
Abstract
The role of angiogenesis after stroke is unclear; if angiogenesis supports long-term recovery of blood flow, then microvessel hyperdensity consequent to angiogenesis should persist in infarcted cortex. Here, we assess the long-term stability of ischemia-induced microvessels after 2-h transient rat middle cerebral artery occlusion (tMCAo) followed by 30, 90, or 165 days of reperfusion. Stereological measures of microvessel density were taken adjacent to and within cortical cysts. Vascular permeability was documented by extravasation of immunoglobulin (IgG) and of fluorescein-dextran. After 30 days reperfusion, a significantly increased microvessel volume density (VV) was restricted to the inner margin of cystic infarcts as compared with the region external to the infarct or contralateral control cortex (F = 42.675, P < 0.001). The hyperdense ischemic vasculature was abnormally leaky to IgG and fluorescein-dextran. Between 30 and 90 days of reperfusion, this vessel hyperdensity regressed significantly and then regressed further but less drastically between 90 and 165 days. Phagocytic macrophages were restricted to the infarct and dynamic changes in their number correlated with microvessel regression. Additional ED-1 labeled inflammatory cells were widely distributed inside and external to the infarct, even after 165 days of reperfusion. These data show that ischemia evoked angiogenesis results, at least in part, in transient populations of leaky microvessels and phagocytic macrophages. This suggests that a major role of this angiogenesis is for the removal of necrotic brain tissue.
Introduction
Angiogenesis is well known to take place after brain ischemia (Krupinski et al, 1994; Plate et al, 1999; Sbarbati et al, 1996; Wei et al, 2001). Stroke evokedangiogenesis is likely to have multiple functions and results from angiogenic growth factors that have pleiotropic effects. For example, vascular endothelial growth factor (VEGF) is a potent mitogen for endothelial cells (Leung et al, 1989) and also exerts direct effects on neurons and acts as a neuroprotectant in rat models of ALS (Storkebaum et al, 2004) and in experimental models of stroke (Hayashi et al, 1998; Manoonkitiwongsa et al, 2004; Sun et al, 2003). However, VEGF can trigger vessel leakiness (Carmeliet, 2003; Paul et al, 2001; Rosenstein et al, 1998; Zhang et al, 2000). This leakiness is necessary for passage of the inflammatory cells that subserve wound healing but it also results in vasogenic edema and inflammation (Croll et al, 2004; Duffield, 2003). A prominent feature of injury-induced expression of multiple angiogenic related factors is their time-dependence (Hayashi et al, 2003). However, relatively little is known about long-term effects of angiogenic factors on newly formed vessels or recruited macrophages or even if these structures or cell types become stably integrated in the injured brain.
Previous work has established that within the first week after human stroke, infarcts are invaded by immature populations of capillaries (Liu, 1988) as well as by macrophages (Chuaqui and Tapia, 1993). More recently in the rat, we showed a quantitative correlation between the quantity of microvessels and of macrophages in cerebral infarcts after 30 days of reperfusion of the transiently occluded middle cerebral artery (tMCAo) (Manoonkitiwongsa et al, 2001a). Thus, an important function of stroke-evoked angiogenesis could be the delivery of macrophages to necrotic brain tissue and the prompt removal of debris (the ‘clean-up' hypothesis) (Manoonkitiwongsa et al, 2001a). The present study further tests the ‘clean-up' hypothesis; here we ask if chronic cerebral infarcts show vessel regression and macrophage loss after tMCAo stroke in the adult rat.
Materials and methods
All protocols were approved by the Animal Research Committee of the Veteran's Affairs Medical Center, San Diego, following all national guidelines for the care of experimental animals. Male Sprague—Dawley rats (Harlan, San Diego, CA, USA), 260 to 320 g, underwent surgery to transiently occlude the left middle cerebral artery (tMCAo) with intraluminal 4-0 nylon suture (Longa et al, 1989). All sutures were pre-blunted in a microforge (Narishige MF83, NY, USA) and only filaments between 280 and 305 μm in diameter were used for occlusion. The surgical exposure and occlusion conditions were performed as previously described (Jackson-Friedman et al, 1997).
