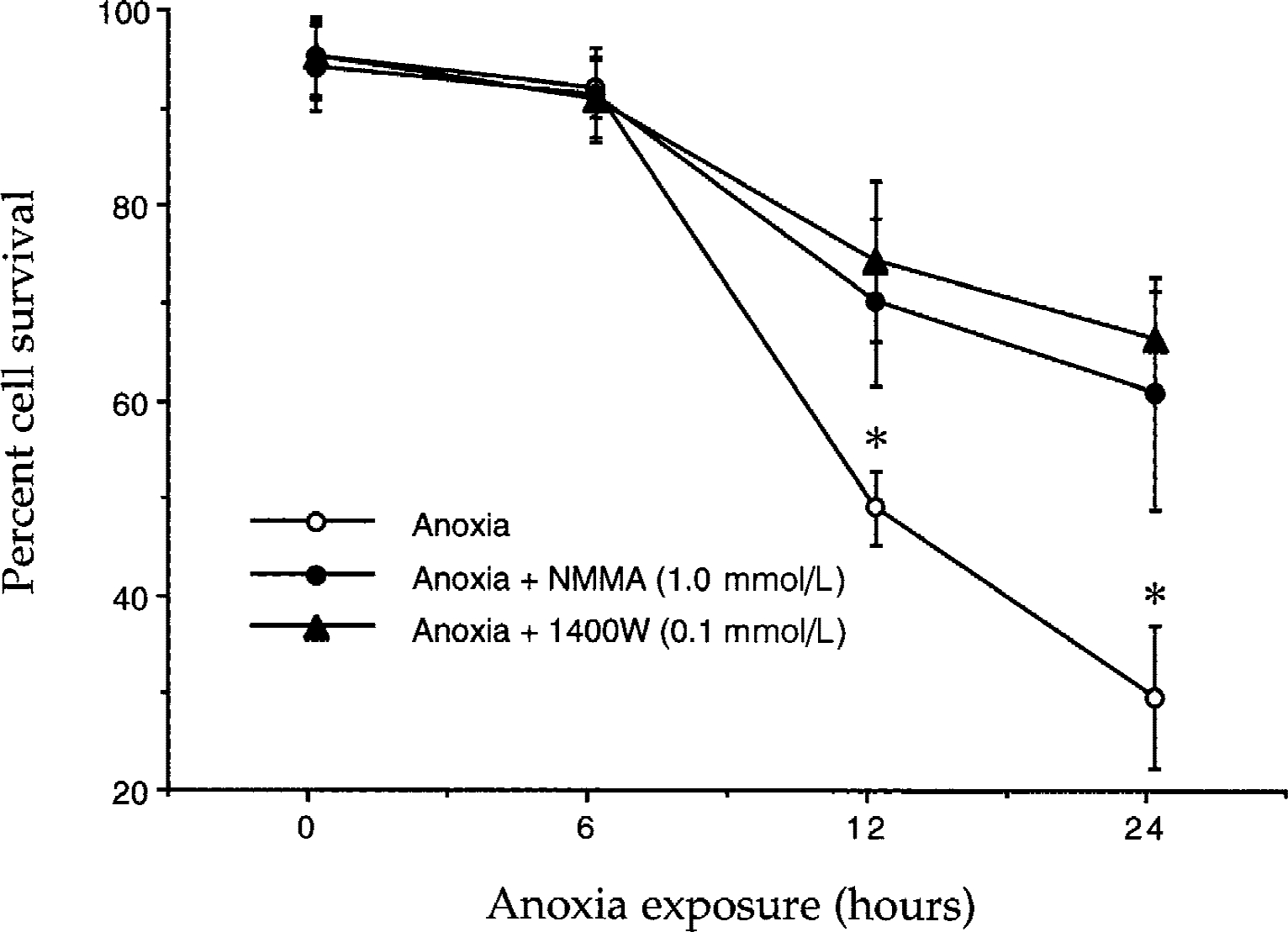
Nitric oxide (NO) production during anoxia is toxic to endothelial cell (EC) cultures. To determine whether NO production during anoxia is cytotoxic to EC cultures, the effect of inhibition of nitric oxide synthase (NOS) on EC viability was examined. Inhibition of EC NOS was performed in an oxygen-free environment with the NOS inhibitors 1400W (100 μmol/L) or N
Nitric oxide exposure induces endothelial cell injury
Because NO is a downstream mediator of anoxic injury in ECs (Fig. 1), the authors assessed the role of exogenous NO on rat brain ECs. Vascular ECs were treated with increasing concentrations of the NO donors NOC-9 (0.1 to 2.0 mmol/L) or SNP (0.1 to 2.0 mmol/L) and cell survival was determined 24 hours later. As shown in Fig. 2A, treatment with the NO donors at concentrations of 0.1 mmol/L and 0.3 mmol/L were not toxic over a 24-hour period after NO exposure. Yet, treatment with the NO donors at concentrations greater than 0.75 mmol/L was toxic. For example, a significant decrease in EC survival was seen in cultures containing 0.75 mmol/L (59% ± 10%), 1.0 mmol/L (41% ± 8%), and 2.0 mmol/L (19% ± 3%) after NO exposure when compared with untreated control cultures (98% ± 2%).
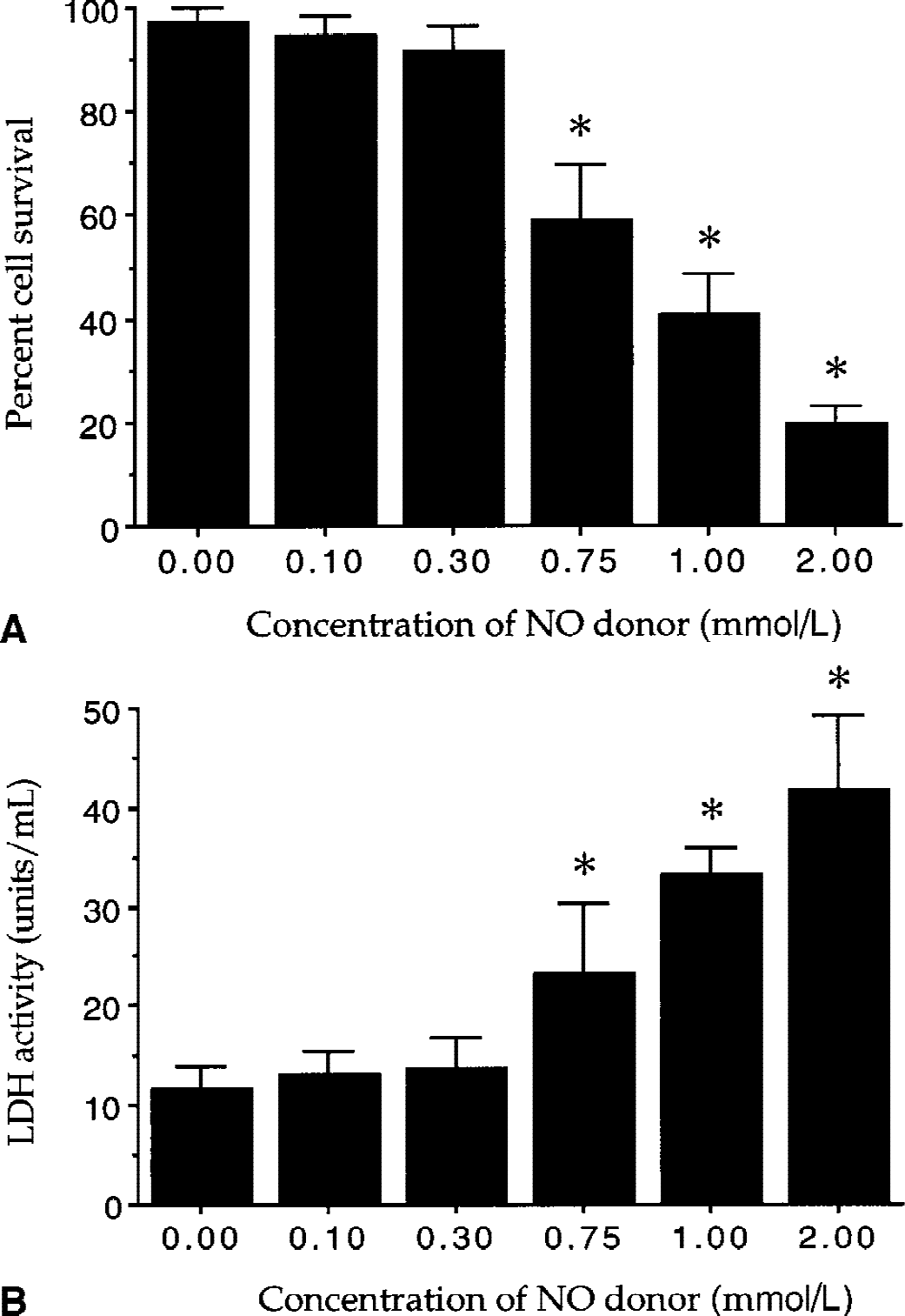
Nitric oxide (NO) exposure is detrimental to endothelial cell (EC) cultures.
