
Abstract
Extracellular regulated kinase (ERK) transduce growth factor signals while c-Jun NH(2)-terminal kinase (JNK) delivers stress signals into the nuclei for regulation of gene expression. These signaling pathways were studied by laser-scanning confocal microcopy and Western blot analysis using phospho-specific antibodies on rat brains that were subjected to 15 minutes transient forebrain ischemia followed by varied periods of reperfusion. Extracellular regulated kinase was activated at 30 minutes and 4 hours of reperfusion in the nuclei and dendrites of surviving dentate gyrus (DG) cells, but not in dying CA1 neurons after ischemia. Tyrosine phosphorylation of Trk kinase, an ERK upstream growth factor receptor, was elevated in the DG tissue, and to a lesser extent in the CA1 region. In addition, phosphorylation of activating transcription factor-2 (ATF-2) and c-Jun was selectively increased in CA1 dying neurons during the late period of reperfusion. These findings suggested that the Trk-ERK signaling pathway might be neuroprotective for dentate granule cells. The activation of ATF-2 and c-Jun pathways in the late period of reperfusion in CA1 dying neurons might reflect damage signals in these neurons. These results suggested that the lack of protective signals acting in concert with the presence of damage signals in CA1 neurons after ischemia might contribute to delayed neuronal death after transient forebrain ischemia.
An insult of 15 minutes forebrain ischemia caused delayed selective neuronal death in CA1 pyramidal neurons at approximately 3 days of reperfusion after the ischemic episode, but leaving CA3-dentate gyrus (DG) and most neocortical neurons intact (Kirino, 1982; Pulsinelli et al., 1982; Smith et al., 1984). This selective and delayed cell death provided a model for studying intracellular signaling pathways in reversible and irreversible neuronal damage.
The mitogen-activated protein kinase (MAPK) family is composed of extracellular regulated kinase (ERK), c-Jun NH(2)-terminal kinase (JNK) and p38 protein kinase. Activation of ERK by growth factors initiates a signal transduction pathway responsible for regulation of gene expression (Hu and Weiloch, 1994; Fukunaga and Miyamoto, 1998). It is known that cyclic AMP responsive element-binding protein (CREB) is a transcription factor regulated by ERK (Brindle et al., 1992; Impey et al., 1998). Stress can also cause the activation of JNK and p38 protein kinase resulting in the delivery of stress signals into the nucleus which leads to the regulation of gene expression (Bogoyevitch et al., 1996). Furthermore, activating transcription factor-2 (ATF-2) is phosphorylated by p38 kinase and c-Jun is regulated by JNK (Bogoyevitch et al., 1996).
Alteration of ERK and JNK phosphorylation has been observed in postischemic brain tissues (Campos-Gonzalez and Kindy, 1992; Hu and Wieloch, 1994; Saluja et al., 1999; Shamloo et al., 1999). However, there is controversy as to whether activation of MAPK pathways is beneficial or detrimental in postischemic neurons (Hetman et al., 1999; Alessandrini et al., 1999). It is unknown whether alteration of these pathways occurs in dying neurons or surviving neurons after brain ischemia. In addition, subcellular redistribution of ERK has not been reported. The objective of the current study was to investigate ERK and JNK pathways in dying CA1 neurons and surviving DG neurons after ischemia by laser-scanning confocal microscopy and Western blot analysis. The results suggested that the Trk-ERK pathway was activated in surviving DG neurons, whereas c-Jun and ATF-2 were activated in dying CA1 neurons in the late period of reperfusion. These findings indicated that the lack of cell survival signals and the presence of cell damage signals might render CA1 neurons vulnerable to transient ischemia.