Animals were prepared with 2 h of tMCAO and the allowed to reperfuse for 30 (n = 8), 90 (n = 20), or 165 (n = 11) days. Animal mortality before the targeted reperfusion time occurred in all groups (n = 16; 75% in the first 48 postoperative hours and the remaining 25% during the first two postoperative weeks), but there was no significant trend in mortality related to reperfusion time. At 30 mins before killing, fluorescein-dextran (2 mDa; Sigma) was administered intravenously by tail vein injection under isofluorane anesthesia. Rats were then administered an overdose of pentobarbital and perfused transcardially with normal saline followed by buffered paraformaldehyde and the brain was removed from the skull. Tissue slices of 600 μm thickness were cut in a coronal plane with a vibratome (Ted Pella). Of the cases that survived for the full reperfusion time, only brains with visible cortical cysts on the coronal brain slice were chosen for further study. This resulted in 3/8 cyst cases for the 30-day group, 4/20 for the 90-day cases, 3/11 for the 165 cases. The remaining surviving animals lacked cysts although their behavioral scores on the first postoperative day confirmed an ischemic insult. An extensive study of the microvessel density in the somatosensory cortex of Sprague—Dawley rats indicated that rat to rat variability was quite low in this measure (Manoonkitiwongsa et al, 2001b); however, it is still possible that rat to rat variations in surface vasculature of the Sprague—Dawley rats used in this study, may have contributed to variable cyst rate as such variations among different strains of rats have been linked to protection from infarction after permanent MCA occlusion (Coyle and Heistad, 1987). Stereological analysis was restricted to mid-parietal slices situated 7 mm posterior to the frontal pole (Manoonkitiwongsa et al, 2001b). The immediately adjacent slice was processed for immunohistochemistry.
For stereologic measures, we selected a 4 × 2 × 0.6 mm3 block of somatosensory cortex that bordered the cyst margin (Figure 1) for embedding into plastic; a homologous block was cut from the corresponding cortical area in the contralateral hemisphere. Each tissue block was postfixed in 2% Karnovsky's solution overnight, washed with phosphate-buffered saline, and then osmicated (0.1 M phosphate-buffered 1% osmium tetroxide for 2 h), dehydrated through graded ethanols and propylene oxide, and infiltrated with Epon (Scipoxy 812; Energy Beam Sciences Inc., Agawam, MA, USA). Semithin sections (1 μm) were cut from the resulting tissue block, counter-stained with 1% toluidine blue solution, and cover-slipped.
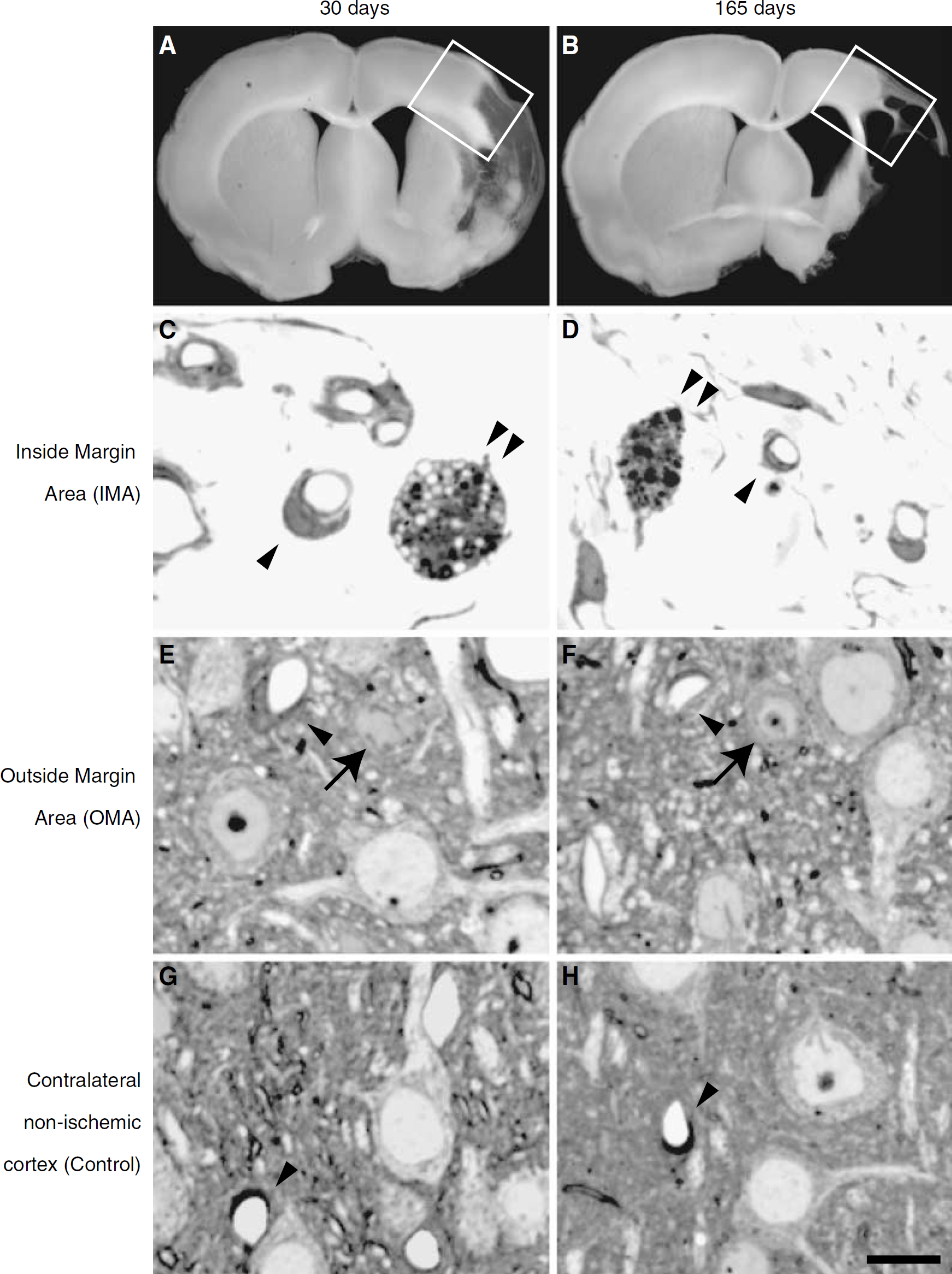
Representative photomicrographs for stereological analysis in toluidine-blue stained 1-μm semithin sections. Blocks including the margin of any visible cystic infarct in cortex were cut from a 600 μm thick slice (