To further examine the effect of NO exposure on EC plasma membrane integrity, the leakage of intracellular LDH into culture medium was measured. Endothelial cells were treated with the NO donors (NOC-9 and SNP) at concentration of 0.1 mmol/L, 0.3 mmol/L, 0.75 mmol/ L, 1.0 mmol/L, and 2.0 mmol/L. The activity of LDH was determined 24 hours after NO exposure.
As shown in Fig. 2B, treatment with the NO donor at concentrations of 0.75 mmol/L, 1.0 mmol/L, and 2.0 mmol/L induced a significant increase in media LDH activity of 23.2 ± 7.2 U/mL, 33.2 ± 2.9 U/mL, and 41.6 ± 7.6 U/mL when compared with untreated control of 11.6 ± 2.4 U/mL. There were no significant changes in LDH activity in EC cultures exposed to NO at concentrations of 0.1 mmol/L (13.0 ± 2.4 U/mL) and 0.3 mmol/L (13.7 ± 3.0 U/mL) when compared with untreated cultures.
Nitric oxide exposure results in DNA degradation and membrane phosphatidylserine externalization
As an active rather than a passive form of cell death, PCD is composed of two distinct components that consist of DNA degradation and membrane PS externalization (Maiese et al., 2000; Maiese and Vincent, 2000b; Vincent and Maiese, 2000). To examine the ability of NO to induce DNA fragmentation in ECs, the authors quantitatively determined DNA fragmentation through the use of a TUNEL assay. Endothelial cells were treated with the NO donors NOC-9 (1.0 mmol/L) or SNP (1.0 mmol/L) and the proportion of DNA fragmentation was assessed 24 hours later. As shown in Fig. 3A, NO exposure induced a significant increase in the percent of DNA fragmentation from 2% ± 2% in untreated cultures to 61% ± 10% over a 24-hour period.
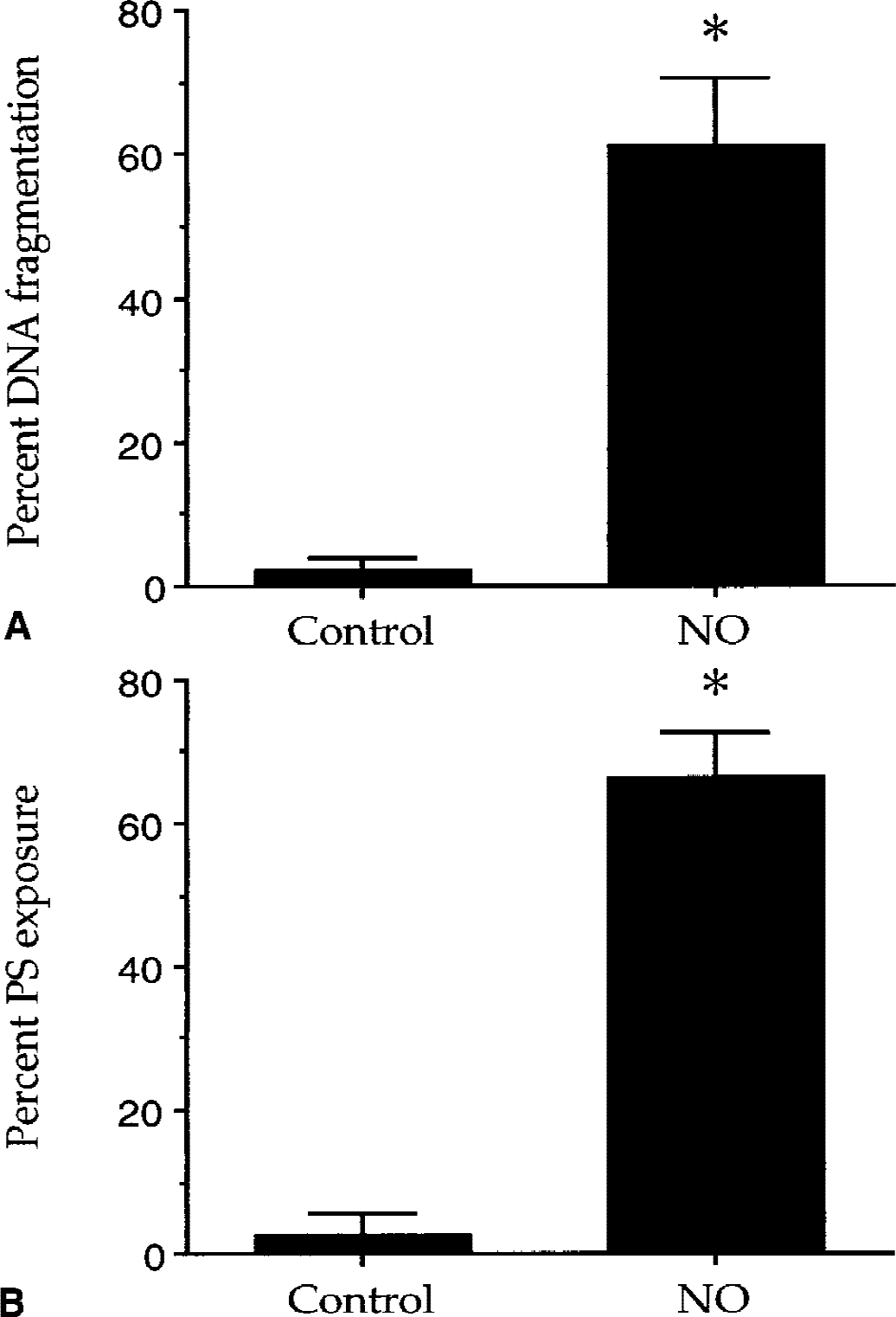
Nitric oxide (NO) induces DNA fragmentation and membrane phosphatidylserine (PS) exposure in endothelial cells (ECs). Cultures of ECs were treated with the NO donors (NOC-9: 1.0 mmol/L or SNP: 1.0 mmol/L). DNA fragmentation
To determine the ability of NO to induce membrane PS exposure in ECs, the authors quantitatively determined the redistribution of membrane PS residues with the use of annexin V labeling. Figure 3B illustrates that NO exposure (1.0 mmol/L NOC-9 or 1.0 mmol/L SNP) significantly increased membrane PS exposure from 3% ± 3% in untreated controls to 66% ± 6% over a 24-hour period. The concentration of NO donors used in these experiments was based on the observation that NO (1.0 mmol/L) decreases EC survival approximately 60% to 70% over a 24-hour period (Fig. 2).