METHODS
Ischemia model
Male Wistar rats (250 to 300 g) were starved overnight. Anesthesia was induced with 3% halothane followed by maintenance with 1% to 2% halothane in an oxygen/nitrous oxide (30%/70%) gas mixture. Catheters were inserted into the external jugular vein, tail artery, and tail vein to allow blood sampling, arterial blood pressure recording, and drug infusion. Loose ligatures encircled both common carotid arteries. Fifteen minutes before ischemia induction and 15 minutes postischemia, blood gases were measured and adjusted to Pa
Immunocytochemistry
Double-label fluorescence immunocytochemistry was performed on coronal brain sections (50 μm) from animals subjected to 15 minutes of ischemia followed by 30 minutes, 4 hours, and 24 hours of reperfusion and on sections from sham-operated control animals. A Monoclonal antibody against phospho-ERK1/2 and polyclonal antibodies against phospho-c-Jun and phospho-ATF-2 were obtained from New England Biolabs (Beverly, MA, U.S.A.). A mouse monoclonal antibody against calbindin was obtained from Sigma (St. Louis, MO, U.S.A.). The brain sections were washed twice in PBS for 5 minutes at room temperature (RT) and then in PBS containing 0.2% Triton X-100 (TX-100) for 30 minutes. Nonspecific binding sites were blocked in 3% BSA in PBS/0.2% TX-100 for 30 minutes. The primary antibodies were diluted 1:200 in PBS/0.1% TX-100 and 1% BSA. After incubation (overnight at 4°C) in the polyclonal primary antibodies, the sections were washed three times in PBS containing 0.1% TX-100 for 10 minutes at RT. The sections were then incubated for 2 hours at 4°C with the monoclonal antibody against calbindin diluted 1:400 in PBS/0.1% TX-100 and 1% BSA, followed by three washes for 10 minutes at RT. The fluorescent secondary antibodies, fluorescein-labeled anti-rabbit and lissamine rhodamine-labeled anti-mouse diluted 1:200, or propidium iodide (15 μg/mL) in PBS containing 0.1% TX-100 and 1% BSA was applied for 1 hour at RT. Immunostaining was eliminated when primary antibodies were omitted. Sections were washed several times in PBS/0.1% TX-100, mounted on glass slides, and coverslipped using Gelvatol (Pharmacia, Peapack, NJ, U.S.A.). The slides were analyzed on a BioRad MRC 1024 laser-scanning confocal microscope (Hercules, CA, U.S.A.). Confocal images that encompassed the hippocampus from one hemisphere were adobe photoshop montages of eight 4x images. For histologic assessment of neuronal damage, Vibratome (Leica) sections from the brains used for immunostaining were stained with 0.2% cresyl fast violet in an acetate buffer at pH 3.5.
Preparation of nuclear extracts and subcellular fractions
A section from sham-operated animals and from ischemic brains after 0 minutes, 4 hours, and 24 hours of reperfusion, Bregma −2.56 through −4.30, was isolated by gross dissection with a scalpel and forceps from an intact brain previously frozen in situ with liquid nitrogen. This section of brain was further divided into 1-mm coronal sections and the CA1 and CA3/DG regions were dissected from the dorsal hippocampus. The transverse hippocampal artery was used as a landmark to separate the CA1 region from the inner blade of the DG. All steps were performed in a cold box at −22°C. Subcellular fractions were prepared from each of the four animals per condition. Sample preparations were carried out at 4°C. Brain tissues (hippocampal CA1 region and CA3/DG region) were homogenized using a Dounce homogenizer (30 strokes) in 15 times the volume of homogenization buffer containing 15 mmol/L Tris base/HCl pH 7.6, 1 mmol/L DTT, 0.25 mol/L sucrose, 1 mmol/L MgCl2, 125 μg/mL pepstatin A, 10 μg/mL leupeptin, 2.5 μg/mL aproptonin, 0.5 mmol/L PMSF, 25 mmol/L EDTA, 1 mmol/L EGTA, 0.1 mol/L Na3VO4, 50 mmol/L NaF, and 2 mmol/L sodium pyrophosphate. The homogenates were then centrifuged at 920x g at 4°C for 10 minutes. The resultant pellet (P1) was used as the crude nuclear fraction. The supernatants were further centrifuged at 10,000× g for 10 minutes to obtain crude synaptosomal fraction (P2). The resulting supernatants were further centrifuged at 165,000× g for 1 hour at 4°C to cytosolic fraction (S3) and microsomal fraction (P3). P1, P2, and P3 were suspended with a homogenization buffer containing 0.1% TX-100. The protein concentration of the samples was determined by the method of Lowry et al. (1951).