The cortical region of persistent tissue within the infarct/cyst margin was labeled ‘Inside Margin Area' (IMA) and was defined by the absence of neurons and by a loose arrangement of cellular processes, phagocytes and blood vessels. The ‘Outside Margin Area' (OMA) was defined as a narrow border zone of neo-cortex containing cortical layers 1 through 6 adjacent to the area of more severe degeneration. For each sample, both IMA and OMA were identified within the same semithin section. These areas, along with contralateral cortical areas, were analyzed in digitized images that were examined at a final magnification of × 980 with a rectangle counting frame of area 0.0145 mm2. Based on previous validation, we targeted 10 counting frames per subject placed in a stratified random pattern around the cyst margin; in some cases more than 10 frames were obtained and in one 30-day case the length of the cyst margin allowed only eight counting frames. In the 30-day cases, the total sampling area (mean ± s.d.) of the IMA and OMA was 0.15 ± 0.02 mm2; for the 90-day cases the IMA total sampling area was 0.16 ± 0.01 mm2 the OMA was 0.17 mm2 (no s.d.), for the 165 day cases the total IMA sampling area was 0.16 ± 0.03 mm2 for the OMA was 0.17 ± 0.02 mm2. All contralateral cortices were sampled with total area of 0.145 mm2. From these sampled areas, we used Image Pro-Plus software as previously described (Manoonkitiwongsa et al, 2001b) to estimate the following stereological measures: numerical density (NA): total number of microvessel profiles per unit area of tissue (# of microvessels/mm2); volume density (VV): total microvessel volume per unit volume of tissue (%); length density (LV): total length of microvessels per unit volume of tissue (mm/mm3); surface density (Sv): total surface of microvessels per unit volume of tissue (mm2/mm3); diameter (D): mean short-axis of the lumena of microvessels (μm). In each of the images, the number of macrophages and neurons also was counted manually (Manoonkitiwongsa et al, 2001a, 2004). Macrophages were characterized by morphologic evidence of phagocytic activity (inclusion of visible ingested material within the cytoplasm). Neurons with pale nucleus and cytoplasm were considered viable neurons. Viable neurons with nucleolus were counted using a modified correction factor to adjust for partial profiles (Manoonkitiwongsa et al, 2004).
We performed immunohistochemistry using the slice immediately adjacent to the one used for plastic embedding. This adjacent slice was cryoprotected in sucrose and cut on a freezing-sliding microtome into 50-μm sections. Immunostaining was performed on free-floating sections with one of three antibodies diluted in blocking buffer (10% normal serum in PBS; 0.2% triton was added in biotinylated antibody incubation steps): (1) mouse anti-ED1, a marker of activated microglia and monocytes (1:1,000, Serotec, Oxford, UK); (2) biotinylated anti-fluorescein to localize retained fluorescein-dextran (1:1000, Vector Labs); (3) biotinylated universal antibodies for immunoglobulin (IgG), to localize retained endogenous serum antibodies (1:1000, Vector Labs, Burlingame, USA). Before staining, endogenous peroxidase was quenched with a 15-mins incubation in 3% (v/v) peroxide/phosphate-buffered saline (PBS Sigma). After overnight incubation in primary antibody and PBS wash steps, sections for ED-1 staining were incubated with biotinylated anti-mouse antibody. Bound biotinylated antibodies were then tagged by incubation with avidin conjugated to biotinylated peroxidase (Vector Labs, ABC kit) followed by peroxidase visualization with a reporter, 3,3′-diaminobenzidine (DAB) (Vector Labs, DAB-peroxidase substrate kit).