Nitric oxide-induced endothelial cell injury requires the induction of caspase 1 and caspase 3-like activities
Because activation of caspase 1 and caspase 3 have been shown to participate in the initiation of neuronal PCD (Armstrong et al., 1997; Rimon et al., 1997; Lin et al., 2000), the authors next investigated the involvement of these caspases in NO-induced EC injury. Caspase 1-and caspase 3-like activities were assessed by measuring the cleavage of colorimetric substrates Ac-YVAD-pNA and Ac-DEVD-pNA after NO exposure. Because the ability of the NO donors (NOC-9 and SNP) at the concentration of 1.0 mmol/L can significantly induce EC death over a 24-hour period, caspase 1 and caspase 3-like activities were measured at 2, 4, 6, 12, and 24 hours after the application of 1.0 mmol/L of each NO donor.
As shown in Fig. 4A, the baseline of caspase 1-like activity in untreated control was 0.13 ± 0.03 μmol/min/g protein. The induction of caspase 1-like activity occurred rapidly within 2 hours to 0.26 ± 0.04 μmol/min/g after NO exposure. Caspase 1-like activity continually increased over a 24-hour period to a maximum level of 0.58 ± 0.14 μmol/min/g at 12 hours after NO application. In a similar manner, a significant increase in caspase 3-like activity was seen in cultures after 4 hours (0.28 ± 0.04 μmol/min/g), 6 hours (0.53 ± 0.07 μmol/min/g), 12 hours (0.70 ± 0.09 μmol/min/g), and 24 hours (0.30 ± 0.04 μmol/min/g) after NO exposure when compared with untreated controls (0.16 ± 0.02 μmol/min/g) (Fig. 4B).
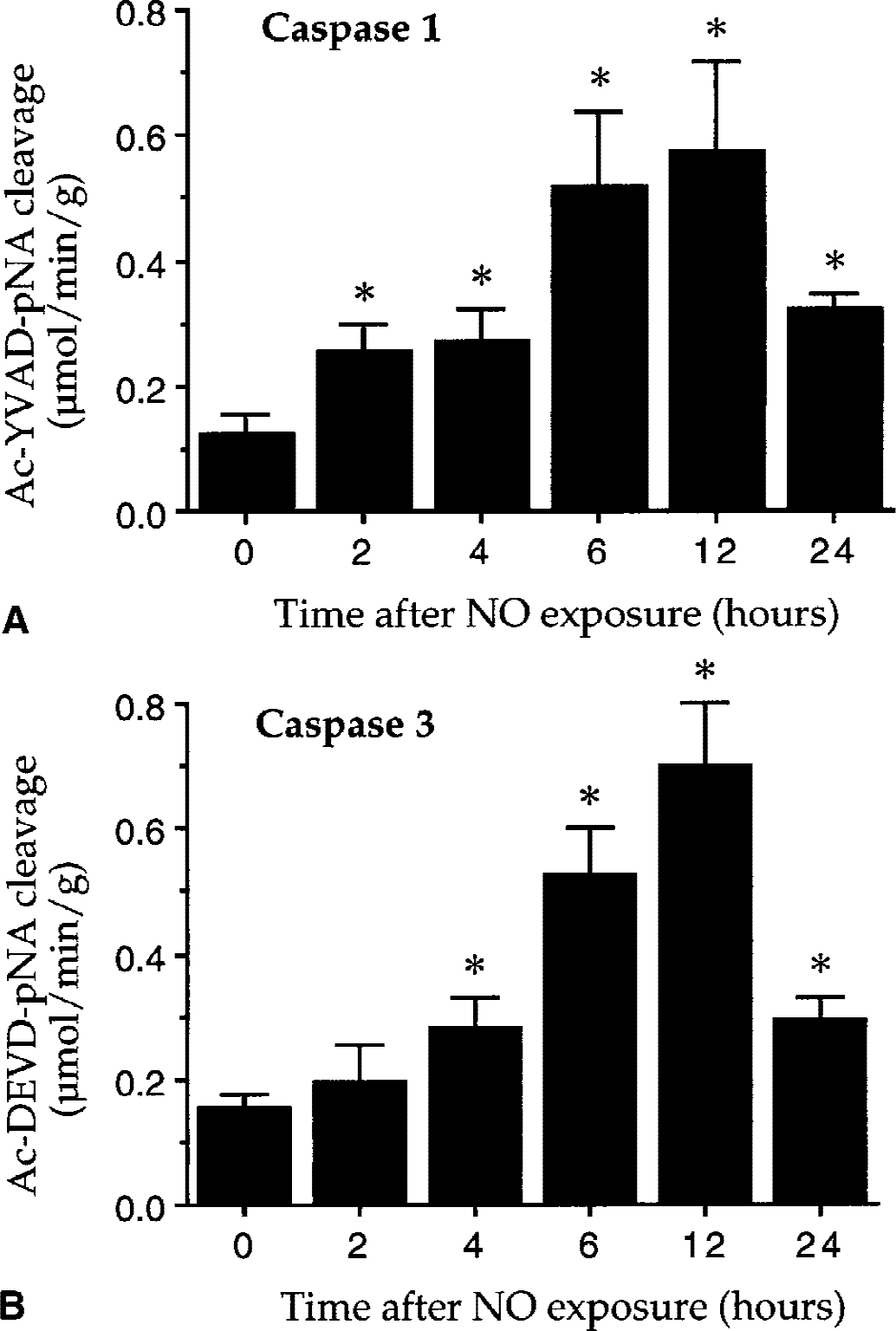
Nitric oxide (NO) enhances caspase 1- and caspase 3-like activities in endothelial cells (ECs). To investigate whether NO-induced EC injury is linked to the alteration of caspase activity, caspase 1- and caspase 3-like activities were determined by measuring the cleavage of colorimetric substrates after NO exposure.
To further address whether the induction of caspase 1-and caspase 3-like activities was required for NO-induced EC injury, the authors examined the effects of inhibition of caspase 1- and caspase 3-like activities on NO-induced endothelial DNA degradation with the use of the irreversible, cell permeable inhibitors YVAD (caspase 1 inhibitor) and DEVD (caspase 3 inhibitor). Caspase inhibitors were added directly to the culture medium 1 hour before NO exposure and DNA fragmentation was determined 24 hours after NO treatment. As shown in Fig. 5, ECs exposed to NO (NOC-9 (1.0 mmol/L) or SNP (1.0 mmol/L)) resulted in DNA fragmentation from approximately 2% in untreated control to 62% over a 24-hour period. Pretreatment of ECs with 10 μmol/L, 50 μmol/L, and 85 μmol/L of the caspase 1 inhibitor YVAD significantly decreased NO-induced DNA fragmentation to approximately 30%, 24%, and 20%, respectively (Fig. 5A). In addition, application of 10 μmol/L, 50 μmol/L, and 85 μmol/L of a caspase 3 inhibitor DEVD 1 hour before NO exposure significantly inhibited NO-induced DNA fragmentation to approximately 20%, 13%, and 10%, respectively (Fig. 5B). Treatment with each inhibitor alone up to a concentration of 85 μmol/L was not toxic.
