
Abstract
To study the importance of metallothionein-I and -II (MT-I+II) for brain inflammation and regeneration, the authors examined normal and MT-I+II knock-out (MT-KO) mice subjected to a cortical freeze injury. Normal mice showed profound neurodegeneration, inflammation, and gliosis around the injury, which was repaired by 20 days postlesion (dpl). However, in MT-KO mice the lesion-associated inflammation was still present as late as 90 dpl. Scanning electron microscopy demonstrated that the number of capillaries was lower, and ultrastructural preservation of the lesioned parenchyma was poorer in MT-KO mice, suggesting an altered angiogenesis. To gain insight into the mechanisms involved, a number of cytokines and growth factors were evaluated. The number of cells expressing the proinflammatory cytokines IL-1β, IL-6, and TNF-α was higher in MT-KO mice than in normal mice, which was confirmed by RNase protection analysis, whereas the number of cells expressing the growth factors bFGF, TGFβ1, VEGF, and NT-3 was lower. Increased expression of proinflammatory cytokines could be involved in the sustained recruitment of CD-14+ and CD-34+ inflammatory cells and their altered functions observed in MT-KO mice. Decreases in trophic factors bFGF, TGFβ1, and VEGF could mediate the decreased angiogenesis and regeneration observed in MT-KO mice after the freeze lesion. A role for MT-I+II in angiogenesis was also observed in transgenic mice expressing IL-6 under the control of the promoter of glial fibrillary acidic protein gene (GFAP-IL6 mice) because MT-I+II deficiency dramatically decreased the IL-6-induced angiogenesis of the GFAP-IL6 mice. In situ hybridization analysis indicated that the MT-III expression was not altered by MT-I+II deficiency. These results suggest that the MT-I+II isoforms have major regulatory functions in the brain inflammatory response to injury, especially in the angiogenesis process.
Metallothioneins are a family of cysteine-rich, ubiquitous intracellular proteins. In the central nervous system (CNS), MT-I+II are expressed coordinately and are increased in macrophages/microglia and astrocytes during various inflammatory and pathologic conditions, including human neurodegenerative disorders, experimentally induced brain injury, epileptic seizures, brain ischemia, astroglial cell death, metal exposure, stress, and myelin deficiency (Agullo et al., 1998; Dalton et al., 1995; Duguid et al., 1989; Gasull et al., 1994; Hidalgo et al., 1990,1997; Neal et al., 1996; Penkowa et al., 1999b,c; Sillevis Smitt et al., 1992; Vela et al., 1997; Zambenedetti et al., 1998).
The physiologic roles of MT-I+II are not fully elucidated however, data describing MT-I+II as tissue protective factors are accumulating (Aschner, 1996,1997; Aschner et al., 1997; Kondo et al., 1997; Lazo and Pitt, 1995; Lazo et al. 1995,1998; Liu et al., 1999; Penkowa et al., 1999a,b; Rossman et al., 1997; Schwarz et al., 1995; Van Lookeren Campagne et al., 1999). MT-I+II can protect against reactive oxygen species causing oxidative damage and stress, ionizing radiation, anti-cancer drugs, and interestingly, MT-I+II may prevent neuronal apoptosis (Aschner, 1998; Lazo and Pitt, 1995; Lazo et al., 1995,1998; Penkowa and Hidalgo, 2000; Penkowa et al., 1999a,2000; Pitt et al., 1997; Schwarz et al., 1995; Tamai et al., 1993; Thornalley and Vasak, 1985).
Furthermore, MT-I+II are putatively involved in regulation of monocyte adherence, invasion, and the respiratory burst (Leibbrandt et al., 1994). In vivo, MT-I+II are important for bone marrow myelo—monocyte survival and resistance after induction of systemic cytotoxicity (Penkowa et al., 1999b); and after brain injury in MT-KO mice, the number of recruited monocytes is significantly increased (Penkowa et al., 1999a). Despite increased levels of monocytes, CNS wound healing in MT-KO mice was dramatically impaired. Thus, MT-I+II may have an important role in both the inflammatory and regenerative responses. However, the mechanisms underlying the effects of MT-I+II deficiency are poorly known and better insights into MT pathophysiology are of significant scientific interest. In the current report the authors examined whether the absence of MT-I+II in vivo had a significant impact in the CNS response to damage by characterizing the angiogenic process, the maturity of infiltrating cells, the expression profiles of several cytokines and growth factors, and the oxidative stress status.
MATERIAL AND METHODS
Animals
Homozygous MT-I+II knock-out (MT-KO) mice generated as previously described (Masters et al., 1994) were purchased from Jackson Laboratories, Bar Harbor, ME, U.S.A. A colony has been established at the Autonomous University of Barcelona. MT-KO mice were routinely genotyped by Southern blot as previously described (Masters et al., 1994). MT-KO mice have no overt abnormalities, and animals live and breed normally in accordance with published studies (Masters et al., 1994). The MT-KO mice were raised and propagated on the 129/Sv genetic background, therefore mice from this strain were used as controls.
Construction and characterization of the glial fibrillary acidic protein-interleukin 6 (GFAP-IL6) transgenic mice has been described previously (Campbell et al., 1993). Briefly, an expression vector derived from the murine GFAP gene was used to target expression of IL-6 to astrocytes. To obtain GFAP-IL6 mice with genetic deficiency of MT-I+II, the authors initiated a series of crosses. First, heterozygous GFAP-IL6 mice were crossed with MT-KO mice. The offspring that were heterozygous for transgenic IL-6 and MT-I+II were identified by polymerase chain reaction and Southern blot (see Results), and crossed again with MT-KO mice. The offspring of this second cross were genotyped as above, and mice with the appropriate genetic background were selected and studied as described below.
Freeze lesion procedure
Normal and MT-KO adult mice were lesioned under tribromethanol anesthesia. The skull over the right fronto-parietal cortex was exposed, and a focal cryoinjury on the surface of the brain was produced with dry ice (–78°C) (Penkowa and Moos, 1995). The animals were housed in cages with free access to food and water. The handling of the animals was approved by the proper Committees of Animal Research and Ethics of Spain and Denmark.
The authors carried out several experiments in which control and MT-KO mice were killed at different times after the cryoinjury. Different procedures were followed for the preparation of the brains depending on the technique used, namely electron microscopy, histochemistry-immunohistochemistry, or in situ hibridization (see below). Normally, 4 to 6 mice per group were used in each experiment.
Electron microscopy
Unlesioned mice and lesioned mice at 7 and 14 days postlesion (dpl) were deeply anesthetized with Brietal and fixed by cardiac perfusion with saline (0.9% NaCl, 3 mL/L 5000 IU heparin) for 2 to 3 minutes, followed by 2% glutaraldehyde in 0.05 mol/L phosphate, buffer (pH 7.4). The brains were dissected and stored in the perfusion fixative for 4 days. Using a stereomicroscope the brain lesions were isolated.
Transmission electron microscopy
Specimens were postfixed in 1% OsO4 in 0.12 mol/L sodium cacodylate buffer (pH 7.4) for 2 hours, dehydrated in graded series of ethanol, transferred to propylene oxide and embedded in Epon according to standard procedures. Ultrathin sections were cut with a Reichert-Jung Ultracut E microtome (Reichert Division der Leica Aktiengesellschaft, Wien, Austria) and collected on one-hole copper grids with Formvar supporting membranes, stained with uranyl acetate and lead citrate and examined and photographed in a Philips CM 100 transmission electron microscope (Philips Electron Optics, Eindhoven, The Netherlands).
Scanning electron microscopy
The brain lesions and underlying undamaged brain parenchyme were divided and prepared for scanning electron microscopy examination by the osmium-thiocarbohydrazide (OTOTO) method (Malik and Wilson, 1975). The samples were rinsed in 0.15 mol/L sodium cacodylate buffer (pH 7.4) and were postfixed in 1% OsO4 in 0.12 mol/L sodium cacodylate buffer (pH 7.4) overnight. After a thorough rinse in distilled water for 1 hour, the samples were treated for 30 minutes with a saturated and filtered solution of thiocarbohydrazide in distilled water. After a second rinse in distilled water, the specimens were returned to 1% OsO4 in distilled water for 30 minutes. The above steps were repeated, and after a final rinse in distilled water the specimens were dehydrated in ethanol and critical point dried (Balzers CPD 030, Balzers, Fürstentum Liechtenstein) using CO2. Specimens were mounted on stubs with colloidal carbon as an adhesive and sputter-coated with chromium (Edvards XE200 Xenosput; Edvards, West Sussex, U.K.). Examination and photography were carried out in a Philips FEG 30 scanning electron microscope (Philips).
Histochemistry and immunohistochemistry
Mice were deeply anesthetized with Brietal and fixed by cardiac perfusion with saline (0.9% NaCl, 3 mL/L 5000 IU heparin) for 2 to 3 minutes followed by Zamboni's fixative (pH 7.4) for 5 to 10 minutes. For immunohistochemical examination, brains were dissected and postfixed for 2 to 4 hours, dehydrated in graded alcohols and xylol, and finally embedded in paraffin before being cut in 3-μm coronal sections. For heat-induced epitope retrieval, sections were boiled in citrate buffer (pH 6 or 9) in a microwave oven for 2 × 10 minutes, or preincubated in Digest-All-3 (code 00-3009; Zymed, South San Francisco, CA, U.S.A.) for 3 to 5 minutes at room temperature, or both, followed by incubation in 10% goat serum (In Vitro, Fredensborg, Denmark, code 04009-1B) or donkey serum (code BP 005.1; The Binding Site, Birmingham, U.K.) in TBS/Nonidet (TBS: 0.05 mol/L TRIS, pH 7.4, 0.15 mol/L NaCl) with 0.01% Nonidet P-40 (TBS/Nonidet) for 30 minutes at room temperature. Sections prepared for incubation with monoclonal mouse-derived antibodies were also incubated with Blocking Solutions A+B from HistoMouse-SP Kit to quench endogenous mouse IgG (code 95-9544; Zymed).
Histochemistry
Biotinylated tomato lectin from the Lycopersicon esculentum (code L9389; Sigma, Vallensbaek, Denmark) 1:500, was used as a marker for cells of the myelomonocytic cell lineages, such as microglia/macrophages, and as a marker for vessels. The lectin was developed using streptavidin-biotin-peroxidase complex (StreptABComplex/HRP; Dakopatts, Glostrup, Denmark; code K377) prepared at manufacturer's recommended dilutions and was performed for 30 minutes at room temperature. The reaction product was visualized using 0.015% H2O2 in 3,3′-diaminobenzidine/Tris buffered saline (DAB/TBS), with DAB as a chromogen. Hematoxylin-eosin stainings of brain sections were performed according to standard procedures.