To assess the significance of differences in the means, we compared ischemic and contralateral cortical regions within a reperfusion time using ANOVA; Tukey's test was used for post hoc comparisons. For evaluation of relationship between two variables, Pearson correlation coefficient was used. Data are expressed as mean ± s.d.
Results
The microvessel volume density 30 days after tMCAo (Figure 1 and Table 1) was significantly increased in the IMA of infarcted tissue compared either to the OMA in parenchyma outside of the infarct, or to the corresponding contralateral cortical area (F = 42.675, P < 0.001). The number, surface area density, and length density of microvessels in the 30-day cases were also significantly greater in the IMA than in the OMA or the homologous contralateral cortical area (Table 1). After 90 or 165 days after 2-h tMCAo, however, there were no significant differences in microvessel parameters (NA, VV, SV, and LV) between the IMA, the OMA, and the contralateral cortical area. Figure 2 shows the evolution of microvessel volume density over time, illustrating that the 30-day increase in excess microvessel density abates partially by 90, and completely by 165 days after tMCAo.
Stereological estimates of microvascular density in cortex 30, 90, and 165 days after tMCAo. The IMA contains infarcted cortical area within the cystic margin, the OMA is the cortical region beyond the cystic margin and the ‘Contra' is the corresponding cortical area of contralateral nonischemic hemisphere. Comparisons tested with ANOVA and Tukey's post hoc procedure
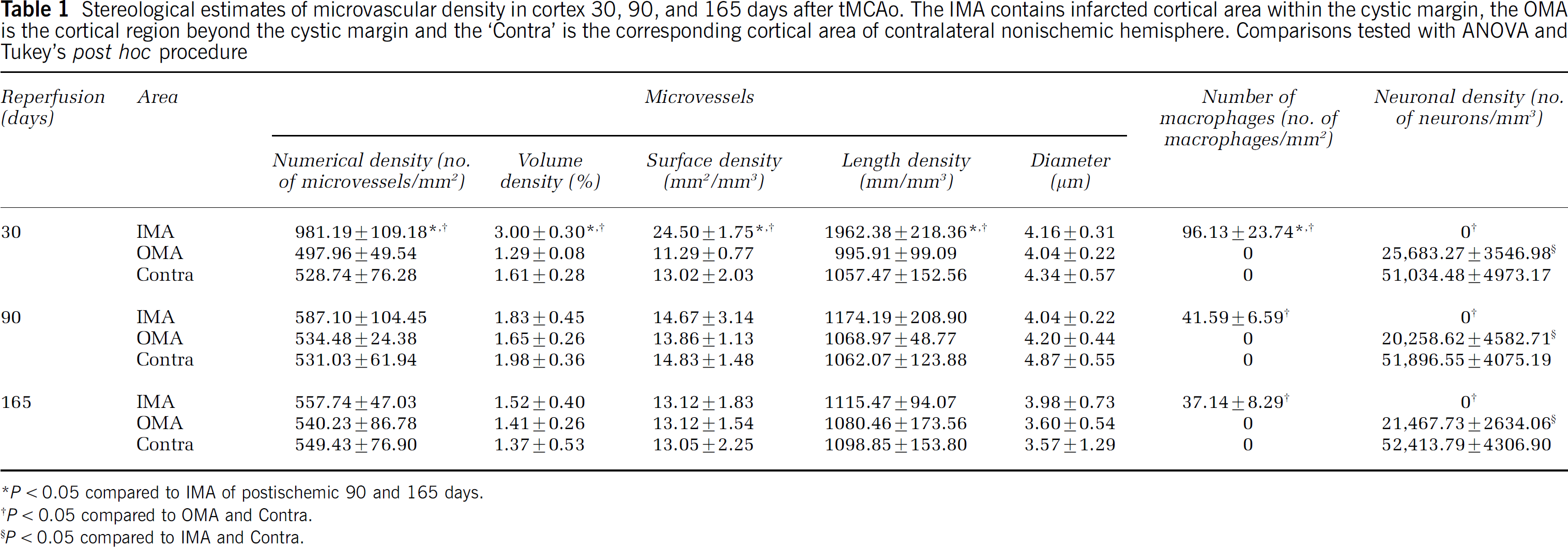
P < 0.05 compared to IMA of postischemic 90 and 165 days.
P < 0.05 compared to OMA and Contra.
P < 0.05 compared to IMA and Contra.