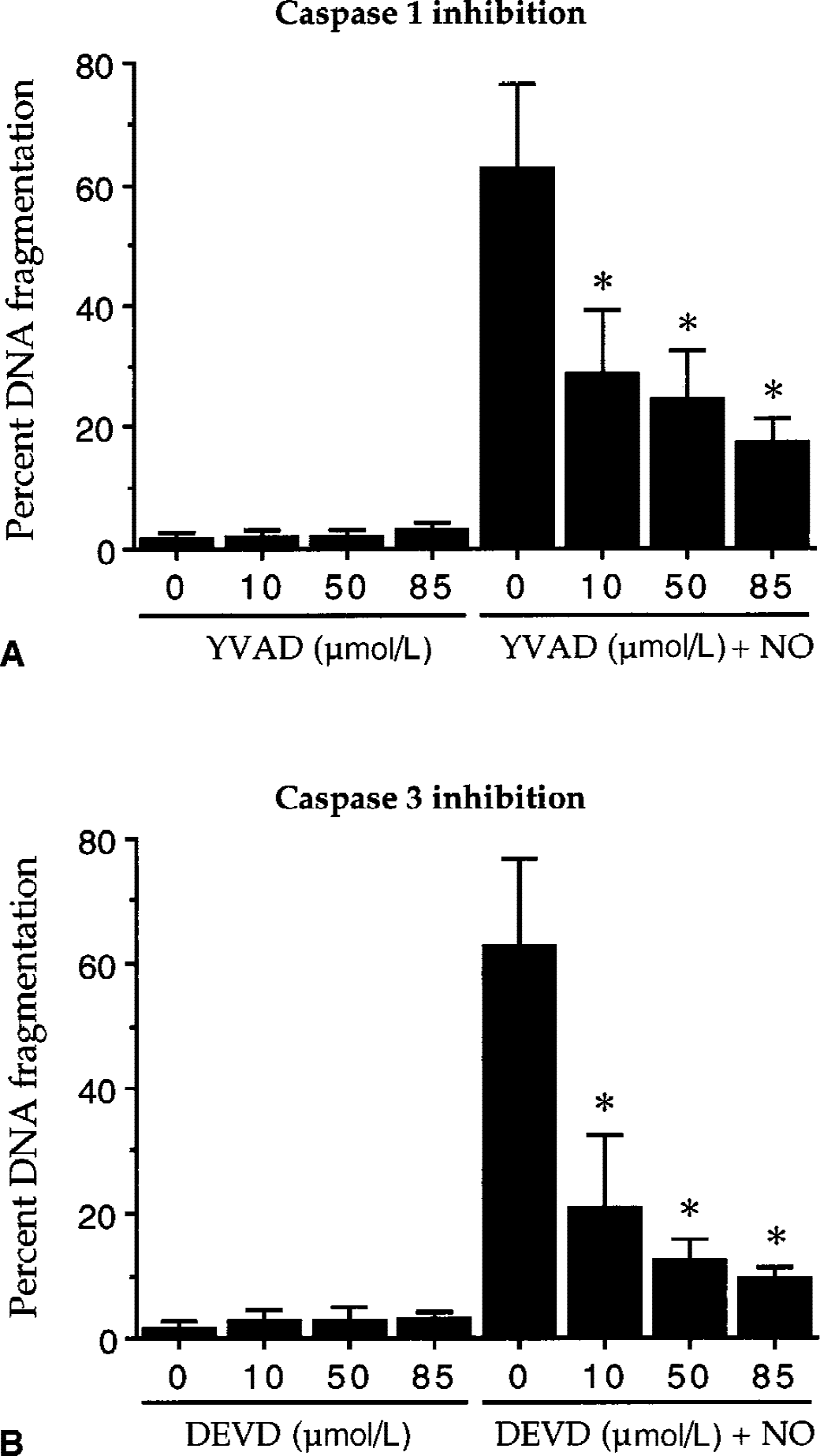
Nitric oxide (NO)-induced DNA degradation requires the induction of caspase 1- and caspase 3-like activities. The ability of caspase 1 and caspase 3 to yield NO-induced DNA fragmentation was examined by TUNEL staining with the use of irreversible and cell permeable caspase inhibitors.
Activation of the mGluR system protects endothelial cells from anoxia and nitric oxide-induced toxicity
To examine the involvement of the mGluR system in ECs during anoxia and NO toxicity, activation of the mGluR system was performed with the use of synthetic pharmacologic ligands DHPG, APDC, and L-AP4. These pharmacologic agonists specifically activate group I, group II, and group III mGluR subtypes, respectively. Each ligand was added directly to the culture medium 1 hour before anoxia or NO exposure (NOC-9: 1.0 mmol/L or SNP: 1.0 mmol/L). A dose response study was performed for each mGluR ligand based on prior dose response studies with neuronal populations (Maiese et al., 1996; Vincent et al., 1997, 1999; Maiese and Vincent, 1999). The concentration of the NO donors (1.0 mmol/L) used was determined on each donor's ability to significantly decrease EC viability during a 24-hour period (Fig. 2).
In Fig. 6, the authors quantitated the ability of mGluR activation to prevent anoxia and NO-induced toxicity. Anoxia or NO exposure (NOC-9: 1.0 mmol/L) or SNP: 1.0 mmol/L) decreased EC survival from approximately 97% to 40% over a 24-hour period. In contrast, activation of the mGluR groups I, II, and III significantly increased EC survival to approximately 83% (DHPG), 72% (APDC), and 74% (L-AP4) during anoxia, and to approximately 66% (DHPG), 69% (APDC), and 70% (L-AP4) during NO toxicity. Antagonism of the mGluR system with AIDA or MPEP did not increase EC survival in the presence of anoxia or NO. Application of each mGluR agonist or antagonist alone at each concentration examined did not alter cell viability over a 24-hour period. In addition, administration of AIDA (750 μmol/L) or MPEP (100 μmol/L) in combination with DHPG (750 μmol/L) during NO exposure reversed protection by DHPG (data not shown). Representative fields of TUNEL-positive labeling in ECs are shown in Fig. 7A.
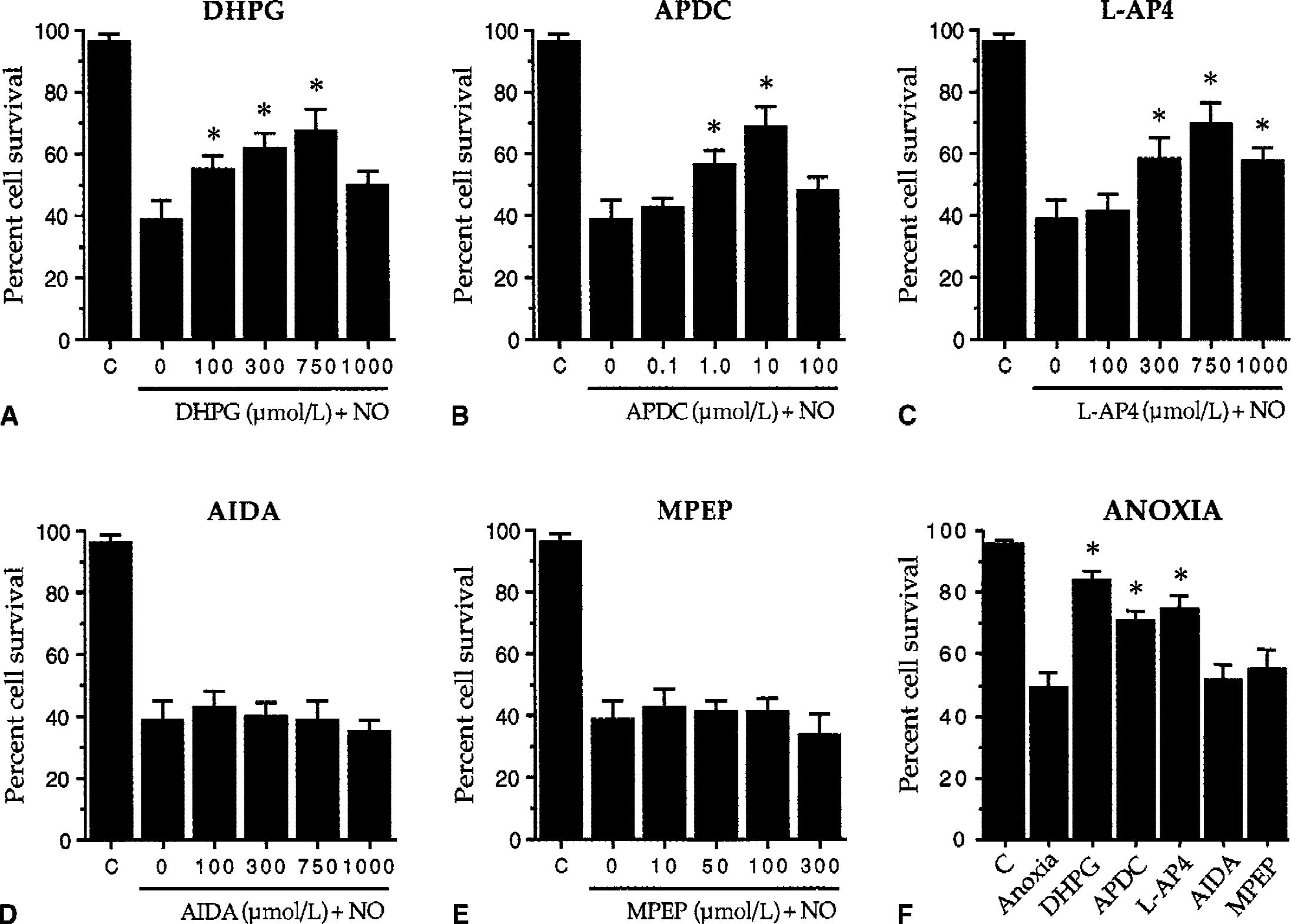