Western blot analysis
Western blot analysis was carried out on 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis according to the method of Laemmli (1970). One sample containing 20 μg of protein was applied to each lane in a slab gel of sodium dodecyl sulfate polyacrylamide gel electrophoresis. After electrophoresis, proteins were transferred to an immobilon-P membrane. The membrane was incubated with a primary monoclonal antibody against phospho-ERK1/2 at a dilution of 1:5000, polyclonal antibodies against phospho-Trk, JNK, and phospho-JNK each at a dilution of 1:1000 for 4 hours at 4°C. All of the antibodies were from New England Biolabs. The membranes were then incubated with horseradish—peroxidase conjugated anti-rabbit or anti-mouse secondary antibodies for 45 minutes at RT. The blots were developed using the Amersham ECL detection method (Piscataway, NJ, U.S.A.). Four animals in each experimental group were used to analyze the levels of the proteins by Western blot. Two samples derived from two animals in each experimental condition were run on the same gel.
RESULTS
Examination of cresyl violet stained sections from postischemic animals that were also used for immunocytochemistry revealed that the neurons in the CA1, CA3 and DG did not show apparent abnormal morphology, relative to sham-operated controls, at any time point up to 2 days of reperfusion. However, a frank neuronal death occurred in more than 95% of CA1 pyramidal neurons and about 10% of cortical neurons in postischemic brains after 3 days of reperfusion, whereas neurons in CA3 and DG did not show any obvious pathologic changes as examined by light microscopy. This pattern of neuronal death was consistent with observations in previous studies using this model (Smith et al., 1984).
Brain sections from sham-operated control (C) and 15 minutes ischemia, followed by 30 minutes and 24 hours of reperfusion, were double-stained with a monoclonal antibody against phospho-ERK1/2 and propidium iodide and examined by laser-scanning confocal microscopy. In Figs. 1A and 1B, red represents propidium iodide, the green indicates fluorescein conjugated antibodies specific for phospho-ERK1/2, and yellow indicates double labeling caused by the overlay of the red and green fluorochromes. No obvious difference in the intensity or morphology of propidium iodide stained nuclei was noted in the hippocampus between sham-operated control and postischemic conditions (Figs. 1A and 1B). Phospho-ERK1/2 was dramatically up-regulated in DG neurons, but not in CA1 neurons, at 30 minutes of reperfusion after 15 minutes of ischemia. Furthermore, the increase in active ERK returned to control levels within 24 hours of reperfusion (Fig. 1A). Higher magnification revealed that a significant increase in active ERK was not only in the dendrites but also in the nuclei (Fig. 1B), which indicated a redistribution of active ERK. Western blots showed a transient increase in active 42 kDa ERK2, but not 44 kDa ERK1, which occurred at 30 minutes and 4 hours of reperfusion in the DG nuclear fraction, and then returned to control level at 24 hours of reperfusion (Fig. 2). However, both 42 kDa and 44 kDa ERK were up-regulated in the cytosolic fraction of DG neurons at 30 minutes (Hu and Weiloch, 1994) and 4 hours of reperfusion after ischemia (Fig. 2). Increased phosphorylation of TrkB, an ERK upstream growth factor receptor, occurred in the membrane fraction from the DG region, whereas moderate changes of TrkB phosphorylation were found in the CA1 membrane fraction (Fig. 3, arrow). Even though the antibody used in the current study against phospho-Trk recognized all Trks isoforms, TrkB is a major neurotrophin receptor that is overexpressed in the hippocampus after brain injury (Lindvall et al., 1992; Medio et al., 1993).