Immunohistochemistry
Sections were incubated overnight with one of the following primary antibodies: Rabbit anti-MT-I+II (IgG) 1:500 (Gasull et al., 1994; Penkowa et al., 1997, Penkowa 1999a, b , c ); Polyclonal rabbit anti-cow GFAP (as a marker for astrocytes) diluted 1:250 (code Z 334; Dakopatts); Mouse antihuman CD-14 (IgG) 1:20 (marking macrophages) (code 18-0121; Zymed); Rat anti-mouse MOMA-1 (IgM) 1:20 (marking only spleen, lymph node, and blood monocytes) (Hybridomus, NL, code HD-212-85-OMA); Mouse antihuman CD-34 (IgG) 1:15 (marking myeloid and lymphoid hematopoietic progenitor cells and proliferating vascular endothelial cells) (code MS-363-A; Neomarkers, Fremont, CA, U.S.A.); Mouse antihuman IL-1β (IgG) 1:50 (code 5375-4329; Biogenesis, Kingston, NH, U.S.A.); Rat anti-mouse IL-6 (IgG) 1:50 (code MAS 584; Harlan Seralab, Sussex, U.K.); Rabbit anti-mouse TNF-α (IgG) 1:100 (code AMC 3012; Biosource, Camarillo, CA, U.S.A.); Rabbit antihuman bFGF (IgG) 1:100 (code sc-79; Santa Cruz, Santa Cruz, CA, U.S.A.); Rabbit antihuman TGF-β1 (IgG) 1:200 (code sc-146; Santa Cruz); Goat antihuman NT-3 (IgG) 1:20 (code AF-267-NA; RD Systems, Minneapolis, MN, U.S.A.); Mouse antihuman VEGF (IgG) 1:20 (code MAB 293; RD Systems, U.S.A.); polyclonal rabbit antihuman iNOS (inducible nitric-oxide synthase, which monitors oxidative stress) 1:100 (code 210-503-R050; Alexis Biochemicals, San Diego, CA, U.S.A.); polyclonal rabbit anti-NITT (a marker for peroxynitrite-induced nitration of tyrosine residues, which monitors oxidative stress) diluted 1:100 (code NITT 12-A; Alpha Diagnostic, San Antonio, TX, U.S.A.); polyclonal rabbit anti-MDA (a marker for malondialdehyde produced as a byproduct of fatty acid peroxidation, which monitors oxidative stress) diluted 1:100 (code MDA 11-S; Alpha Diagnostic).
The primary antibodies were detected using biotinylated anti-mouse IgG 1:200 (code B8774; Sigma), or biotinylated anti-rat IgM (μ chain specific) 1:50 (code 112-065-020; Jackson ImmunoResearch Laboratory, West Grove, PA, U.S.A.), or biotinylated anti-rat IgG 1:1000 (code RPN 1002; Amersham, Buckinghamshire, U.K.), or biotinylated anti-rabbit IgG 1:400 (code B3275; Sigma), or biotinylated anti-sheep/goat IgG 1:20 (code RPN 1025; Amersham, U.K.) followed by streptavidin-biotin-peroxidase complex. These secondary and tertiary steps in the immunoreaction were performed for 30 minutes at room temperature. Afterwards, sections were incubated with biotinylated tyramide and streptavidin-peroxidase complex (tyramide signal amplification, TSA indirect) (code NEL700A, NEN; Life Science Products, Boston, MA, U.S.A.), prepared according to manufacturer's recommendations. The immunoreaction was visualized using DAB as a chromogen.
To evaluate the extent of nonspecific binding of the antisera in the immunohistochemical experiments, 1:100-1:1000 of normal rabbit or mouse serum or just the preincubation agent (normal goat or donkey serum) was substituted for the primary antibody step described above. A further control for some cytokines and growth factors was to preabsorb the primary antibodies with their corresponding antigenic proteins. The authors used the following for this purpose: human IL-1β (code 201-LB-005; RD Systems, Abingdon, U.K.); mouse IL-6 (code 406-ML-005; RD Systems, U.K.); mouse TNF-α (code 410-MT-010; RD Systems, U.K.); human bFGF (code 234-FSE-025; RD Systems, U.K.); human TGFβ1 (code 240-B-002; RD Systems, U.K.); and human NT-3 (code 267-N3-005; RD Systems, U.K.); human VEGF (code 298-VS-005; RD Systems, U.K.). Results were considered significant only if these controls were negative.
Immunofluorescence
To study the spatiotemporal expression pattern of MT-I+II in relation to growth factors b-FGF, TGFβ1, and VEGF, the authors performed double immunofluorescence staining of monoclonal mouse anti-horse MT-I+II 1:50 (code M0639; Dakopatts) incubated simultaneously with either b-FGF or TGFβ1 or VEGF. These primary antibodies were incubated overnight at 4°C and were detected by using goat anti-mouse IgG linked with TexasRed (TXRD) 1:50 (code 1030-07; Southern Biotechnology, U.S.A.) and goat anti-rabbit IgG linked with fluorescein (FITC) 1:50 (code 4050-02; Southern Biotechnology, Birmingham, AL, U.S.A.).
Also, b-FGF and VEGF were incubated simultaneously and were detected as mentioned above. NT-3 and either mouse anti-porcine vimentin 1:50 (as a marker of astrocytes and mesenchymal cells including macrophages) (code M0725; Dakopatts) or mouse antihuman neurofilament 1:250 (code M762; Dakopatts) were incubated simultaneously overnight at 40°C and detected by using donkey anti-mouse IgG linked with TXRD 1:50 (code 715-075-150; Jackson ImmunoResearch) and donkey anti-goat/sheep IgG linked with FITC 1:60 (code AF360; Binding Site).
The sections were embedded in 20-μL Fluorescent mounting (code S3023; Dakopatts) and kept in darkness. To evaluate the extent of nonspecific binding of the antisera in the immunohistochemical experiments, the primary antibody was omitted. Results were considered significant only if these controls were negative.
RNase protection assay
RNase protection assay for the detection of cytokine RNA was performed as described previously (Hobbs et al., 1993; Campbell et al., 1994).
In situ hybridization
Mice were killed and their brains were removed quickly, frozen in liquid nitrogen, and stored at −80°C. Later, serial coronal sections (20-μm thick) were obtained with a cryostat and mounted on slides coated with poly(L)lysine in which an in situ hybridization analysis of the MT-I and MT-III isoforms was carried out as previously described (Carrasco et al., 1998).
For MT-I mRNA studies, the authors used the mouse cDNA, which was kindly provided by Dr. R.D. Palmiter, University of Washington, WA, U.S.A. For MT-III mRNA studies and to avoid crosshybridization with MT-I and MT-II mRNA, the authors used a specific DNA fragment of 153 bp that contained the coding region for the terminal 15 amino acids and the 3′ untranslated region until the poly G stretch of MT-III mRNA, which was generously provided by Dr. G.K. Andrews, Department of Biochemistry, Kansas City, Kansas, U.S.A. All sections to be compared were prepared simultaneously and exposed to the same autoradiographic film. MT-I or MT-III mRNA levels in the border of the lesion were determined semiquantitatively in three sections per animal by measuring the optical densities and the number of pixels in defined areas with a Leica Q 500 MC system. The MT-I and MT-III mRNA values shown are expressed in arbitrary units (number of pixels × optic density).
Some slides were coated with Hypercoat LM-1 emulsion (Amersham) according to the instructions of the manufacturer. The slides were exposed for 3 weeks at 4°C into a light-tight box, and then developed in D-19 (Kodak-Pathé, Paris, France). The slides were counterstained with hematoxylineosin for microscope observation.
Cell counts
Positively stained cells, defined as those cells with cytoplasmic or nuclear staining, or both, were counted from a 1-mm2 area of 3-μm-thick sections of cortex of unlesioned mice hemispheres, and from the lesioned mice hemispheres, both from the ipsilateral cortex (lesioned cortex side) and contralateral cortex (unlesioned cortex side), for statistical evaluation of the results. Counting from the lesioned hemispheres, an area was chosen at the borderzone of the deepest part of the lesion in all of the mice. Cell counts were performed in at least 3 mice per group and 2 sections within each brain. Counts were done in a blinded manner.
Statistical analysis
Results were evaluated by two-way analysis of variance, with strain and freeze lesion as main factors. Separate two-way analyses of variance were carried out for the lesioned and unlesioned hemispheres of the lesioned mice. When the interaction was significant it was interpreted as the consequence of a specific effect of the MT-I+II deficiency during the lesion. When only two groups were compared the Student's t-test was used.
RESULTS
Cryoinjury to the CNS elicits an inflammatory response
It is well known that cryoinjury to the CNS produces a vigorous inflammatory response, with recruitment of cytokine expressing bone marrow- and microglia-derived macrophages and reactive astrocytes (Ghirnikar et al., 1998; Hopkins and Rothwell, 1995). These responses were previously described chronologically from 1 to 20 dpl in the freeze lesion model used in this study (Penkowa et al., 1999a) in both normal and MT-KO mice. In contrast to normal mice, MT-KO mice showed a prolonged inflammatory response, and at 20 dpl no wound repair had occurred. In the current report the authors confirm and extend these previous results. Thus, MT-KO mice, as late as 90 dpl, had a prominent lesion filled with round or amoeboid monocytes/macrophages and reactive astrocytes, although the size of the lesion was certainly smaller than at 20 dpl (Fig. 1). The MT-KO mice used were from a colony grown in the authors' laboratory from mice purchased from Jackson Laboratories. These mice were routinely genotyped by Southern blot as previously described (Masters et al., 1994) and, as expected, did not show an abnormal phenotype in normal conditions. The results clearly indicate that the CNS response to damage is impaired in the MT-KO mice.