Activation of the metabotropic glutamate receptor (mGluR) system protects endothelial cells (ECs) against anoxia and nitric oxide (NO) toxicity. To investigate the role of each group of the mGluR system during NO toxicity (NOC-9: 1.0 mmol/L or SNP: 1.0 mmol/L), EC viability was assessed in cultures with and without pretreatment with the mGluR ligands. Cell survival was determined 24 hours after NO exposure using the trypan blue dye exclusion method. Pretreatment with the mGluR agonists DHPG
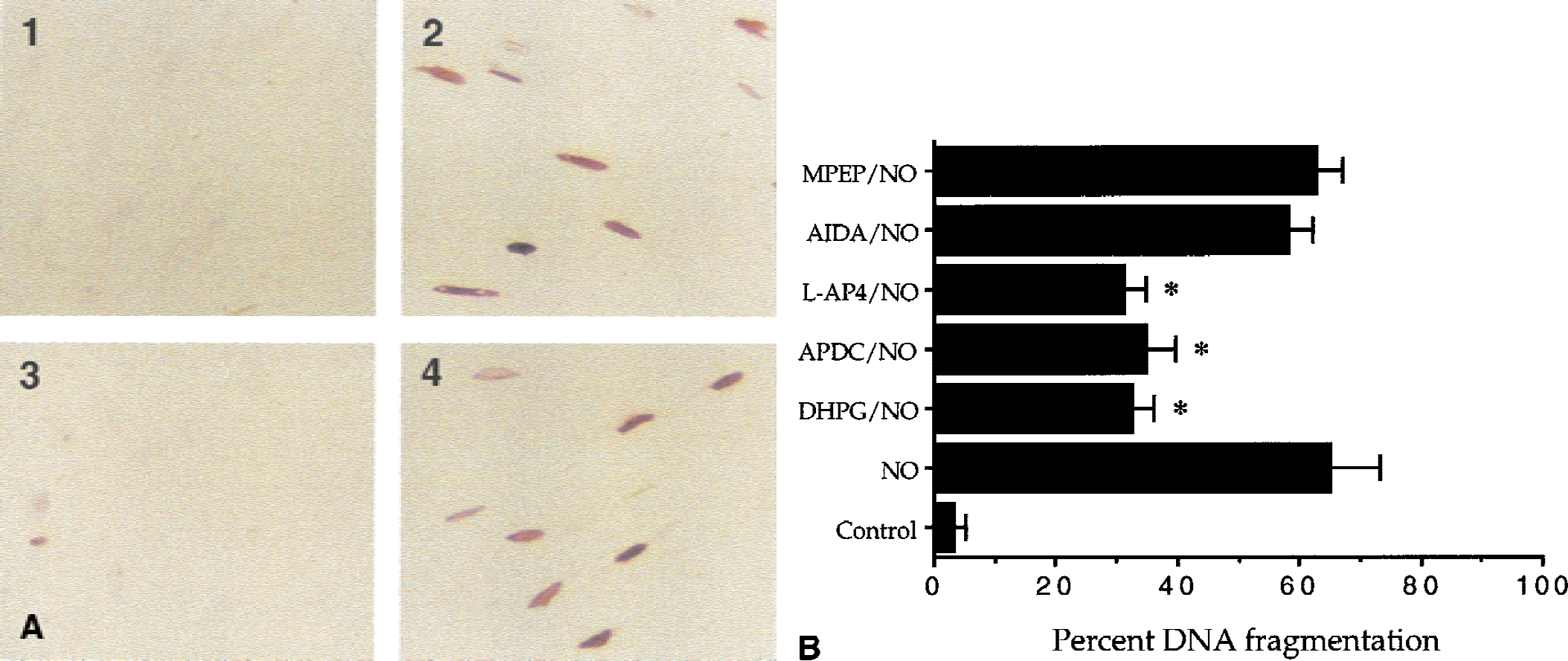
Activation of the metabotropic glutamate receptor (mGluR) subtypes prevents nitric oxide (NO)-induced DNA fragmentation.
After a 24-hour period of NO exposure (NOC-9, 1.0 mmol/L), several TUNEL-positive cells are seen in cultures exposed to NO only (Fig. 7A(2)) as compared with untreated control cultures (Fig. 7A(1)). Pretreatment of ECs 1 hour before NO with the agonist DHPG (750 μmol/L, Fig. 7A(3)) decreased the number of TUNEL-positive cells during NO toxicity. Antagonism of the mGluR system with AIDA (750 μmol/L, Fig. 7A(4)) did not prevent NO-induced DNA fragmentation.
Figure 7B represents a quantitation of the mGluR system to protect against NO-induced DNA fragmentation in ECs. Activation of each group of the mGluR system significantly decreased NO-induced DNA fragmentation from approximately 65% (NO only) to 32% (DHPG), 35% (APDC), and 32% (L-AP4). Antagonism of the mGluR system with AIDA or MPEP did not prevent DNA fragmentation after NO exposure.
Activation of the mGluR system prevents the induction and the progression of nitric oxide-induced membrane phosphatidylserine exposure
Membrane PS residue exposure usually precedes DNA degradation and is believed to signal the onset and progression of PCD (Rimon et al., 1997; Maiese et al., 2000; Maiese and Vincent, 2000a, b). As a result, the authors developed a reversible assay using annexin V labeling to detect membrane PS residue exposure over time in living cells (Vincent and Maiese, 1999a). To investigate the ability of the mGluR system to modulate the early stages of membrane PS exposure during NO toxicity, ECs were pretreated with DHPG (750 μmol/L) 1 hour before NO exposure (NOC-9, 1.0 mmol/L) and membrane PS exposure was assessed 7 hours after NO treatment. As shown in Fig. 8, several ECs are positive for membrane PS exposure after NO administration (Fig. 8B). In contrast, pretreatment with the group I mGluR agonist DHPG prevented NO-induced membrane PS exposure in ECs (Fig. 8D).