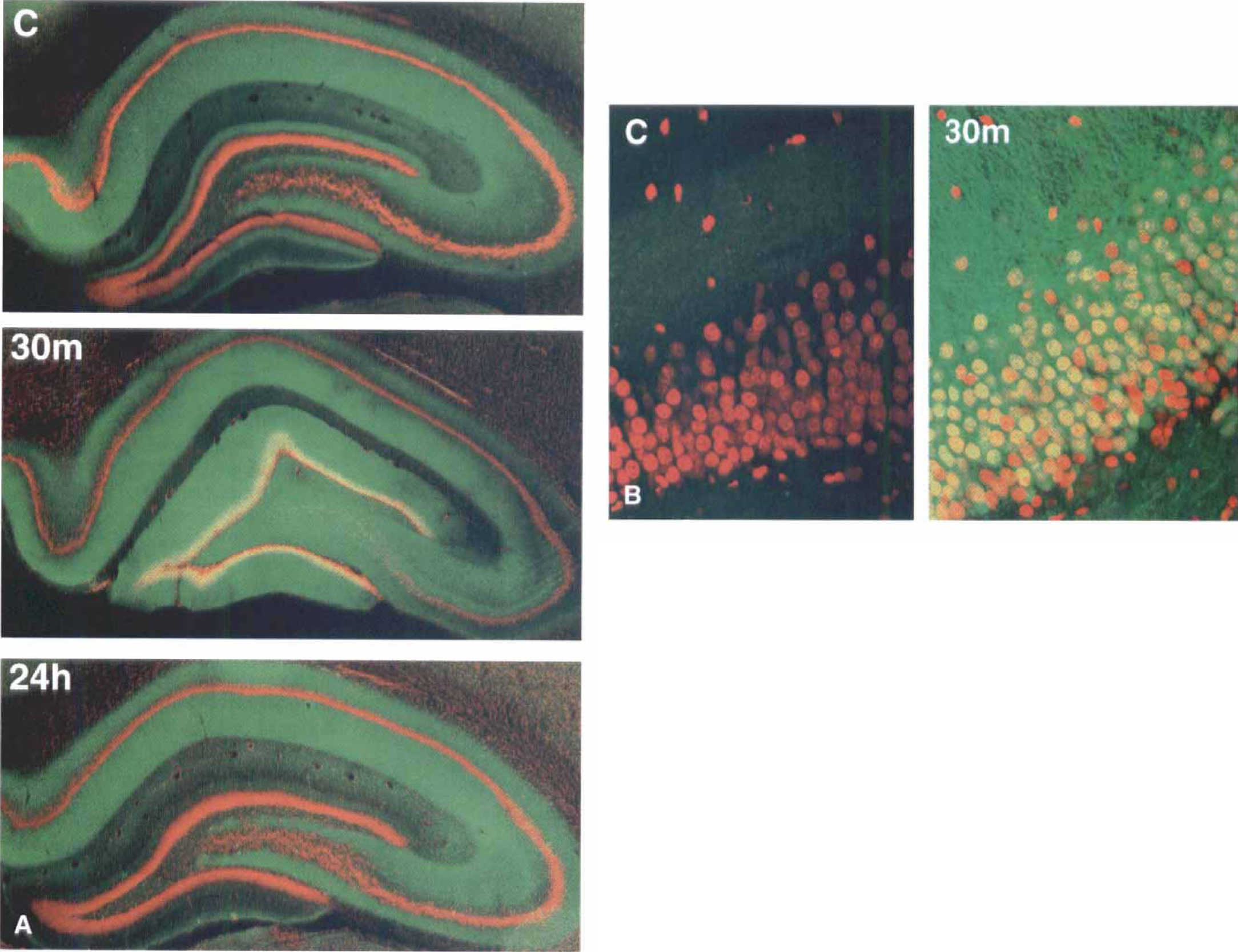
Phosphorylation of mitogen-activated protein kinase (MAPK) is highly increased in dentate gyrus (DG) neurons at 30 minutes of reperfusion.