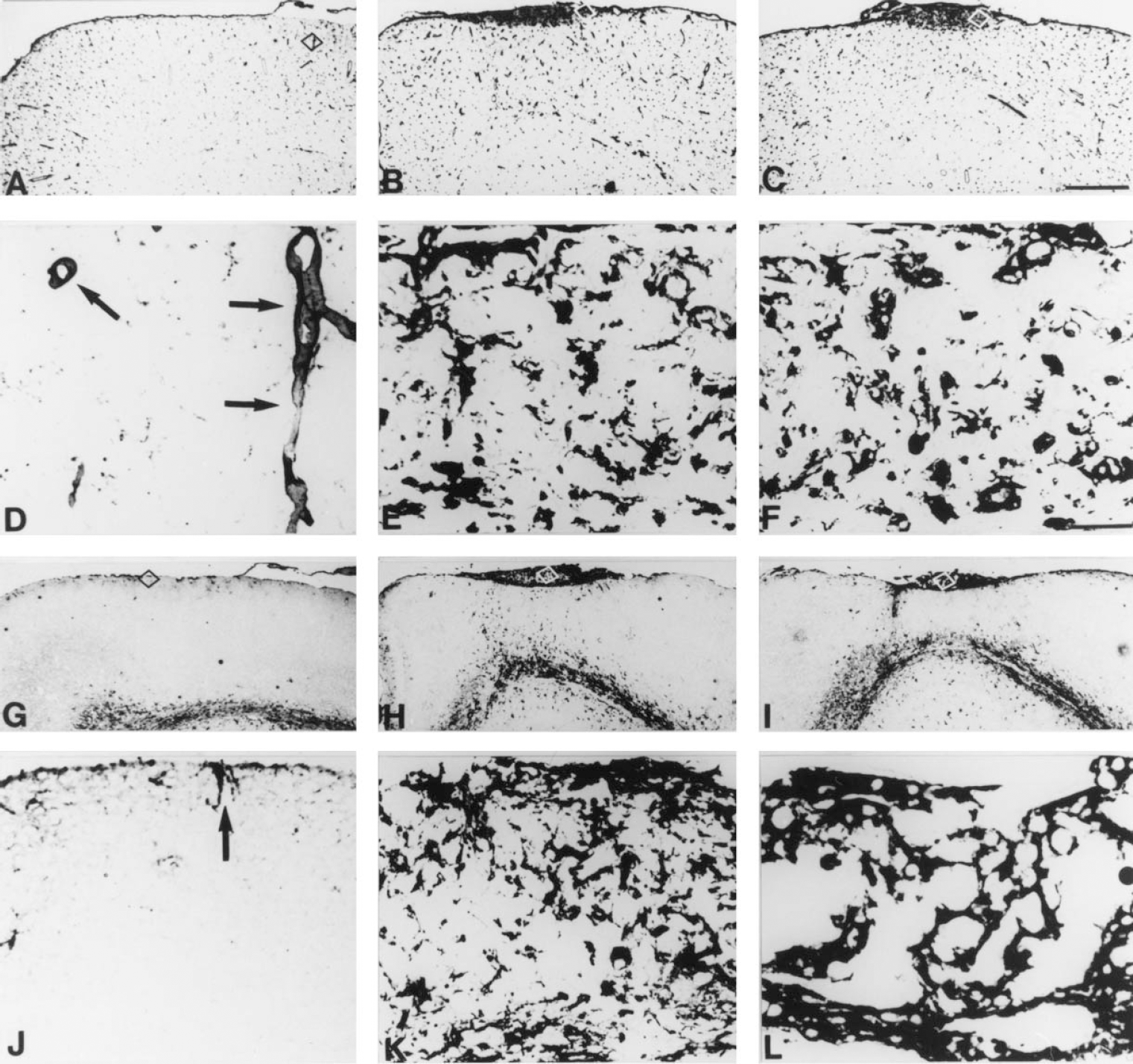
Lectin staining and glial fibrillary acidic protein (GFAP) immunostainings of normal and MT-KO mice at 60 and 90 days postlesion (dpl).
Decrease in capillaries and poor ultrastructural preservation of the lesioned area in MT-KO mice
Using scanning electron microscopy at low power magnification of the brain tissue the lesion appeared as a concave area, which was slightly more pronounced in MT-KO mice than in normal mice at 7 and 14 dpl (Fig. 2A and 2B). Examination of the vessels in the lesioned area revealed that the vasculature was more damaged in MT-KO mice than in normal mice at both 7 and 14 dpl (Fig. 2C and 2D). The normal mice displayed intact capillaries in a noncollapsed condition with a maintained and continuous vessel wall. In MT-KO mice the capillaries were fewer and the vessel walls were less preserved and discontinuous.
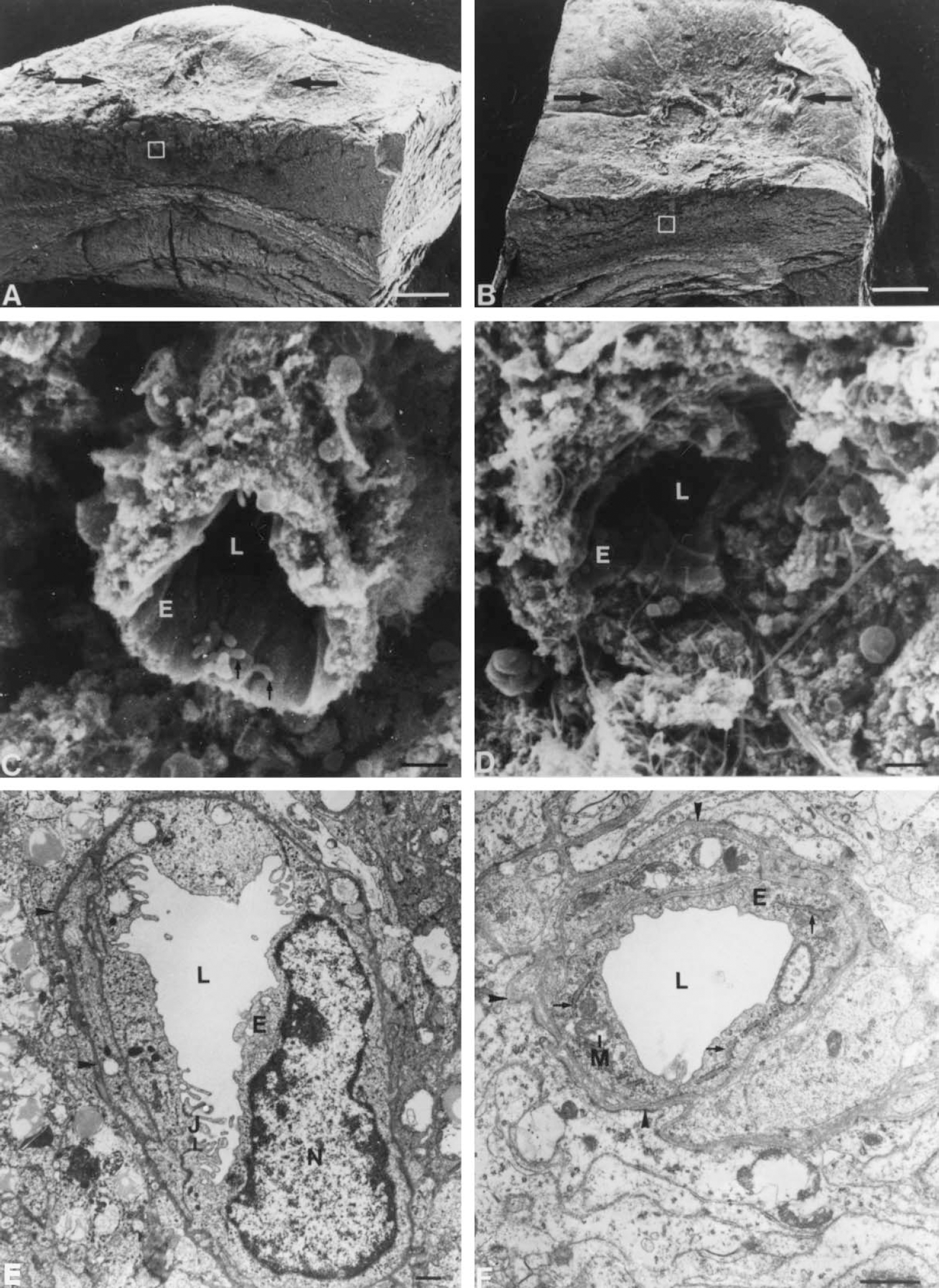
Angiogenesis and the general ultrastructure of the freeze lesioned cortex are abnormal in MT-KO mice. Scanning electron micrographs displaying the cortical surface and the transected surface of a freeze lesion in normal mice
Transmission electron microscopy of the newly sprouting capillaries within the lesion did not reveal any morphologic differences between vessels of normal and MT-KO mice (Fig. 2E and 2F). Endothelial cells were joined with junctional complexes and contained mitochondria and ribosome studded endoplasmic reticulum in all mice examined. The abluminal cell membranes of the endothelial cells were traced by a continuous basal lamina, and perivascular cells were seen equally in all mice. However, transmission electron microscopy confirmed that the number of capillaries in any given section was higher in normal mice than in MT-KO mice at 7 and 14 dpl, whereas the general ultrastructural preservation of the brain parenchyme within the lesions was more deteriorated in MT-KO mice than in normal mice.
Infiltration by macrophages and their progenitor cells is greater in MT-KO mice
The authors first explored whether the maturity of the macrophages infiltrating the lesion differed between 129/Sv and MT-KO mice. The response of hematogenous macrophages and their bone marrow progenitor cells to the lesion was observed by using immunoreactivity (IR) for CD-14 and CD-34, respectively (Figs. 3 and 4). CD-14 is expressed by blood monocytes/macrophages and resident microglia, whereas CD-34 IR labels hematopoietic myeloid and lymphoid progenitor cells and proliferating vascular endothelial cells/angiogenesis (Kishimoto et al., 1997; Kuzu et al., 1992; Serke and Huhn, 1996; Silvestri et al., 1992).