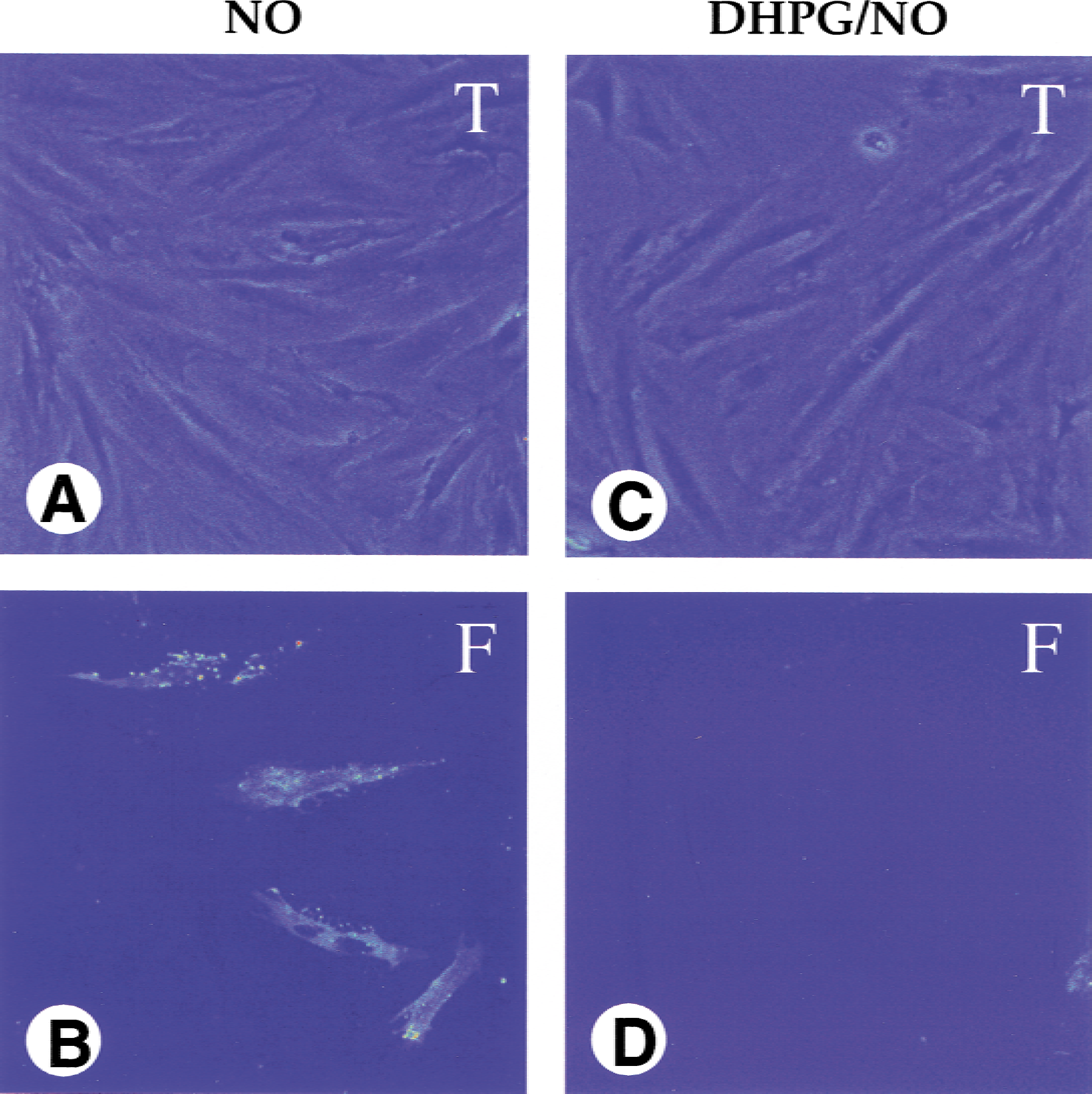
Activation of group I metabotropic glutamate receptor (mGluR) subtypes prevents nitric oxide (NO)-induced membrane phosphatidylserine (PS) exposure. Representative fields of endothelial cells (ECs) illustrate the effect of activation of group I mGluR on NO-induced membrane PS exposure in EC cultures. Annexin V staining was performed 7 hours after NO exposure (NOC-9: 1.0 mmol/L). The same ECs were imaged by transmitted light (T) and fluorescent light (F) using 490-nm excitation and 585-nm emission wavelengths to locate the annexin V-phycoerythrin label. NO-induced membrane PS exposure was seen in the cell culture exposed to NO only
The authors next examined the time course of membrane PS exposure in ECs in the presence or absence of the mGluR ligands during NO toxicity. A progressive increase in the percent of annexin V labeling was observed over a 24-hour period during NO toxicity alone (Fig. 9). A significant increase in the proportion of membrane PS exposure was initially observed in cultures 3 hours after NO exposure (14% ± 5%) when compared with untreated control cultures (3% ± 2%). The proportion of ECs with membrane PS exposure continued to increase significantly at 5 hours (19% ± 4%), 7 hours (31% ± 5%), 12 hours (51% ± 5%), and 24 hours (71% ± 6%) after NO exposure. Activation of the group I mGluR system with DHPG (750 μmol/L, 1 hour before NO) significantly inhibited NO-induced externalization of membrane PS residues to 3% ± 2%, 5% ± 3%, 7% ± 3%, 14% ± 3%, and 20% ± 3% at 3, 5, 7, 12, and 24 hours after NO exposure. At these time points, significant reduction of NO-induced membrane PS exposure was also seen during activation of group II and group III mGluRs with APDC and L-AP4, respectively. Antagonism of the mGluR system with AIDA or MPEP did not prevent membrane PS exposure after NO administration. A slight increase in membrane PS exposure was observed in untreated control cultures from 3% ± 2% to 5% ± 3% over a 24-hour period. This degree of membrane PS exposure in untreated cultures represents mild mechanical induction of membrane PS exposure during the staining and washing procedures (Vincent and Maiese, 1999a).
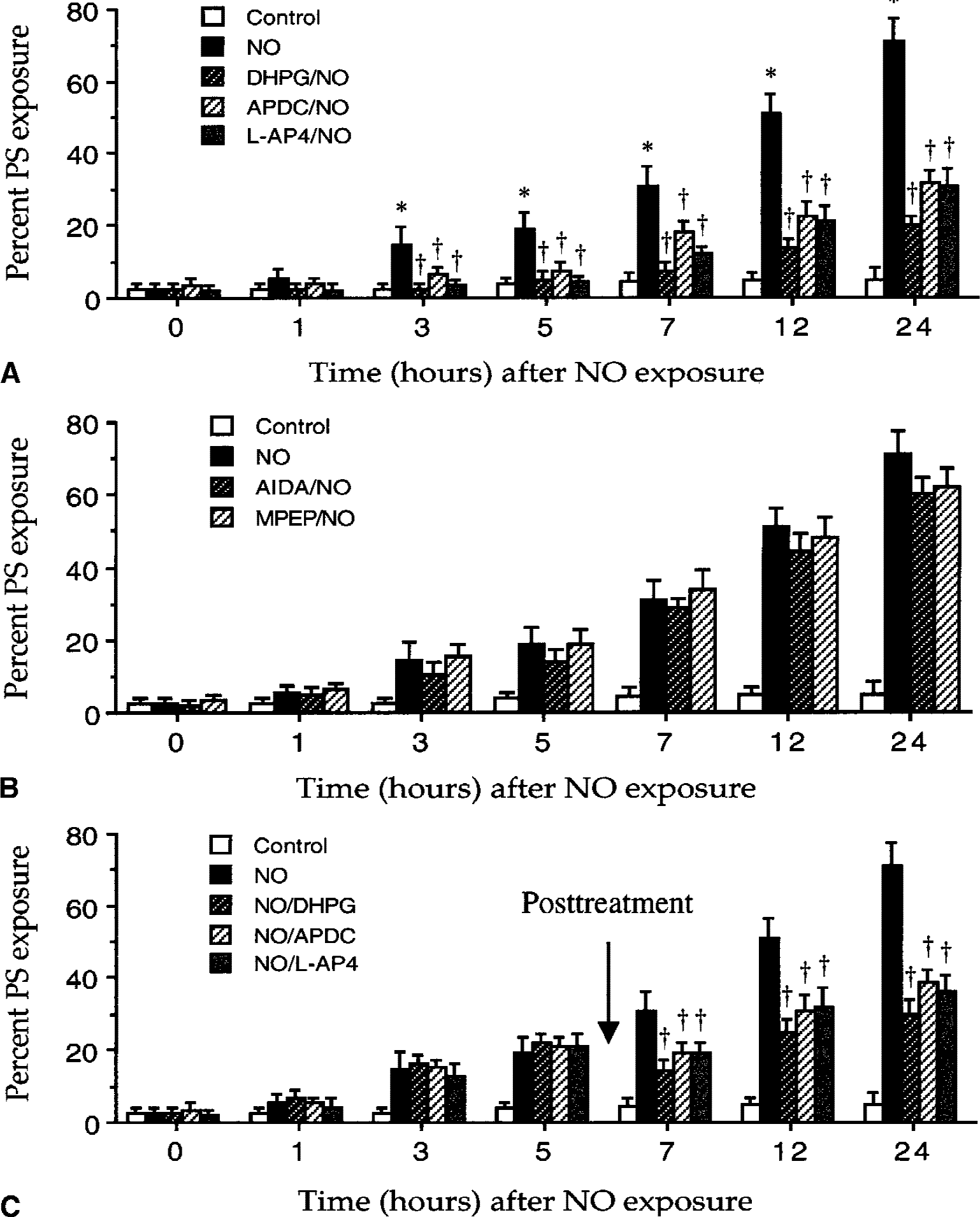
Activation of the metabotropic glutamate receptor (mGluR) system inhibits the induction and the progression of membrane phosphatidylserine (PS) exposure during pretreatment and posttreatment.