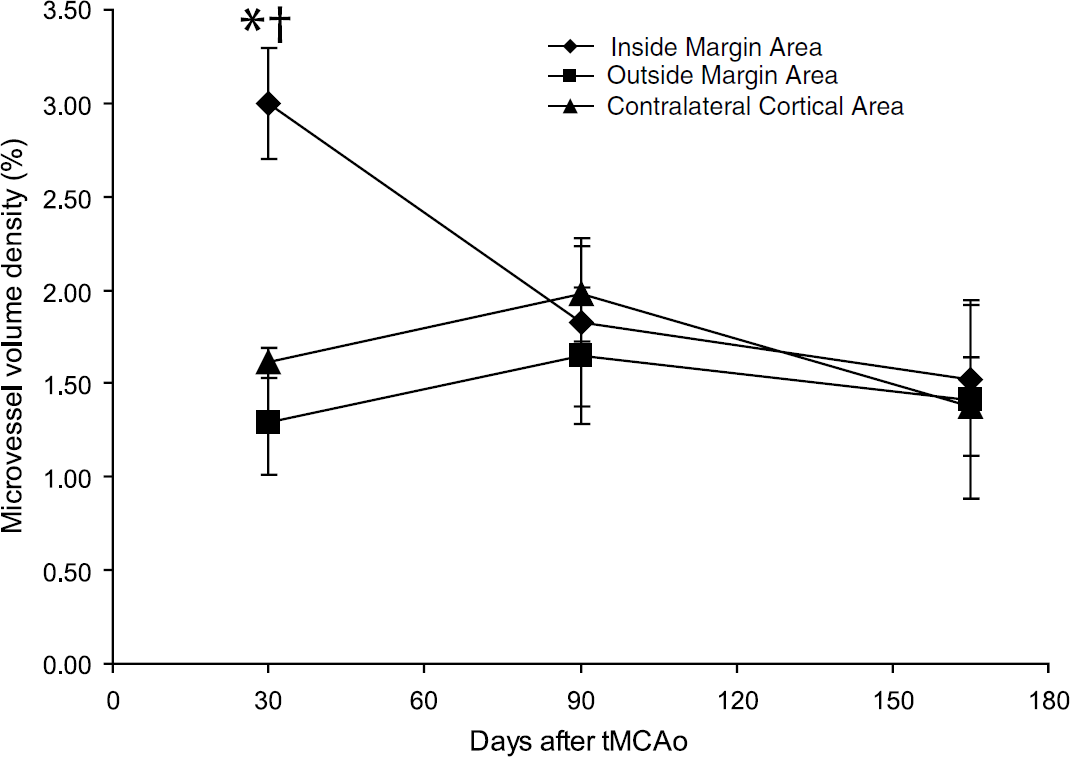
Microvessel volume density in the cortex 30, 90, and 165 days after tMCAo. The stereological parameter VV (density of microvessels in cortex) was measured using a systematic, nonbiased method previously validated (Manoonkitiwongsa et al, 2001a). The microvessel volume density was significantly elevated in the IMA region 30 but not 90 or 165 days after tMCAo, compared with contralateral control measures (P < 0.001, ANOVA with Tukey's post hoc procedure). *P < 0.001 compared with IMA of postischemic 90 and 165 days. †P < 0.05 compared with OMA and Contra.
No morphologically intact neurons were observed in the IMA, consistent with the criteria for identification of IMA as infarcted tissue (Figure 1C and 1D and Table 1). There was a significant reduction in neuronal density in the OMA, near the infarct, compared with the contralateral cortical area (Figure 1E and 1F and Table 1, P < 0.05 at all time points). Qualitative comparisons indicated that cysts expand over time in this model (Figure 1).
Macrophages with a distinctly phagocytic morphology were found only in the IMA, consistent with earlier findings (Manoonkitiwongsa et al, 2001a). The number of such phagocytic macrophages in the IMA region was highest 30 days after ischemia but significantly decreased 90 and 165 days after ischemia. Since some phagocytic macrophages were still found 165 days after ischemia in the IMA, however, we infer tissue clean-up may continue very late after tMCAo (Figure 3 and Table 1). To assess the relation between microvessel density and phagocytic macrophage numbers, we compared numbers of phagocytes to vessel volume density in the IMA. Combining the data from all three reperfusion groups, volume density showed positive linear correlation with increased number of macrophages (Pearson r = 0.71, R2 = 0.50, P < 0.05). There were similar correlations for numeric, surface, and length densities (data not shown). The data was insufficient to allow correlations within a reperfusion group. No increase in phagocyte number or change in microvascular parameters were seen outside the IMA, that is, in the OMA of neocortical parenchymal tissue outside of the infarct (Figure 3 and Table 1).