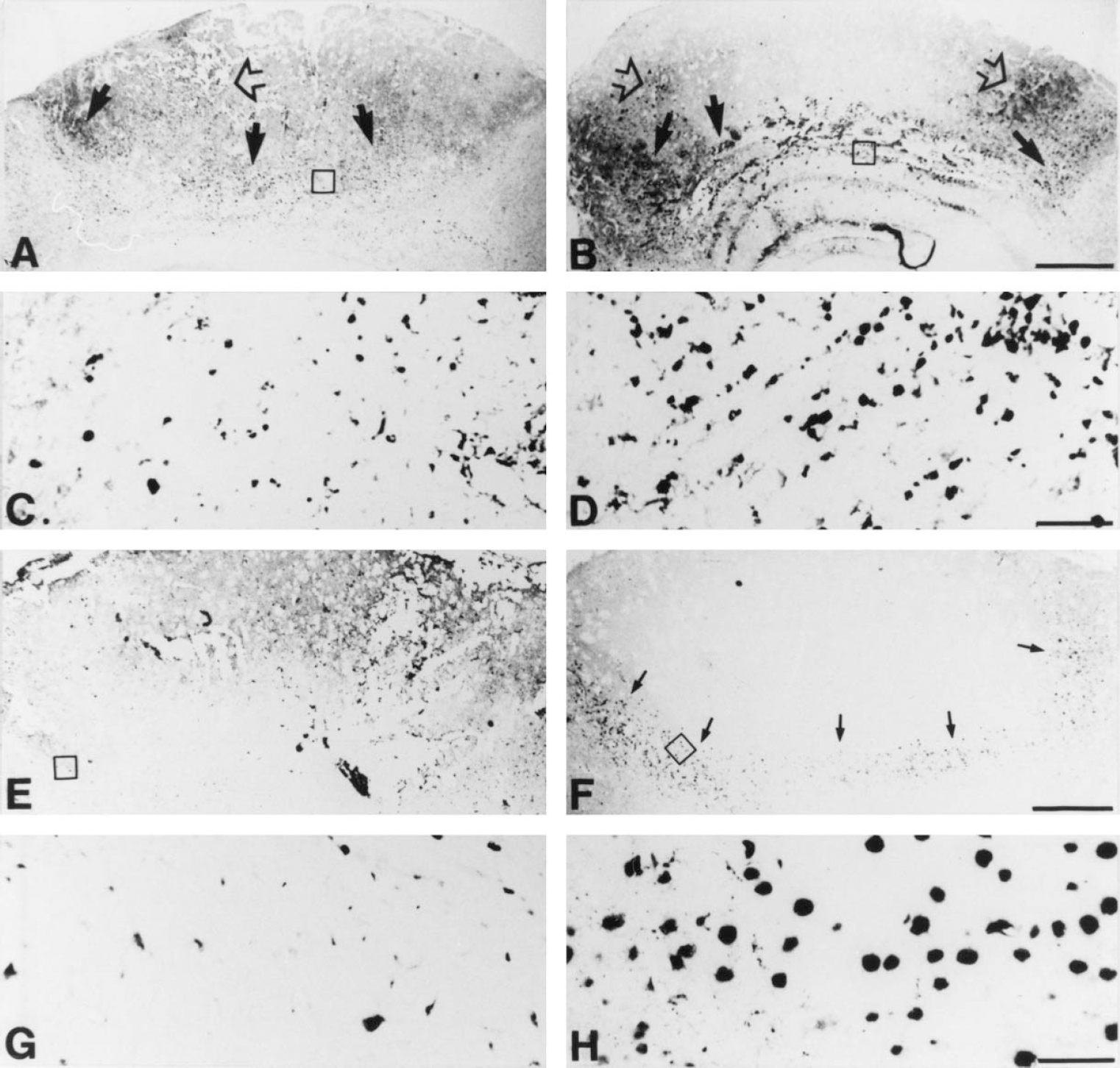
Immunostainings for CD-14+ macrophages and CD-34+ myeloid and lymphoid hematopoietic progenitor cells at 3 days poslesion (dpl).
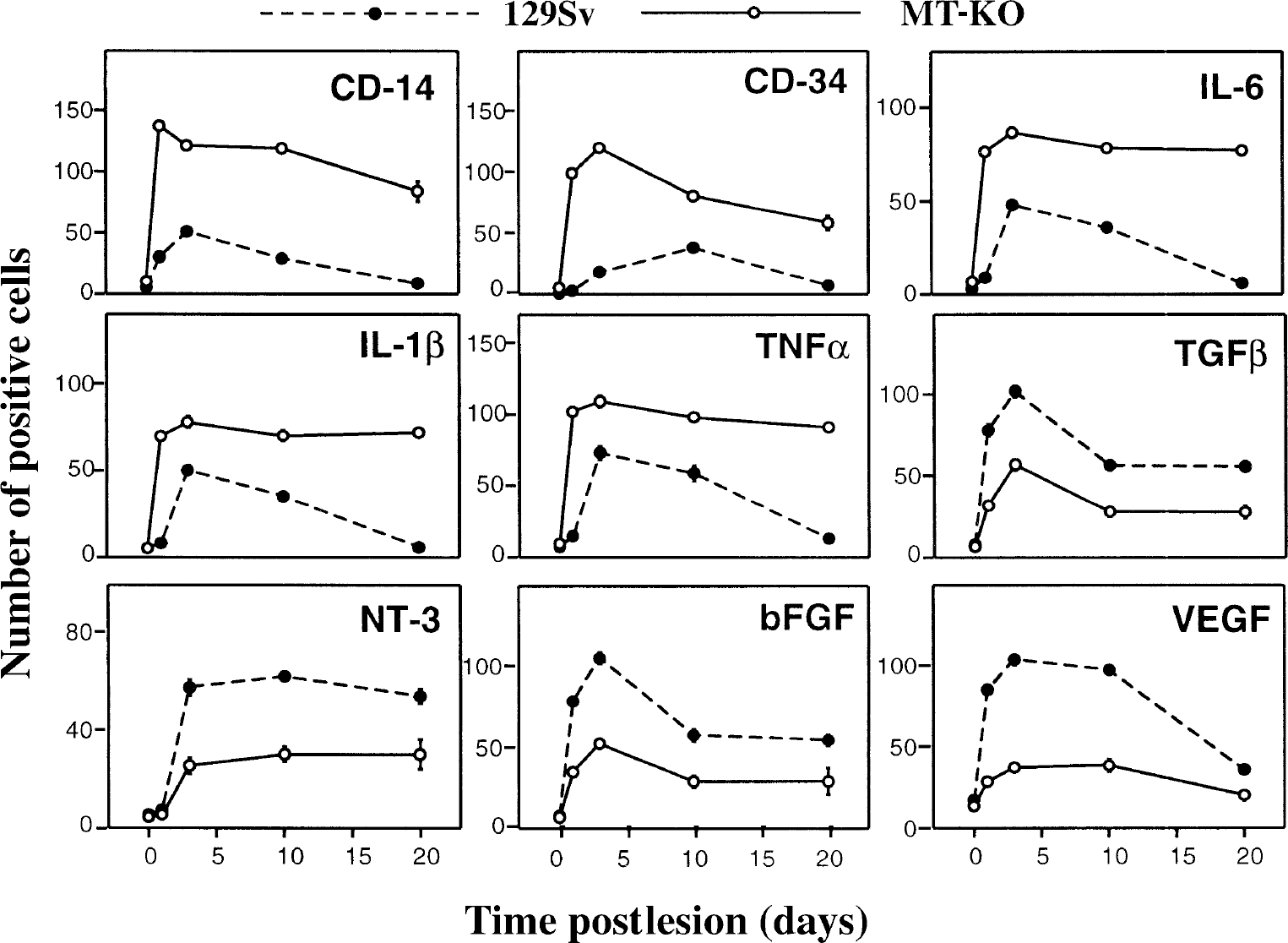
For Statistical purposes, cell counts in the border of the lesion of normal and MT-KO mice were carried out in a blinded manner at the different time points postlesioning studied. The criteria followed for defining a positively stained cell were as exemplified in Fig. 5E. Results shown are mean ± SE (n = 4 mice per each time and strain). Results were evaluated with two-way analysis of variance with lesion and strain as main factors. Both were significant (P < 0.001) for all the cytokines and growth factors studied.
Representative controls for immunohistochemistry experiments.
In unlesioned mice, CD-14+ and CD-34+ cells in the brain were hardly detectable (data not shown). In cryolesioned mice, a progressive appearance of these cells in the lesioned area was prominent, which was always greater in the MT-KO mice (P < 0.001; Fig. 4). Hematogenous CD-14+ macrophages appeared around and inside of the lesioned area and, as determined by morphology, no significant differences were observed between normal and MT-KO mice, in that all CD-14+ cells examined were mononuclear and round or amoeboid and thus resembled the typical macrophage. Immunoreactivity for MOMA-1, which is specific for peripheral monocytes of the spleen, lymph nodes, and liver (Kraal and Janse, 1986), confirmed the results obtained using CD-14 IR (data not shown).
A few CD-34+ cells had entered the lesioned area at 1 dpl, whereas at 3 to 10 dpl the number of CD-34+ cells had significantly increased at the lesion site (Figs. 3 and 4). From 10 dpl some CD-34+ vessels emerged in the tissue encircling the lesion. At 20 dpl, the number of CD34+ hematopoietic myeloid and lymphoid cells had clearly decreased compared with that seen at 3 to 10 dpl, and the lesioned tissue had regenerated significantly. However, the amount of CD-34+ vessels had increased by 20 dpl in the area subjected to the injury, which indicated a proliferative state of the vessel endothelium, or angiogenesis, or both. In MT-KO mice, the number of CD-34+ round hematopoietic cells dramatically increased and CD-34+ vessels decreased at all time points examined compared with normal mice (Fig. 4). CD-34+ vessels were hardly detectable in MT-KO mice from 1 to 20 dpl.
Cytokine-growth factor expression is altered by MT-I+II deficiency
After identifying clear differences between normal and MT-KO mice regarding their inflammatory and angiogenic responses, the authors then examined for possible differences in the expression of a number of cytokines and growth factors thought to be important in the regulation of these responses. The authors analyzed by immunoreactivity IL-6, IL-1β, TNF-α, TGF-β, NT-3, bFGF, and VEGF. In all cases, the omission of the first antibody and the preabsorption of the primary antibody eliminated the staining (Fig. 5A to 5F). The authors also carried out cell counts of positive cells for each factor in the border of the lesion as stated in Materials and Methods (see Fig. 5E for identifying positive versus negative staining), which allowed statistical evaluation of the results.
IL-6 expression
In all unlesioned mice IL-6 IR was observed in meningeal cells, ependymal cells, choroid plexus, and in a few perivascular cells of the cortex. The number of IL-6+ glial cells was slightly higher in MT-KO mice than in normal mice (data not shown).
After the cryoinjury IL-6 IR was transiently increased in macrophages and reactive astrocytes in normal mice, whereas in MT-KO mice IL-6 IR was further and continuously increased (P < 0.001) (Figs. 4 and 6). At 20 dpl, in normal mice the transiently increased IL-6 IR had returned to the levels of unlesioned mice and was only observed in some macrophages and astrocytes of the glial scar, in a few perivascular cells, and in meninges, whereas no signs of returning to the basal condition were observed in MT-KO mice.