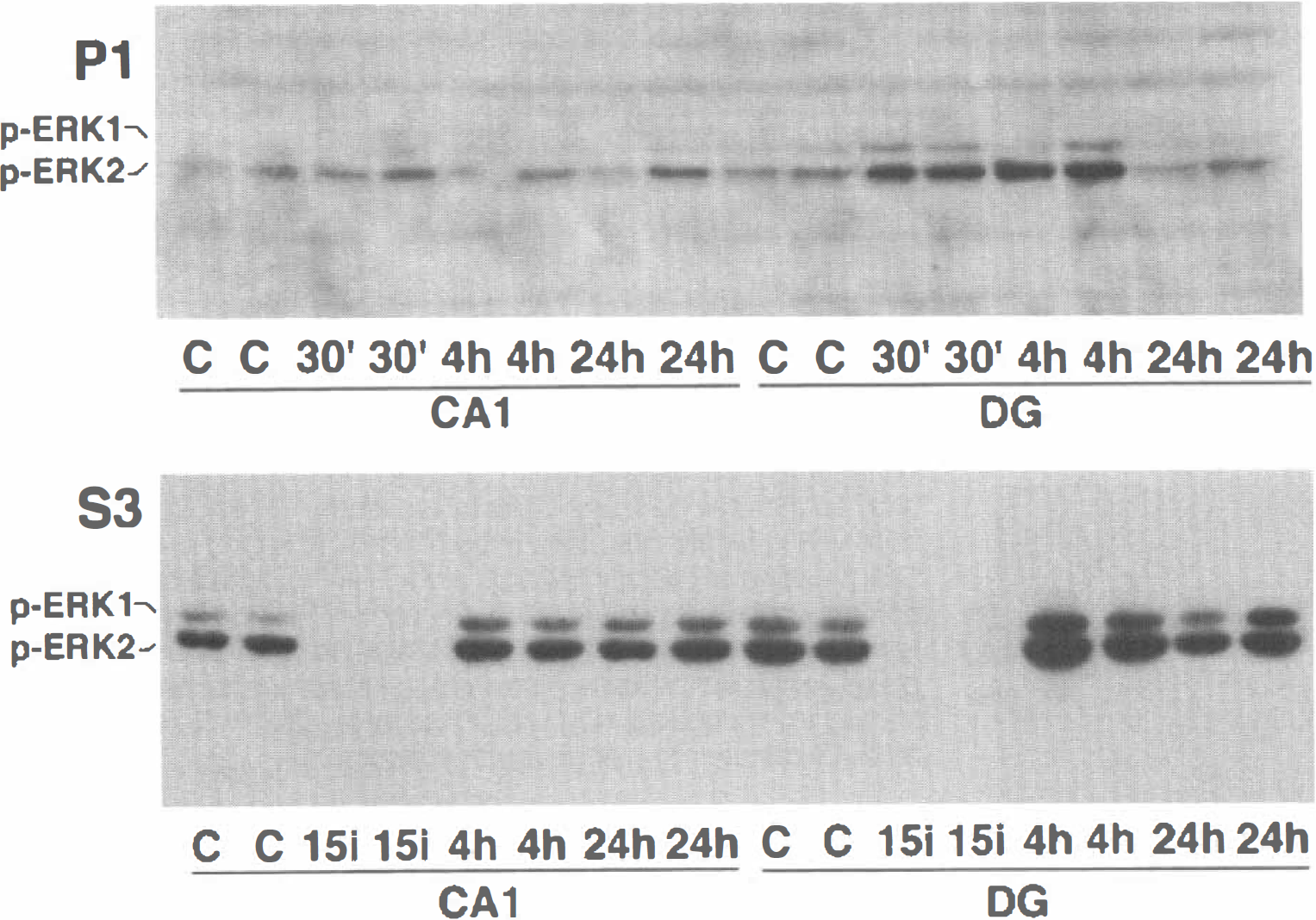
Phospho-extracellular regulated kinase (ERK) is markedly increased in the nuclear fraction of dentate gyrus (DG) regions and the cytosolic fraction of both CA1 and DG regions. Samples of hippocampal CA1 and DG were from sham-control rats (C) and rats subjected to 15 minutes of ischemia (15i) followed by 4 hours (4h) and 24 hours (24h) of reperfusion. Each lane represents a sample from one rat. Two separate samples in each experimental group were run on sodium dodecyl sulfate polyacrylamide gel electrophoresis. The immunoblots were labeled with antibodies against phospho-ERK and visualized with chemiluminescence.