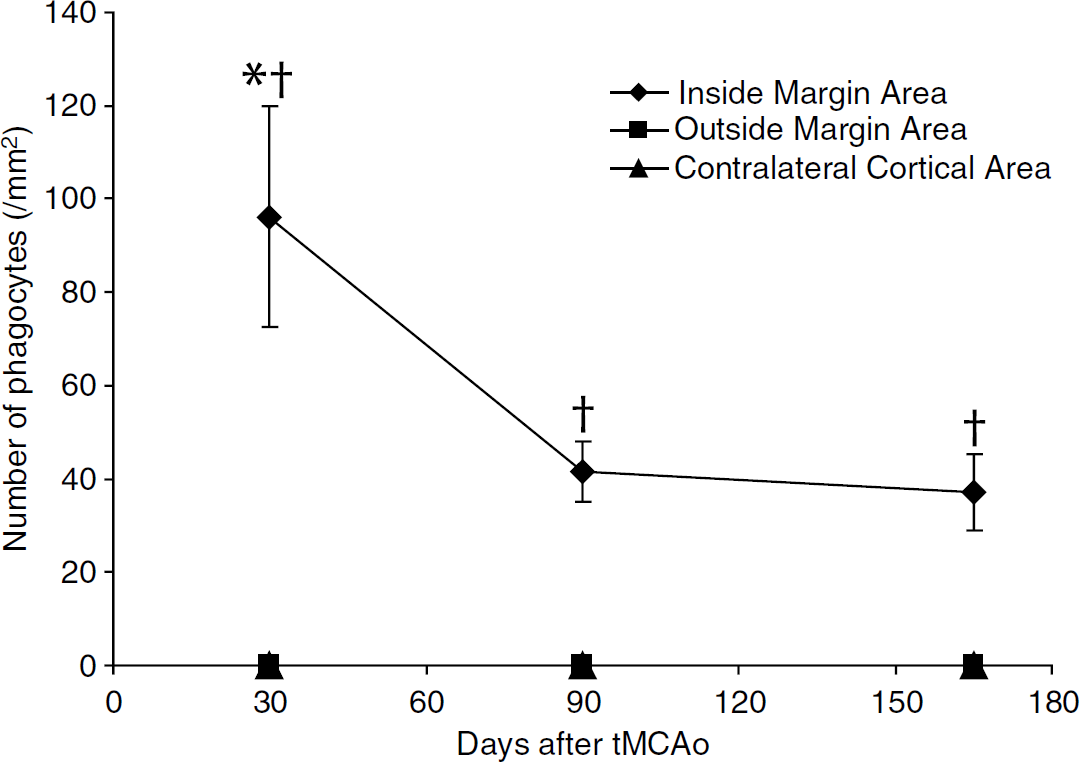
Phagocyte counts around cystic infarction 30, 90, and 165 days after tMCAo. Phagocytes were identified as vacuolated cells containing cellular debris; activated microglia and monocytes were not included to assure reproducibility. Phagocyte number was significantly increased 30 compared with 90 or 165 days after tMCAo. *P < 0.05 compared with IMA of postischemic 90 and 165 days. †P < 0.05 compared with OMA and Contra.
Immunoglobulin immunostaining allowed identification of extravasated serum proteins. At 30 days after tMCAo there was significant extravasation in and around the infarcts (Figure 4A). The area with IgG immunoreactivity was confined to a thin boundary zone surrounding cystic infarction 90 and 165 days after tMCAo (Figure 4B illustrates 165 days). To evaluate acute blood—brain barrier leakage, fluorescein-dextran was injected intravenously 30 mins before killing. The extravasation of fluorescein-dextran, as identified by antifluorescein immunoreactivity, was seen in and near the necrotic infarct 30 days after tMCAo (Figure 4C). However, 165 days after tMCAo, anti-fluorescein immunoreactivity was barely detected (Figure 4D). Taken together these data suggest that the immunoreactive IgG observed in the 165-day cases may represent the persistence of IgG that was extravasated at an earlier time point.