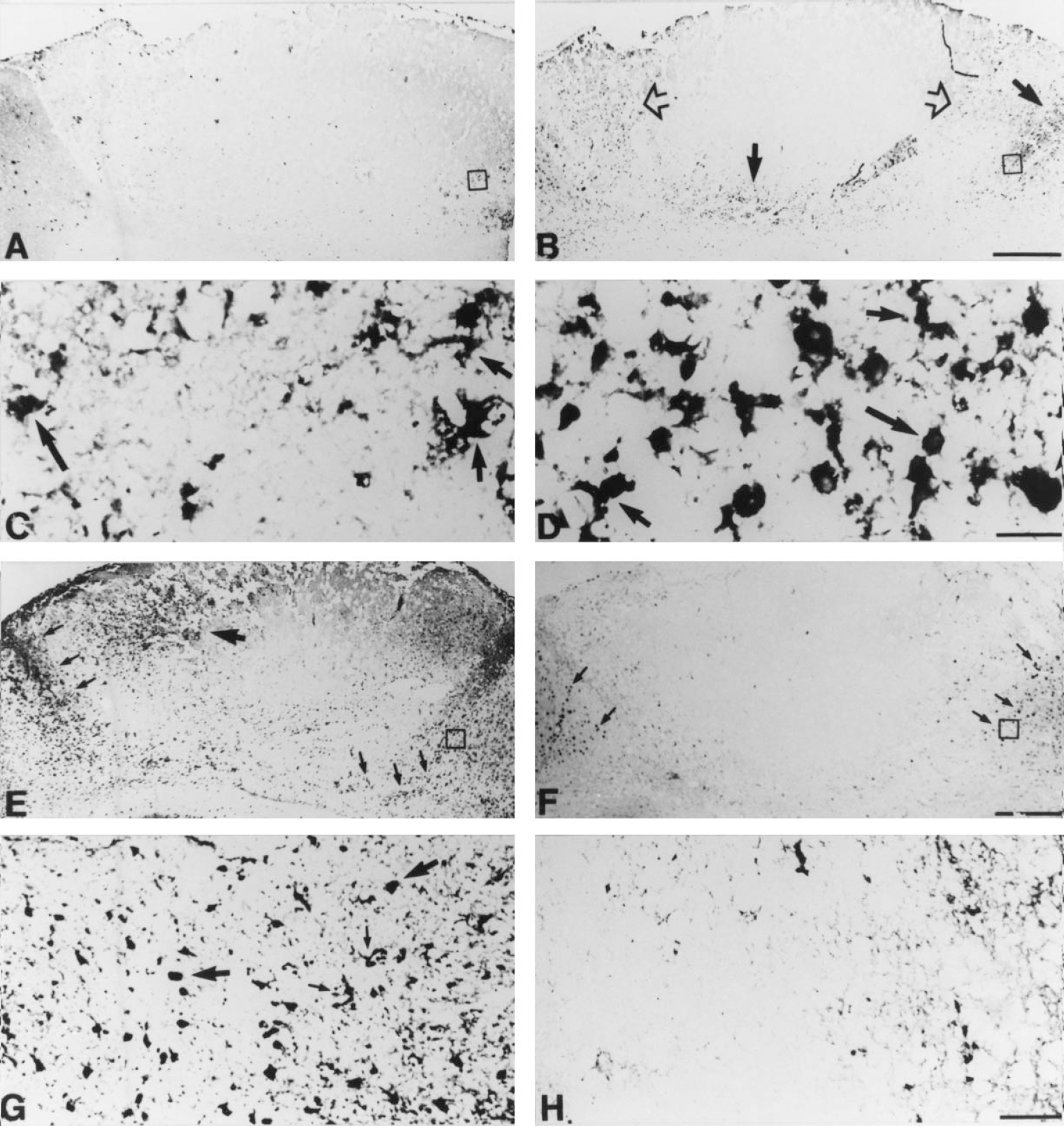
IL-6 and bFGF immunostainings at 3 days postlesion (dpl).
IL-1β expression
In all unlesioned mice, IL-1β IR was similar and was observed in meninges, ependyma, choroid plexus, and in a few astro- and microglial cells of the cortex (data not shown). After cryoinjury IL-1β IR was increased in macrophages, reactive astrocytes, and at late stages, perivascular cells of the glial scar of both normal and MT-KO mice (data not shown). Cell counts indicated that, in most respects, the results were comparable with those for IL-6 (Fig. 4).
TNF-α expression
In all unlesioned mice, TNF-α IR was confined to meningeal cells, ependymal cells, choroid plexus, and a small number of astrocytes, microglia, and perivascular cells of the cortex (data not shown). After cryoinjury, the cellular sources and the pattern of responses of TNF-α IR were similar to those of IL-6 and IL-1β (Fig. 4).
TGF-β1 expression
In all unlesioned mice examined, TGF-β1 was expressed in ependymal cells, choroid plexus, meningeal cells, and faintly in white matter ramified microglia, grey matter astrocytes, and some cortical neurons (data not shown).
After cryoinjury TGF-β1 IR increased in all mice, but in contrast to the proinflammatory cytokines, normal mice showed a greater increase in TGF-β1 levels compared with MT-KO mice (P < 0.001; Fig. 4). In normal mice, TGF-β1 IR increased significantly in macrophages and to a lesser degree in astrocytes at 1 dpl, peaked at 3 dpl, and remained high at 10 to 20 dpl. At 20 dpl, TGF-β1 IR was observed in macrophages and astrocytes of the glial scar, in meninges, and in some cortical neurons. Moreover, the TGF-β1 expressing macrophages and reactive astrocytes were also expressing MT-I+II (Fig. 7A).
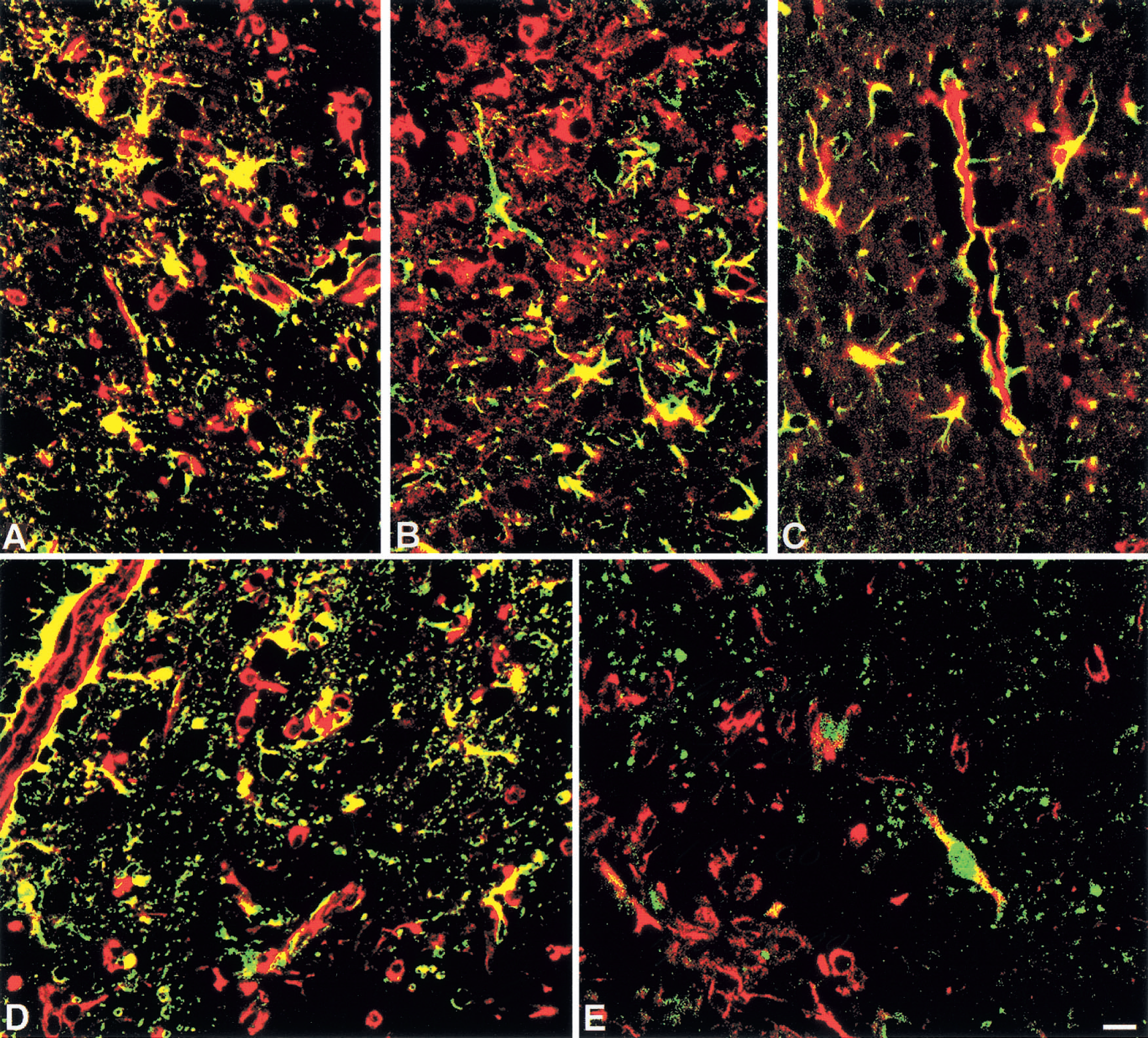
Double immunoflourescence histochemistry from normal mice at 10 days postlesion (dpl).
NT-3 expression
In all unlesioned mice the NT-3 expression was comparable and was found in a few ramified microglia, perivascular macrophages, grey matter astrocytes and neurons (data not shown).
After cryoinjury in normal mice, NT-3 IR was essentially unchanged at 1 dpl, whereas at 3 to 10 dpl NT-3 IR was clearly increased in macrophages inside of the lesion and in reactive astrocytes and survived neurons surrounding the injured area; at 20 dpl NT-3 expression was still significantly increased in macrophages and astrocytes of the glial scar and in neurons encircling the scar tissue (Fig. 4). In MT-KO mice, NT-3 expression also increased after cryoinjury, but the NT-3 response was reduced compared with normal mice at all time points examined (P < 0.001; Fig. 4). Primarily round or amoeboid macrophages and astrocytes expressed NT-3, whereas neuronal NT-3 IR was less pronounced in MT-KO mice.
b-FGF expression
In all unlesioned mice, b-FGF IR was comparable and was observed in meninges, ependyma, choroid plexus and some dispersed astro- and microglial cells of the cerebral cortex, hippocampus, thalamus, and basal nuclei (data not shown).
After cryoinjury, b-FGF was increased in macrophages and reactive astrocytes of normal and MT-KO mice (P < 0.001); however, at all time points examined, b-FGF levels of MT-KO mice were significantly reduced compared with those of normal mice (P < 0.001; Figs. 4 and 6). In normal mice, b-FGF expression was increased at 1 dpl, peaked at 3 dpl, and was still increased at 10 to 20 dpl in macrophages and reactive astrocytes. By 20 dpl, the b-FGF IR was seen in macrophages and astrocytes of the glial scar, perivascular cells, and in meningeal cells. Interestingly, b-FGF expressing macrophages and astrocytes were those containing MT-I+II (Fig. 7B). Furthermore, b-FGF was colocalized with VEGF at 10 and 20 dpl (Fig. 7D and 7E). In MT-KO mice at 1 to 20 dpl, increased b-FGF IR was observed in some macrophages and astrocytes at the lesion site. However, the number of macrophages and especially of astrocytes expressing b-FGF in MT-KO mice was clearly lower than in normal mice (Figs. 4 and 6).