Activation of the mGluR system down-regulates caspase 1- and caspase 3-like activities during nitric oxide exposure
Induction of PCD in ECs is dependent upon, at least in part, the generation of caspase 1- and caspase 3-like activities (Figs. 4 and 5). Because activation of the mGluR system can increase EC viability, decrease DNA fragmentation, and inhibit membrane PS exposure in the presence of NO, the authors further examined whether protection through the mGluR system against NO toxicity is also dependent upon the modulation of caspase 1-and caspase 3-like activities. During agonism and antagonism of the mGluR system, the authors assessed caspase 1- and caspase 3-like activities 12 hours after NO exposure (NOC-9: 1.0 mmol/L or SNP: 1.0 mmol/L). The 12-hour time point was selected because this represents maximum caspase 1- and caspase 3-like activities after NO application (Fig. 4). Activation of group I, group II, and group III of the mGluR subtypes significantly prevented the induction of both caspase 1- and caspase 3-like activities after NO exposure (Fig. 10A and 10B). For example, administration of DHPG (750 μmol/L) decreased caspase 1-like activity from 0.58 ± 0.20 μmol/min/g (NO only) to 0.18 ± 0.04 μmol/min/g (DHPG/NO) and decreased caspase 3-like activity from 0.65 ± 0.18 μmol/min/g (NO only) to 0.27 ± 0.02 μmol/min/g (DHPG/NO). Antagonism of the mGluR system with AIDA (750 μmol/L) or MPEP (100 μmol/L) did not significantly alter caspase 1 and caspase 3-like activities when compared with ECs treated with NO only (Fig. 10A and 10B).
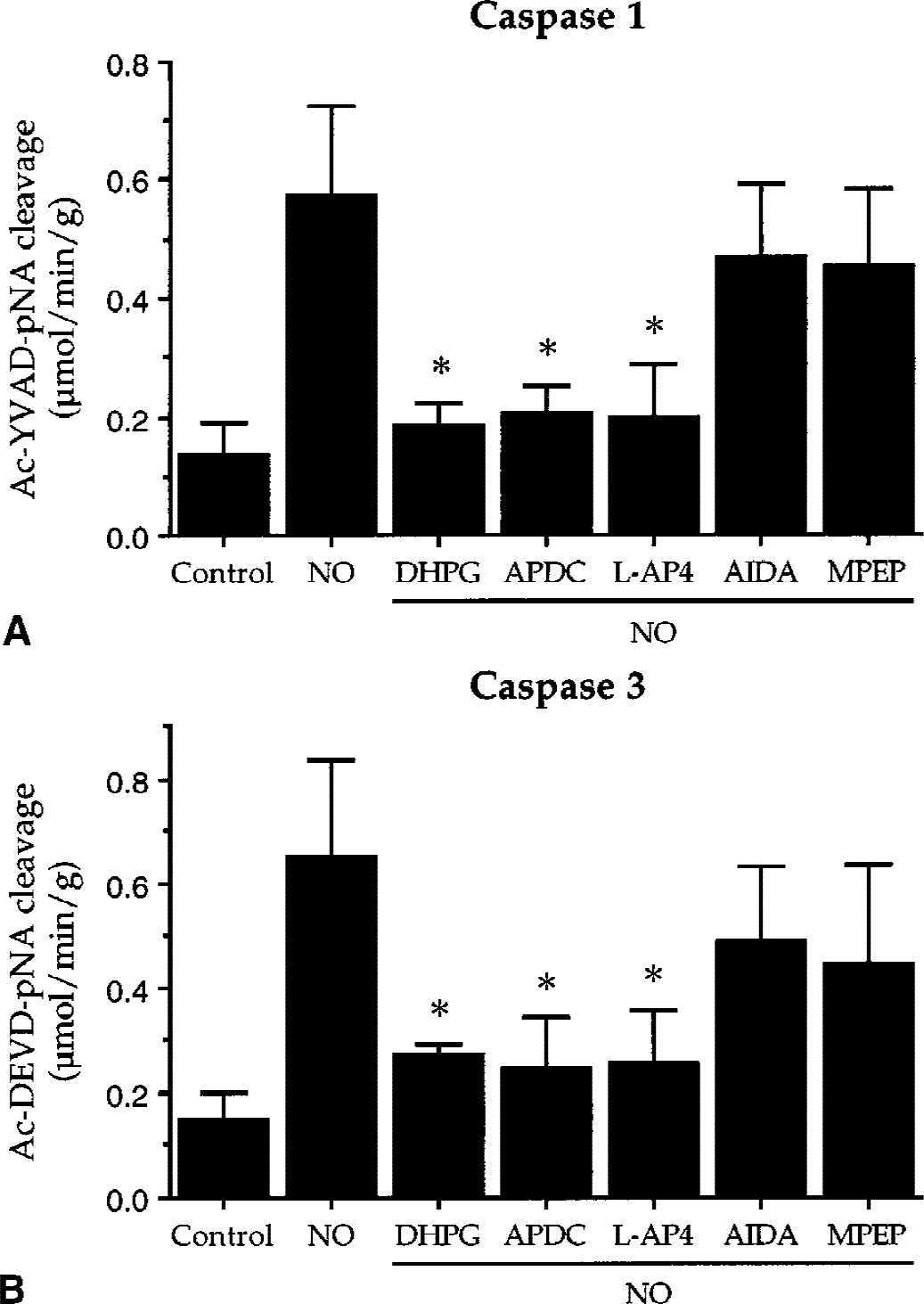
Activation of the metabotropic glutamate receptor (mGluR) system attenuates the induction of caspase 1- and caspase 3-like activities after nitric oxide (NO) exposure. The mGluR agonists, DHPG (750 μmol/L), APDC (10 μmol/L), L-AP4 (750 μmol/L), and the mGluR antagonists, AIDA (750 μmol/L) and MPEP (100 μmol/L), were applied 1 hour before NO treatment (NOC-9: 1.0 mmol/L or SNP: 1.0 mmol/L).
DISCUSSION
The EC lining of the cerebral vascular system plays an important role in organ physiology and pathology of a variety of diseases. In particular, preserving the integrity of the endothelium is crucial for the maintenance of an antithrombotic state and the homeostasis of the central nervous system. Because vascular ECs are primary cellular targets for a variety of insults such as ischemic injury, a clear understanding of the mechanisms that mediate free radical endothelial injury may contribute to the development of therapeutic regiments for cardiovascular and neurodegenerative disorders.
Nitric oxide is known to play a vital role in diverse physiologic responses such as blood vessel relaxation, platelet adhesion, and immune responses (Lopez-Farre et al., 1998). Yet, production of NO after pathologic insults may be cytotoxic. For example, NO can become a toxic mediator during ischemic-reperfusion injury in the heart (Liu et al., 1997), lung (Ischiropoulos et al., 1995), and brain (Togashi et al., 1998). The dual role of NO generation in mediating both protection and injury in a disease state was exemplified through studies using iNOS knockout mice. Mice devoid of iNOS production demonstrated enhanced susceptibility to the growth of Listeria monocytogenes, but became tolerant to lipopolysaccharide-induced injury (MacMicking et al., 1995; Wei et al., 1995). Thus, the role of NO in pathologic conditions is complex and may depend upon both the stimulus presentation and organ status.