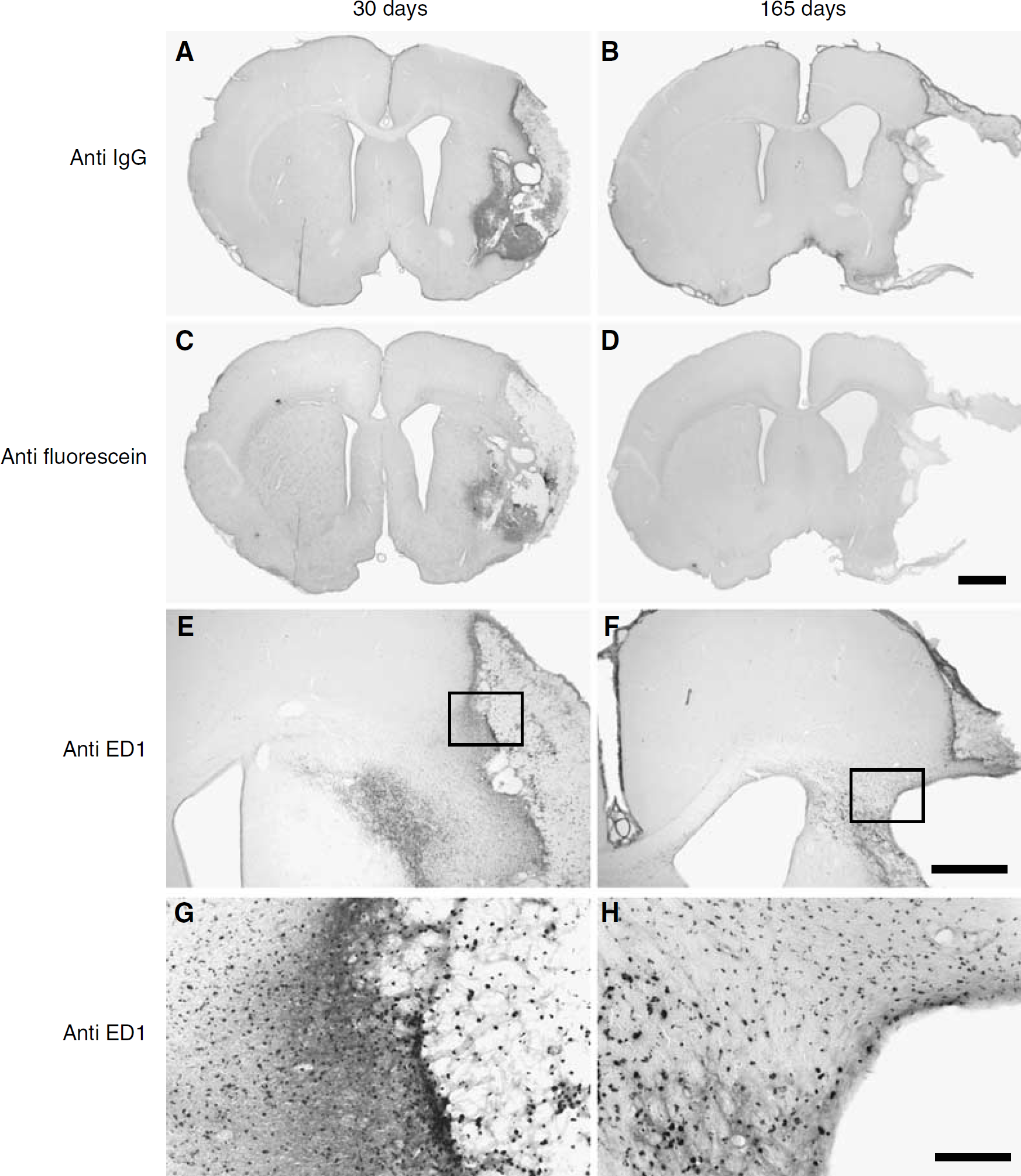
Localization of immunoreactivity for IgG, fluorescein-dextran, and ED1 30 and 165 days after tMCAo. Extravasation of serum protein was detected in the cystic necrotic area 30 days after tMCAo using anti-IgG (
To document the inflammatory cellular response, we stained for ED1 as a marker of reactive microglia/macrophages/monocytes (Damoiseaux et al, 1994; Lehrmann et al, 1997). Immunoreactive ED1 cells were found within the infarcted cyst and also outside the cyst in the OMA 30 and 165 days after tMCAo (Figures 4E and 4F). Inside the cyst, ED1 positive cells were associated with phagocytes (Figures 4E and 4G) but outside the cyst, ED1 positive cells were present even though no phagocytes were seen, suggesting that the ED1 cells in the OMA may correspond to reactive microglia or monocytes in different stages of activation. Interestingly, ED1 positive cells were also found in the basal part of the corpus callosum (Figures 4F and 4H), perhaps in response to secondary degeneration from neuronal denervation after brain infarction.
Discussion
This study showed that the ischemic angiogenic response in cerebral infarcts results in a transient population of microvessels. Thirty days after ischemia these vessels are leaky to acutely injected fluorescein-dextran and are intermingled with phagocytic macrophages and extravasated IgG. The regression of these microvessels over longer survival times is accompanied by a partial loss of these phagocytic macrophages. As the cerebral infarcts were devoid of morphologically identifiable neurons by 30 days after ischemia, the function of these microvessels cannot be for local neuro-protection.