VEGF
In all unlesioned mice, VEGF expression was similar and was seen in a few vessels, perivascular cells, ependyma, choroid plexus, and meninges (data not shown). After cryoinjury in normal mice, VEGF expression was increased in many large and small vessels at the lesion site and in macrophages and reactive astrocytes. The number of VEGF+ macrophages and reactive astrocytes peaked at 3 to 10 dpl and afterwards declined, but remained higher at 20 dpl than in unlesioned mice (Fig. 4). Vascular VEGF IR peaked at 10 dpl and approximately maintained that level at 20 dpl. Interestingly, VEGF was coexpressed with MT-I+II (Fig. 7C). In lesioned MT-KO mice, VEGF IR also increased at 1 to 20 dpl, but not as much as in normal mice (P < 0.001) (Fig. 6). Moreover, cryoinjured MT-KO mice showed VEGF IR in macrophages and reactive astrocytes, but VEGF expression was less pronounced and vague in vascular endothelial cells.
To study coexpression of b-FGF and VEGF during brain regeneration, double immunofluorescence histochemistry was performed as shown in Fig. 7D and 7E. In normal mice at 3 to 20 dpl, many of the VEGF+ macrophages and astrocytes were coexpressing b-FGF. VEGF+ vascular endothelium was not coexpressing b-FGF, but astrocytic end feet adjacent to vessel walls and perivascular cells were coexpressing VEGF and b-FGF (Fig. 7D). In MT-KO mice, few of the VEGF+ macrophages and astrocytes were coexpressing b-FGF. Only a few vessels of MT-KO mice showed VEGF IR in the vascular endothelium and these were negative for b-FGF. Some of the astrocytic end feet adjacent to vessel walls and perivascular cells of MT-KO mice were VEGF+ but negative for b-FGF (Fig. 7E).
RNase protection analysis of cytokine mRNA levels confirm the immunohistochemical results
As a further control for the above cytokine findings, cytokine mRNA levels were measured by an RNase protection assay in mice killed at 1 and 3 dpl (Fig. 8). It became clear that, in accordance with the IR results described above, the freeze lesioning caused a significant up-regulation of all the cytokines analyzed, namely IL-6, IL-1β, TNF-α, but also lymphotoxin-α and IL-1α. Densitometric analysis of each cytokine, once normalized to a per RPL32 content (a ribosomal protein), revealed that MT-KO mice had a greater up-regulation than control mice for all cytokines except IL-1α (Table 1).
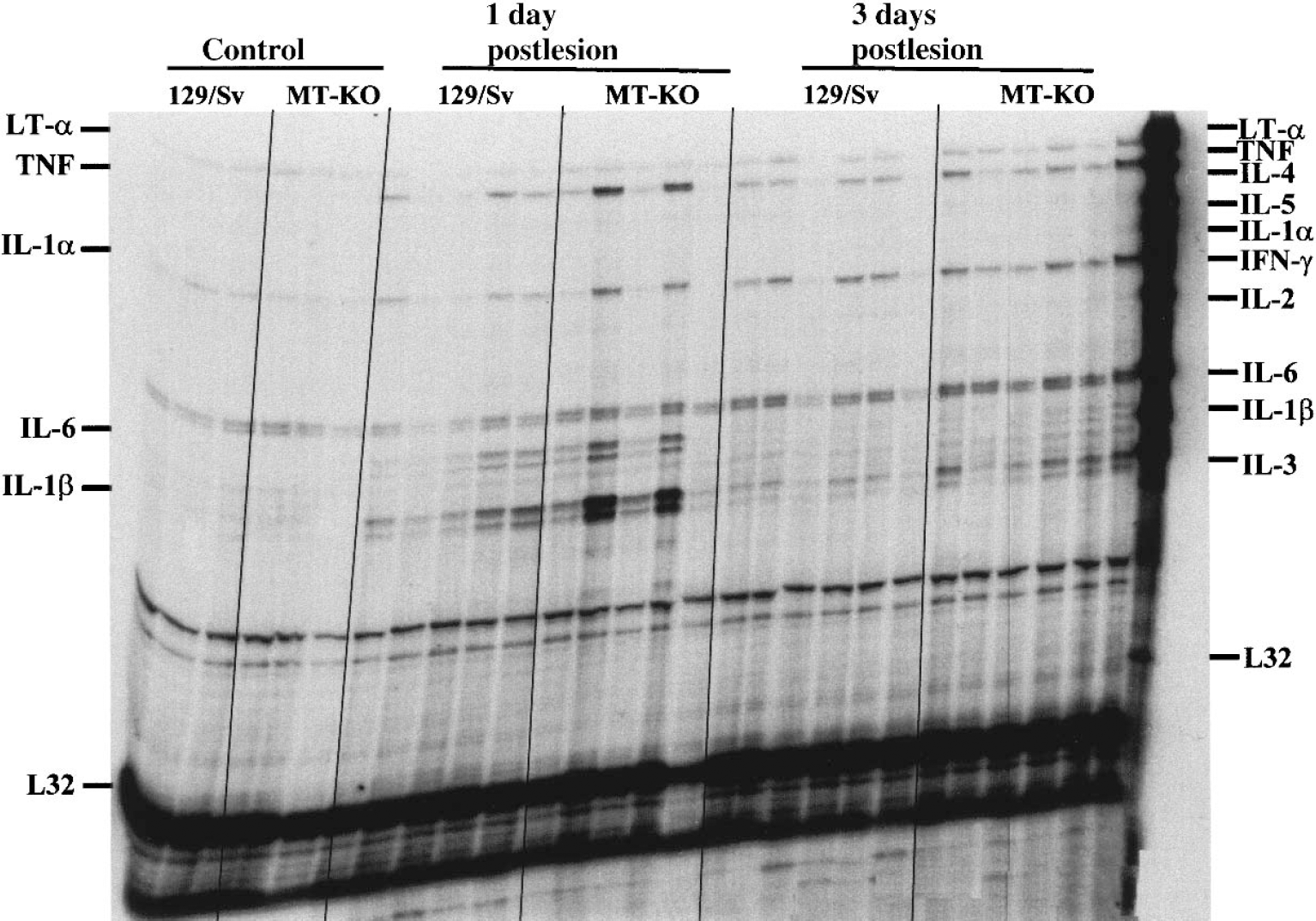
Analysis of brain cytokine expression during the cortical freeze lesion. mRNA levels of a number of cytokines were measured with an RNase protection assay. Unlesioned (control) and freeze lesioned (1 and 3 days postlesion) mice were killed and their ipsilateral cortex immediately frozen in liquid nitrogen. The results show that only some cytokines were up-regulated by the freeze lesion, namely lymphotoxin-α, TNF-α, IL-1α, IL-6, and IL-1β. The densitometric analysis of the bands (shown in Table 1) demonstrates that MT-KO mice have a greater response for these cytokines, except IL-1α, which confirms the immunocytochemistry data.
Analysis of the brain cytokine expression during the cortical freeze lesion
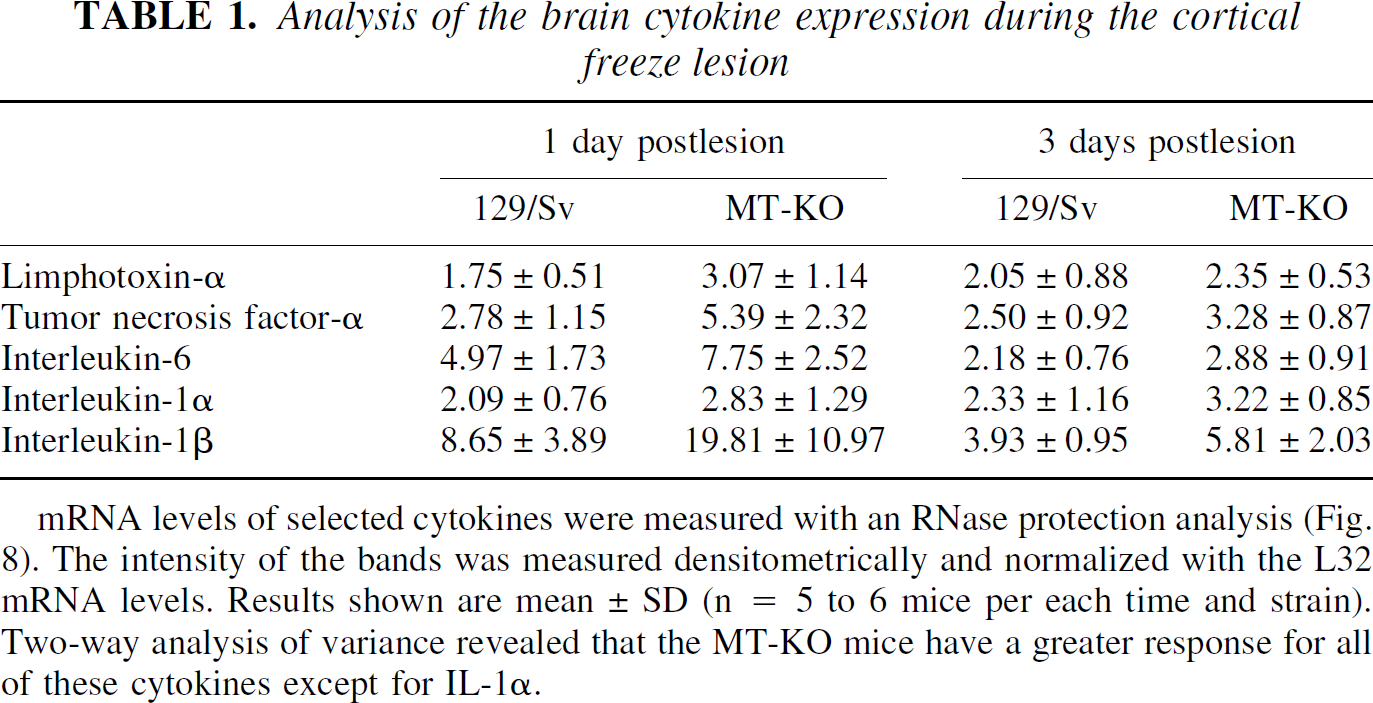
mRNA levels of selected cytokines were measured with an RNase protection analysis (Fig. 8). The intensity of the bands was measured densitometrically and normalized with the L32 mRNA levels. Results shown are mean ± SD (n = 5 to 6 mice per each time and strain). Two-way analysis of variance revealed that the MT-KO mice have a greater response for all of these cytokines except for IL-1α.