The current results demonstrate that inhibition of iNOS activity significantly increases EC viability during anoxia, suggesting that NO generation through NOS is cytotoxic to rat cerebrovascular ECs. In addition, inhibition of all three isoforms of NOS activity by L-NMMA also significantly increased EC survival after anoxic exposure, further supporting the observation that NO is detrimental to ECs in the authors' culture systems. Interestingly, no increase in EC viability was noted during inhibition of all three forms of NOS when compared with the inhibition of iNOS alone. This may suggest that anoxic injury in ECs is primarily mediated by the induction of iNOS activity.
Because NO is a downstream mediator of anoxic injury in ECs, the authors next evaluated the ability of exogenous NO to alter EC survival. Experimental models have illustrated that NO can be a trigger for the induction of neuronal PCD in a variety of cell types, such as cortical neurons and hippocampal neurons (Brune et al., 1999; Vincent and Maiese, 1999b). The current work illustrates that cytotoxicity of NO in EC cultures occurs with NO generator concentrations greater than 0.75 mmol/L. In addition, the principal pathway that leads to EC injury is through PCD.
Exposure of NO at elevated concentrations not only results in the loss of plasma membrane asymmetry leading to membrane PS exposure but also induces genomic DNA degradation.
Induction of cysteine protease activity is believed to be a central component in mediating cellular injury (Enari et al., 1998), cytotoxic insults (Jordan et al., 1997), and chronic degenerative disorders (Friedlander et al., 1997). In particular, induction of caspase 1 and caspase 3-like activities can lead to neuronal PCD (Rimon et al., 1997; Maiese and Vincent, 1999; Vincent and Maiese, 2000). In this study, NO exposure significantly increased caspase 1 and caspase 3-like activities in ECs. The induction of caspase 1 and caspase 3-like activities peaked rapidly within 12 hours after NO treatment. This increase in cysteine protease activity represents at least one mediator of NO-induced EC injury, because treatment with the caspase 1 inhibitor YVAD and the caspase 3 inhibitor DEVD prevented PCD in ECs. These results demonstrate that NO-induced PCD in ECs requires, at least in part, the induction of caspase 1- and caspase 3-like activities.
Activation of the mGluR system has been demonstrated to be neuroprotective against a number of insults. For example, group I mGluRs are linked to the activation of phospholipase C and may provide important pharmacologic therapeutic targets for a variety of disorders such as pain, epilepsy, ischemia, and chronic neurodegenerative diseases (Maiese et al., 1996; Bordi and Ugolini, 1999; Vincent et al., 1999). Group II mGluRs have been shown to be neuroprotective against toxicity induced by N-methyl-d-aspartic acid (NMDA), kainic acid, and staurosporine (Vincent et al., 1997; Klodzinska et al., 1999; Maiese et al., 2000). Group III mGluRs are neuroprotective against excitotoxicity, NO toxcity, and can serve as experimental anticonvulsants (Suzuki et al., 1999; Vincent et al., 1999). Thus, the mGluR system has demonstrated efficacy in preventing neuronal injury and remains under continued investigation. In contrast, the role of the mGluR system in cerebrovascular ECs is without definition. In the current study, the authors illustrate that the mGluR system serves as a vital protectant against EC injury. Activation of each group of the mGluR system offers cytoprotection against anoxia and NO-induced endothelial injury. Activation of group I, group II, and group III mGluRs significantly increased EC survival in the presence of anoxia or NO. Yet, inhibition of the mGluR system did not prevent anoxic or NO-induced EC injury. These results illustrate a significant cytoprotective effect of the mGluR system in ECs. To the authors' knowledge, this is the first study to provide direct evidence for the role of mGluRs in cerebrovascular ECs during anoxia and NO-induced injury.
The demonstration that the mGluR system can provide protection to ECs is novel for several reasons. First, cytoprotection by the mGluR system in ECs occurs at two distinct levels during PCD. Programed cell death is an active process that is composed of two independent components that consist of genomic DNA degradation and membrane PS exposure (Vincent and Maiese, 1999a; Maiese and Vincent, 2000a, b). Redistribution of membrane PS residues from the inside of plasma membrane to the cell surface is an early marker for PCD and precedes genomic DNA degradation (Rimon et al., 1997). Protection against PCD during activation of the mGluR system is both rapid and broad in nature by addressing the separate components of PCD.
Agonism of each group of the mGluR system prevented the early exposure of membrane PS residues and also inhibited the later stages of PCD that involve the destruction of genomic DNA.
In addition, posttreatment paradigms illustrate a “window of opportunity” to prevent the further progression of membrane PS residue exposure once an injury in ECs has been initiated. Second, the mGluR system provides both immediate and long-term cytoprotection. Immediate protection is afforded through the maintenance of intact genomic DNA. Long-term protection results through the inhibition of membrane PS residue exposure which can allow cells to be recognized by phagocytes for subsequent destruction (Savill, 1997). A third unique aspect of the protective ability of the mGluR system involves maintenance of the normal anticoagulant state of ECs.
Unperturbed ECs prevent both the initiation and propagation of the coagulation process (Martin et al., 1995). Yet, exposure of membrane PS residues during injury can lead to the loss of anticoagulant membrane components in ECs (Bombeli et al., 1997). As a result, preserving membrane PS asymmetry through activation of mGluR subtypes may contribute to the maintenance of ECs anticoagulant state and inhibition of atherosclerosis.
The subsequent downstream pathways that mediate the cytoprotective effects of the mGluR system against NO-induced PCD appear to be closely linked to the modulation of cysteine protease activity but will require further investigation. Similar to others, the authors have demonstrated that NO generation in neurons can elicit cysteine protease activity and directly stimulate caspase 1- and caspase 3-like activities (Brune et al., 1999; Maiese and Vincent, 1999; Lin et al., 2000). This signal transduction system of cysteine protease generation by NO also appears to be maintained in ECs per the current results. In addition, activation of the mGluR system mediates protection against PCD in neurons (Vincent et al., 1997, 1999) and now in ECs through the direct inhibition of both caspase 1- and caspase 3-like activities. Caspase 1 has been linked to the modulation of membrane PS residues through cytoskeletal proteins such as fodrin (Cryns et al., 1996) and caspase 3 can lead to the direct degradation of DNA through the enhancement of DNase activity (Enari et al., 1998). As a result, the mGluR system may maintain both genomic DNA integrity and membrane PS asymmetry through the inhibition of cysteine protease activity. Yet, additional pathways for the cytoprotective effects of the mGluR system in ECs should be addressed with future investigations. In neurons, protection by the mGluR system is also dependent upon the signal transduction pathways of protein kinase C and protein kinase A (Abdul-Ghani et al., 1996; Maiese et al., 1996), the modulation of intracellular calcium (Maiese et al., 1999), the regulation of intracellular pH (Vincent et al., 1999), and the down-regulation of endonuclease activity (Vincent et al., 1999). It is conceivable that several of these pathways may play a significant role by the mGluR system for the intact maintenance of the EC environment.
The ability of mGluR system to protect cerebrovascular ECs from NO-induced PCD may have important therapeutic implications for chronic neurodegenerative disorders given that the neuronal microenvironment is tightly regulated by the brain endothelium. In addition, prevention of NO-induced endothelial PCD by the mGluR system during acute vascular injury may provide significant protection against thrombotic and ischemic injury. Further investigation into the ability of the mGluR system to prevent anoxic and NO generated PCD in ECs may open new therapeutic foundations for the treatment of acute and chronic neurodegenerative disorders.