An emerging theme in studies of the response of the adult brain to stroke is that ischemic damage occurs at widely variant length scales, from highly localized pannecrosis at the site of cystic infarction to distant zones of gliosis (Katsman et al, 2003). The present study confirms observations of microvessel hyperdensities in the ischemic infarction zone (Manoonkitiwongsa et al, 2001a) and is consistent with previous studies that indicate that angiogenesis induced by ischemia is not a widespread reaction of parenchymal microvessels in the entire ischemic zone distal to MCA occlusion (Marti et al, 2000; Zhang et al, 2000, 2002). This is reminiscent of the localized angiogenesis observed after myocardial infarct where it has been postulated that a spatially restricted pattern of thrombospondin-1 in the infarct margin limits the spread of both ischemic inflammation and angiogenesis (Frangogiannis et al, 2005). In this regard, it is of interest that the earliest upregulation of endogenous VEGF messenger RNA after ischemia is in the ischemic infarct in rodents (Zhang et al, 2002) with little extension into parenchyma bordering the infarct. Immunocytochemical studies localize VEGF immunoreactive protein to endothelial cells—among other cell types—in infarcts after acute strokes (Kovacs et al, 1996). In other models of stroke, ischemic angiogenesis has been shown in large cortical surface vessels, a distinct vascular territory from the one examined in the present study (Wei et al, 2001; Zhang et al, 2000), and it is of interest that the pia-arachnoid through which such vessels course has also been identified as a site of VEGF upregulation after stroke (Marti et al, 2000). It should be noted that in the present study we did not find the elevations in microvessel number that have been documented in penumbral regions in mouse cortex after tMCAo (Hayashi et al, 2003). In that study, elevated microvessel counts were found 21 days after stroke and were identified by quantitation of laminin stained microvessels. Thus, the discrepancy between the present work and this previous study might stem from the use of different species or from methodological differences in vessel labeling and quantitation or both.
Recent studies suggest that angiogenesis triggered by exogenous VEGF or by ischemia in peripheral vascular beds requires additional factors for long-term stabilization (Kermani et al, 2005; Pettersson et al, 2000). The regression of supernumerary vessels in the infarcted tissue seen in the present study at the longer survivals of 90 and 165 days may reflect the well-documented downregulation of ischemictriggered expression of VEGF (Hayashi et al, 2003) in the brain 1 week after stroke. Instability of vessels whose formation is largely triggered by VEGF would be consistent with findings that vessels formed in response to exogenous VEGF are not stable and regress according to tissue dependent time courses (Pettersson et al, 2000). Furthermore, recent studies with VEGF antagonists point to a continuing vascular dependence on VEGF in normal adult peripheral capillaries and in the choroid plexus (Baffert et al, 2006; Kamba et al, 2006). In addition to a postulated vessel-stabilizing role for VEGF (Maharaj et al, 2006), vessel stabilization can also be dependent on neurotrophin signaling, as was recently showed in ischemic skeletal muscle (Kermani et al, 2005). The vascular regression observed in the present study, then, may reflect a coordinated reduction in these or other vessel stabilizing molecules.
The idea that ischemic angiogenesis in the brain could provide supplementary blood flow derives largely from studies of ischemia in other tissues such as the heart or skeletal muscle (Losordo and Dimmeler, 2004) in which improved blood flow after therapeutic angiogenesis resulted in new, functional collaterals. Furthermore, studies in human stroke patients have shown a correlation between elevated microvessel density in brain parenchyma and delayed mortality (Krupinski et al, 1994). A problem in evaluating ischemic angiogenesis within the brain parenchyma is to know whether supernumerary vessels support normal blood—brain barrier function. The failure of blood—brain barrier function is one cause of functional deficiencies in ischemic brain vasculature early after human stroke and it may contribute to long-term deficits since stainable extravasated albumin has been identified in infarction zones up to 4 weeks after stroke in humans (Liu, 1988). Experimental studies of microvessels in ischemic brain infarcts indicate that ischemic vessels are leaky after acute stroke (Veltkamp et al, 2005; Zhang et al, 2000) and experimental studies of cardiac arrest indicate that such leakiness can persist for as long as 1 year after the ischemic insult (Pluta, 2005). The present study extends the time window for active vascular leakage after the tMCAo model of stroke and shows that even after 30 days there are infarct subregions that leak high molecular weight fluorescein dextran. Of interest, VEGF antagonism after focal cerebral ischemia in the mouse reduces infarct formation at least in part by reduction of edema (van Bruggen et al, 1999); this may suggest that a decline in ischemic VEGF levels could contribute to the concomitant decline in the quantified microvessel density and to the qualitative observations of reduced fluorescein leakage at long reperfusion times, observed in the present study. While extravasated serum proteins can persist in the parenchyma up to 165 days after transient ischemia (Figure 4B), the paucity of fluorescein-dextran leakage at this time (Figure 4D) makes it unlikely that this staining reflects ongoing leakage. This persistent deposition of extravasated IgG, left over from the acute phase, may contribute to the prolonged inflammation after stroke that we and others have observed (Lehrmann et al, 1997).
The regression of microvessels associated with ischemic cyst formation is in striking contrast to the persistence of vessels formed after hindlimb or myocardial ischemia (Hariawala et al, 1996; Isner, 2000; Takeshita et al, 1994). Whether this difference in part results from blood flow failures in cystic vasculature, as this is one of the identified triggers for vessel regression (Carmeliet, 2000), remains to be determined.