Oxidative stress
In all unlesioned mice, IR for iNOS, NITT, and MDA was comparable and was only observed in a few meningeal and scattered glial cells (data not shown). After the lesion, normal mice showed increased numbers of cells stained positively for iNOS, NITT, and MDA at 1 dpl, peaking at 3 dpl, and by 10 dpl the number of cells had decreased; and at 20 dpl, iNOS, NITT, and MDA IR were almost similar to those of unlesioned mice (Fig. 9). Macrophages, but also many reactive astrocytes and some neurons surrounding the lesion site, contained iNOS, NITT, and MDA. In MT-KO mice, IR for iNOS, NITT, and MDA was increased at all time points examined relative to normal mice (Fig. 9). Thus, at 1 dpl, an increased number of macrophages, reactive astrocytes, and neurons showed iNOS, NITT, and MDA IR at the lesion site; this IR peaked by 3 to 10 dpl and remained significantly greater than that of unlesioned mice at 20 dpl in MT-KO mice.
Oxidative stress responses at 3 and 20 days postlesion (dpl) in normal and MT-KO mice.
MT-I+II deficiency does not alter MT-III expression during the freeze lesion
To establish whether MT-III, a MT isoform that is also expressed in the CNS, showed a compensatory response in the absence of MT-I+II, a freeze lesion experiment was performed in which an in situ hybridization of MT-I and MT-III mRNA of normal and MT-KO mice was carried out. As expected (Penkowa et al., 1999a), the MT-I isoform in normal mice was significantly increased in the areas surrounding the lesion (not shown). Quantitative measurements carried out in defined areas of the border of the lesion showed that the up-regulation was significant throughout the experimental period studied, from 1 to 13 dpl, whereas no up-regulation was observed in the contralateral cortex (Fig. 10). In contrast to MT-I, the MT-III isoform was downregulated by the 1 dpl and subsequently up-regulated progressively, showing a threefold induction in the border of the lesion at 13 dpl (Figs. 10 and 11).
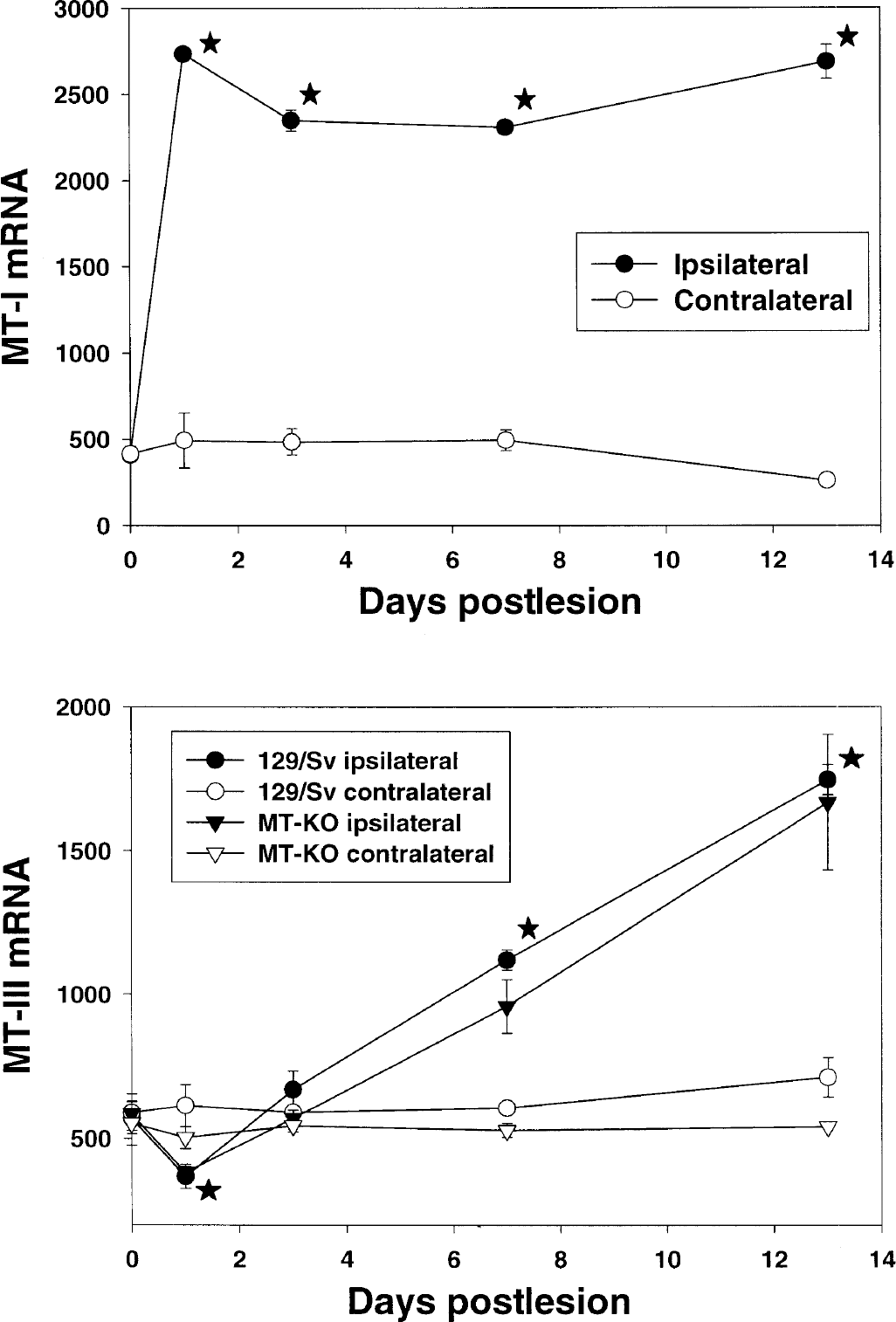
In situ hybridization analysis of MT-I and MT-III mRNA of normal and MT-KO mice. As expected, the cortical freeze lesion increased significantly the MT-I signal in the border of the lesion in the ipsilateral, but not contralateral, cortex of normal mice. The MT-III isoform was initially (1 day postlesion) downregulated, but afterwards it increased progressively and up to a threefold induction was observed at 13 days postlesion (see also Fig. 11). The response of the MT-III isoform did not differ between normal and MT-KO mice. Results are mean ± SE (n = 3 mice per each time and strain). *P < 0.05 versus the unlesioned mice.
Microautoradiographic localization of the MT-III expression. The disappearance of the signal within the lesion is because of the death of the cells. At 1 day postlesion (dpl)
MT-I+II deficiency also decreases angiogenesis in transgenic GFAP-IL6 mice
To establish further the putative role of MT-I+II in angiogenesis, the authors examined transgenic mice with expression of IL-6 in the CNS, under the control of the glial fibrillary acidic protein gene promoter (GFAP-IL6) and simultaneous MT-I+II deficiency. These mice were generated by successive crossings of GFAP-IL6 mice (Campbell et al., 1993) and MT-KO mice (Masters et al., 1994). The offspring were identified by Southern blot (MT-I+II) and by polymerase chain reaction (GFAP-IL6). Figure 12 shows a typical polymerase chain reaction result. The GFAP-IL6 mice showed a dramatically increased angiogenesis in the CNS, especially in the cerebellum (Brett et al., 1995), which was confirmed in the current report (Fig. 13). GFAP-IL6 mice heterozygous for MT-I+II showed a slightly decreased angiogenesis in the cerebellum, whereas those homozygous for the null mutation for MT-I+II showed a clear reduction of the IL-6-induced angiogenesis (Fig. 13A to 13D). In situ hybridization analysis demonstrated that the regulation of the MT-III isoform was again not altered by the simultaneous MT-I+II deficiency (data not shown). Thus, these results agree with the notion of MT-I+II as factors involved in angiogenesis.
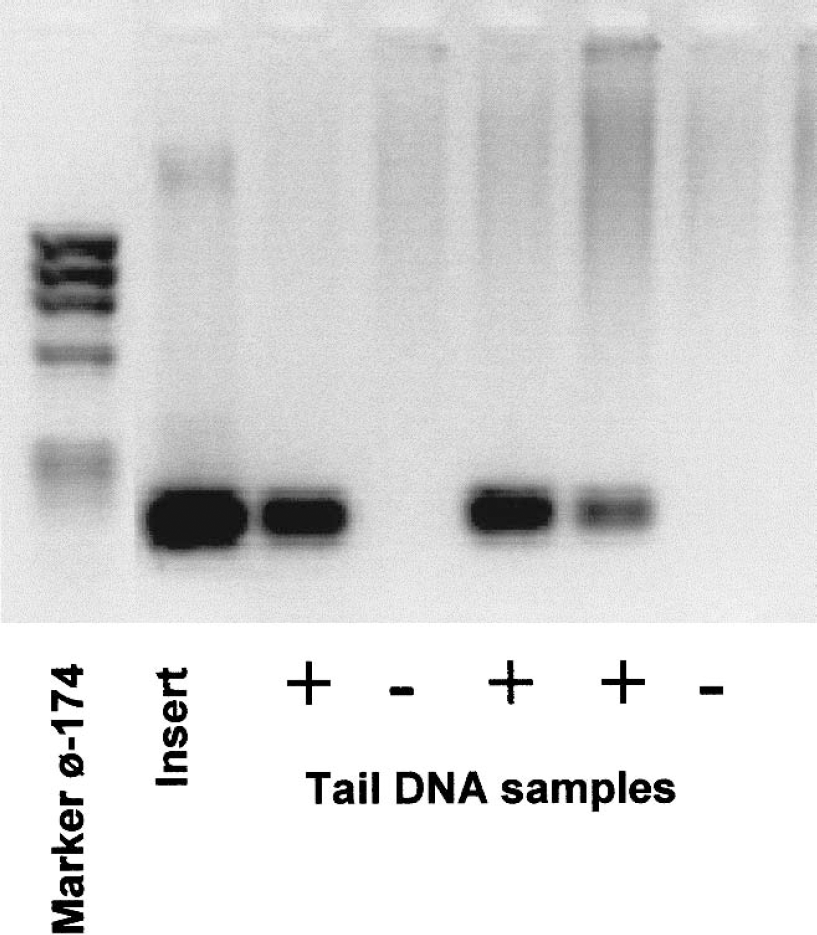
Polymerase chain reaction analysis of GFAP-IL6 mice. SV-40 sequences were detected in tail DNA by using the primers jhsv5 (5′-GATCCAGACATGATAAGATA-3′) plus jhsv3 (5′-CCGAAAAAACCTCCCACACC-3′). Polymerase chain reaction products were electrophoresed on a 2% agarose gel, and a unique product of 196 pb was generated by the GFAP-IL6 positive mice.