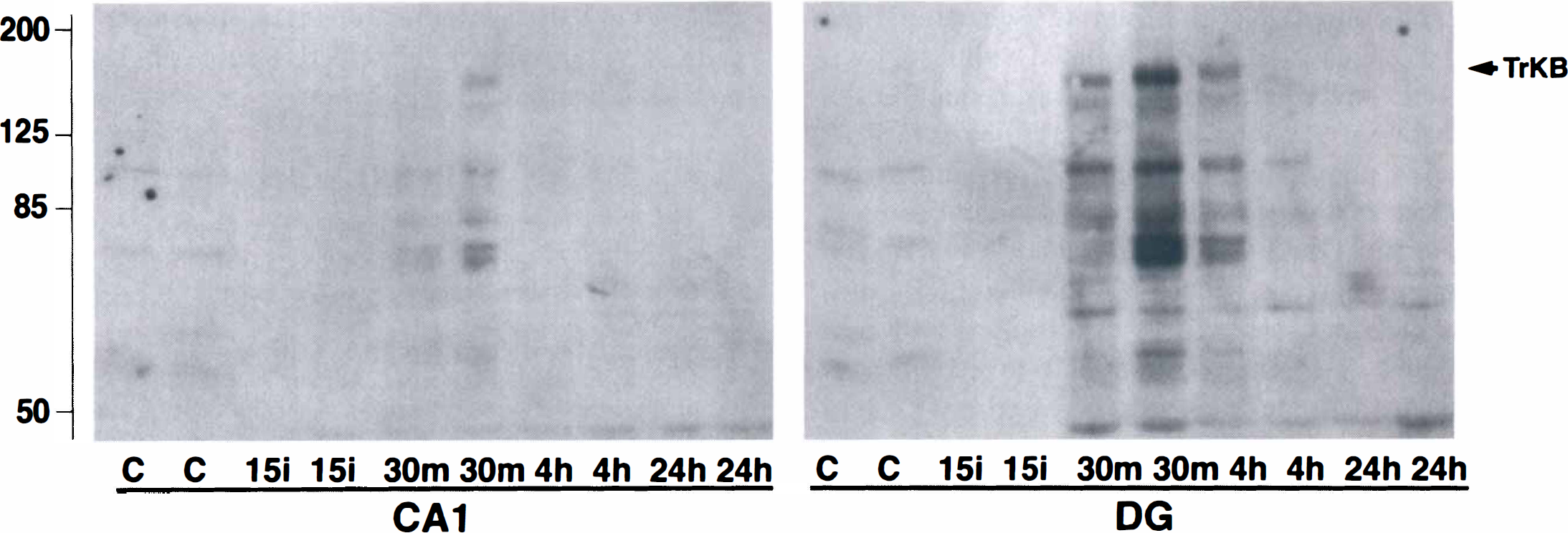
Phospho-Trk increases in the membrane fraction of dentate gyrus (DG) regions relative to CA1 region after ischemia. Samples of hippocampal CA1 and DG were from sham-control rats (C) and rats subjected to 15 minutes of ischemia (15i) followed by 30 minutes (30m), 4 hours (4h), and 24 hours (24h) of reperfusion. Each lane represents a sample from a single animal. Two separate samples in each experimental group were run on sodium dodecyl sulfate polyacrylamide gel electrophoresis. The immunoblots were labeled with antibodies against phospho-Trk and visualized by chemiluminescence.
In contrast to Trk and ERK, which were activated in the early period of reperfusion in surviving DG neurons, phosphorylation of ATF-2 and c-Jun were up-regulated in dying CA1 neurons in the late period of reperfusion. Brain sections of control and postischemia were either double-labeled with phospho-ATF-2 (green) and calbindin (red), or phospho-c-Jun (green) and calbindin (red) then examined by laser-scanning confocal microscopy (Fig. 4). Moderate increases in phospho-ATF-2 and phospho-c-Jun were found in both CA1 neurons and DG granule cells after 4 hours of reperfusion (Hu et al., 1999). However, after 24 hours of reperfusion, increased phosphorylation of ATF-2 and c-Jun primarily was seen in dying CA1 neurons (Fig. 4).