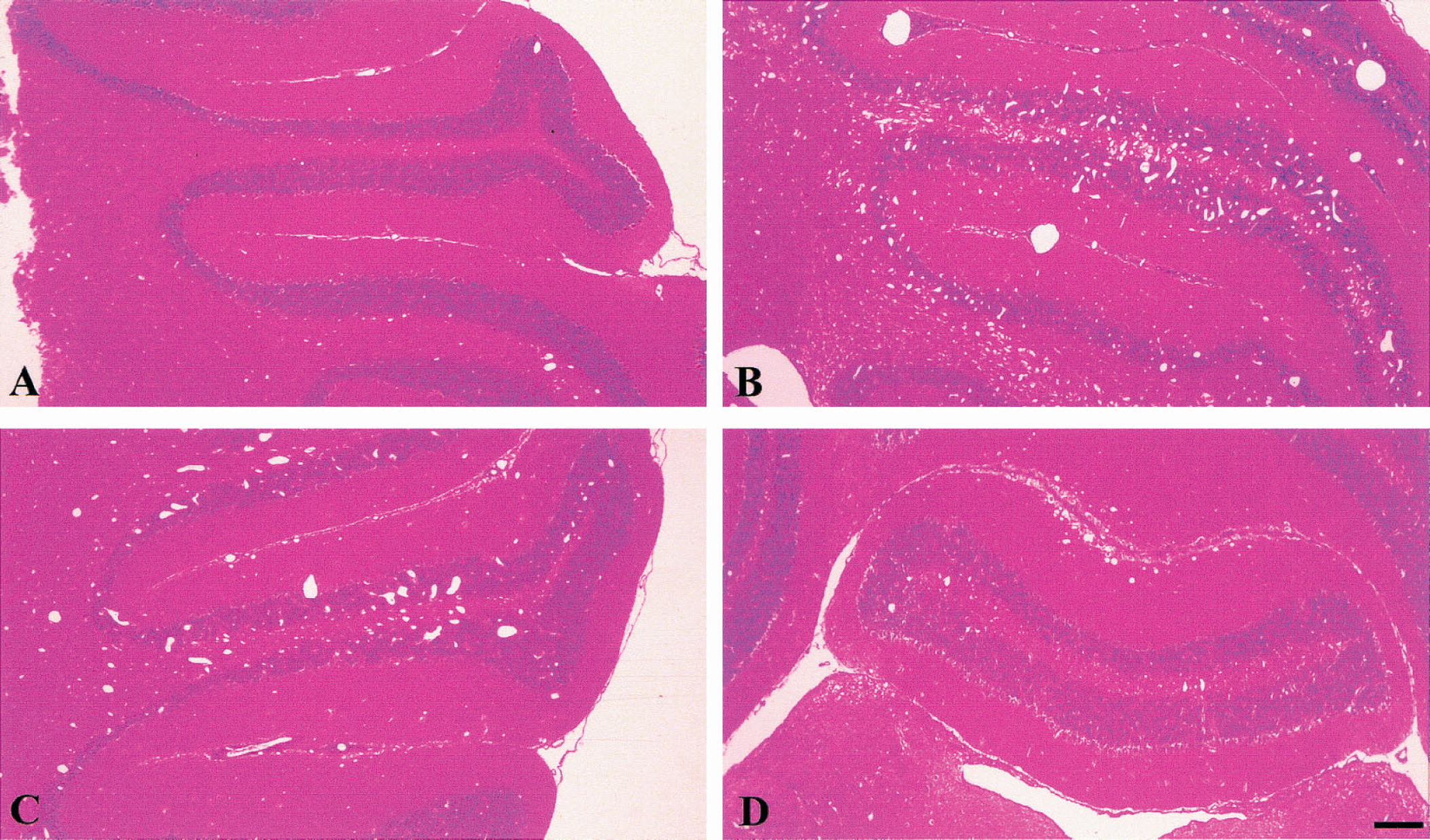
Metallothionein-I+II deficiency also decreases angiogenesis in the GFAP-IL6 mice.
DISCUSSION
In this report we have evaluated the putative importance of MT-I+II in an experimental CNS damage model, the cortical freeze injury. Previous studies have shown that MT-I+II IR and MT-I mRNA levels increase significantly in microglia/macrophages and reactive astrocytes situated around the freeze lesion site, and that MT-KO mice show a prolonged inflammatory response (Penkowa and Moos, 1995; Penkowa et al., 1999a). In the current study, we demonstrated that an altered angiogenesis is likely involved in this impaired response of MT-KO mice, in which altered monocyte/macrophage profiles, altered cytokine-growth factors expression, and increased oxidative stress are all factors presumably underlying the deficient response to CNS damage of MT-KO mice.
In the current study we confirmed and extended previous results (Penkowa et al., 1999a) that MT-KO mice showed a prolonged inflammatory response after a cortical freeze injury, as verified by GFAP and lectin stainings. We demonstrated by scanning electron microscopy and transmission electron microscopy that the vasculature of the lesioned area was more damaged, and the ultrastructural preservation of the parenchyma was poorer in MT-KO mice compared with control mice. To identify putative mechanisms underlying this decreased angiogenesis in MT-KO mice, we have analyzed several possibilities. We first evaluated the maturity of the monocyte adherence and invasiveness, as shown in a human monocyte-derived cell line THP-1 (Leibbrandt et al., 1994), but the current results indicated that MT-I+II apparently are not required for monocyte adhesion to the vascular endothelium or migration into the brain. Moreover, MT-KO mice actually showed higher levels of mature CD-14+ and premature CD-34+ cells; thus, it seems unlikely that the prolonged inflammatory response of MT-KO mice are related to the maturity of the infiltrating monocyte/macrophage cells. Rather, MT-I+II could putatively have a role in regulation or suppression of the inflammatory response of these cells.
The increased number of myeloid progenitor cells and monocytes/macrophages in lesioned MT-KO mice could alternatively be related to the unbalanced cytokine profile. Certainly, during the freeze lesion the expression of proinflammatory cytokines IL-6, IL-1β, and TNF-α was dramatically increased, whereas that of the growth factors TGF-β1, NT-3, b-FGF, and VEGF was decreased in the absence of MT-I+II. Proinflammatory cytokines have several functions in the CNS and are potentially deleterious or beneficial, depending on their concentration, site, duration of action, and the presence of other monocytes/macrophages infiltrating the lesioned area by analyzing the IR for CD-14 (blood monocytes) and CD-34 (bone marrow myeloid and lymphoid progenitor cells) and their morphology. Mature and well differentiated macrophages have neurite growth-promoting roles and are required for CNS tissue repair and recovery (Giulian et al., 1989; Rapalino et al., 1998; Rabchevsky and Streit, 1997). After the cortical freeze lesion, the number of CD-14+ and CD-34+ cells was significantly increased in normal and MT-KO mice, although the latter clearly showed a much greater response, and the majority of the cells stained had a round morphology. Because the number of ramified microglial cells was similar in normal and MT-KO mice before and after lesioning (data not shown), the results suggest that the MT-I+II absence influences the blood-derived macrophages rather than microglia-derived macrophages. Decreases in MT levels by antisense downregulation may lead to changes in cytokines or related molecules (Rothwell and Hopkins, 1995; Merrill and Benveniste, 1996). The growth factors b-FGF, TGF-β1, and VEGF, which in normal mice are coexpressed in a spatiotemporal pattern with MT-I+II, are neuroprotective factors involved in angiogenesis and tissue regeneration (Gómez-Pinilla et al., 1992; Kalaria et al., 1998; Logan et al., 1994; Morisaki et al., 1999; Neufeld et al., 1999; Unsicker et al., 1992; Yoshida et al., 1997). Thus, the altered cytokine-growth factors shown here in MT-KO mice during the cortical freeze lesion could likely contribute to the altered angiogenesis and thus to the decreased tissue repair. In lesioned normal mice, angiogenesis-promoting factors b-FGF, TGF-β1, and VEGF were expressed simultaneously with neovascularization as verified by electron microscopy (not shown) and by using CD-34 and VEGF IR. Increased VEGF expression and angiogenesis are essential for normal tissue regeneration to occur (Neufeld et al., 1999) and are also observed during pathologic conditions in the brain, such as reduced perfusion, infarcted brain, insufficient vascularity, or in Alzheimer's disease and brain tumors (Kalaria et al., 1998; Neufeld et al., 1999). In MT-KO mice, angiogenesis-promoting factors, vascular expression of CD-34, and neovascularization were decreased as part of the reduced brain regeneration. Because b-FGF, TGF-β1, and VEGF were coexpressed with MT-I+II in normal mice, it is likely that absent expression of MT-I+II may be directly related to the lack of angiogenesis seen in MT-KO mice. In agreement with this, vascular endothelial cells have been shown to up-regulate MT-I+II during pathologic conditions, such as after cerebral ischemia and in brains of Alzheimer's disease patients (Zambenedetti et al., 1998; Van Lookeren Campagne et al., 1999). Furthermore, as shown in the current study, GFAP-IL-6 mice made deficient for MT-I+II had a dramatic reduction of the IL-6-induced angiogenesis. Although a role for MTI+II in angiogenesis appears to be important during neuropathologic states, this is not the case in the normal CNS because no abnormalities were observed in unlesioned mice.
The exact mechanisms underlying the unbalanced cytokine profiles and the impaired angiogenesis of MT-KO mice are still unknown. It is very likely, however, that the metal binding properties, or antioxidant functions, or both, of MT-I+II per se could have a significant impact in the CNS physiology. Also, the argument could be made that compensatory responses of MT-III could have a role on the phenotype of the MT-KO mice, but the current results find that possibility unlikely. We have evaluated the oxidative stress status of MT-KO mice in the cortical freeze lesion model by determining iNOS, MDA (an index of lipid peroxidation), and NITT (an index of protein tyrosine nitration caused by increased nitric oxide, or superoxide radical levels, or both) expression levels. The results demonstrated that MT-KO mice have an increased lesion-associated oxidative stress, which considering the well-known damaging effects of reactive oxygen species suggests that MT-I+II per se have a significant impact on the capability of the CNS to respond to injury. In accordance, preliminary data indicated that exogenous MT-I+II administration has protective effects in the damaged brain (unpublished observations).
Thus, many diverse alterations in the inflammatory-immunologic responses may contribute to the decreased brain regeneration seen in MT-KO mice after cortical freeze injury. The profound effects of MT-I+II in vivo on regenerative actions including angiogenesis in the brain, myelomonocytic recruitment, impaired cytokine and neurotrophic growth factor expression after cortical freeze injury are shown and suggest a fundamental role for MT-I+II in brain inflammation and immunopathology. A clear understanding of these processes, combined with development of methods to manipulate them, is likely to offer significant therapeutic potential in the control of brain pathology and neurologic diseases.
Footnotes
Acknowledgments
The authors acknowledge Hanne Hadberg, Pernille S. Thomsen, Jordi Canto, and Carrie Kincaid for their excellent technical assistance. The authors thank Grazyna Hahn, Birgit Risto, and Keld Stub for their excellent photographic assistance.