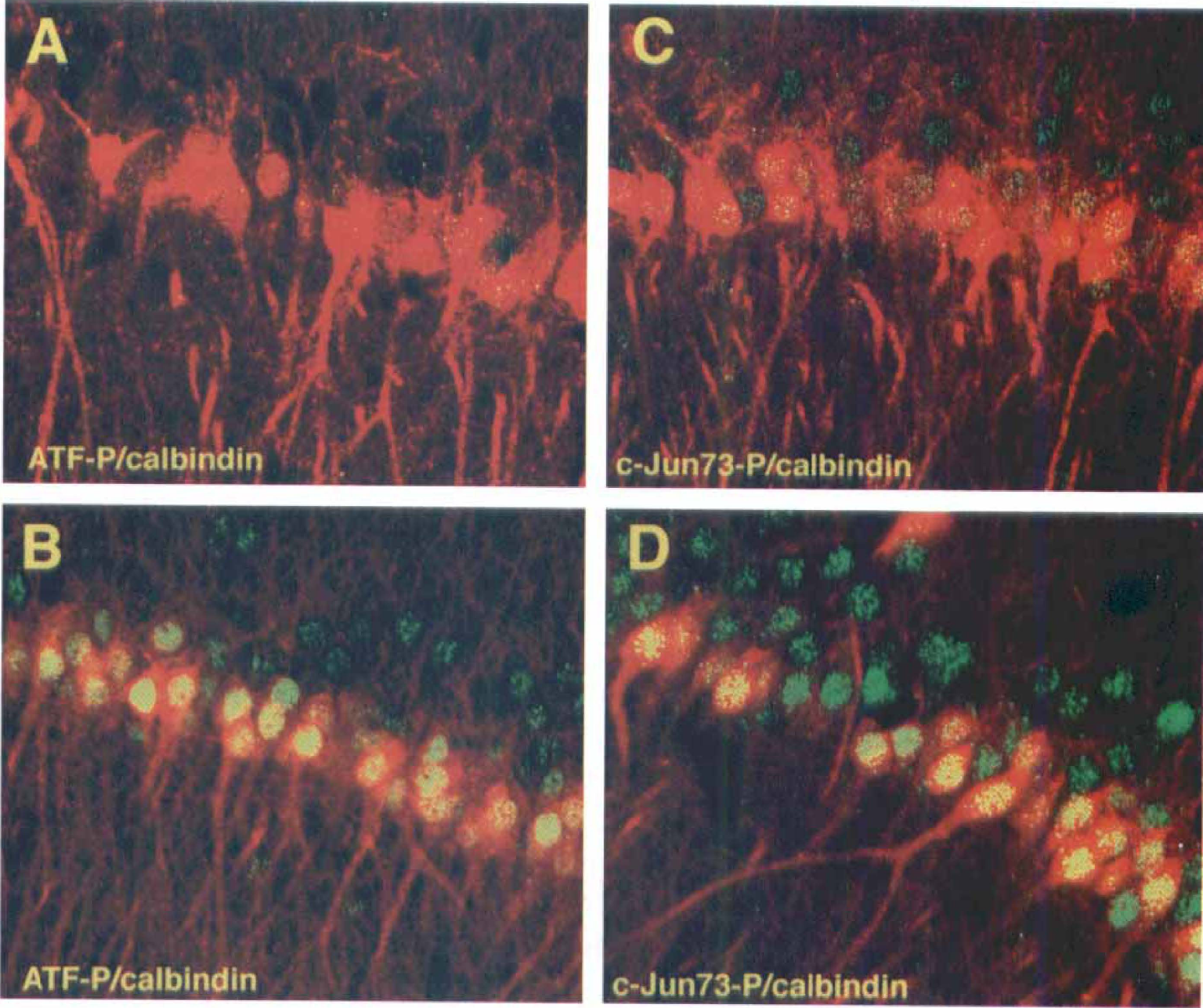
Increased expression of Phospho-ATF-2 and Phospho-c-Jun kinase in the nuclei of CA1 pyramidal neurons at 24 hours of reperfusion.
DISCUSSION
In the current study, we investigated regional, temporal, and subcellular phosphorylation of Trk, ERK, ATF-2, and c-Jun using phospho-specific antibodies. In addition to the roles of ATF-2 and c-Jun in cellular stress, the involvement of Trk and ERK in cell growth and repair gave value to studying these signal transduction molecules in ischemic neuronal cell death. We used an ischemic model that produces irreversible damage in CA1 pyramidal neurons and reversible damage in dentate granule cells (Smith et al., 1984). We found that Trk and ERK were up-regulated in surviving DG neurons during the early phase of reperfusion. In contrast, ATF-2 and c-Jun were phosphorylated in CA1 dying neurons in the late phase of reperfusion. In DG neurons after ischemia, phosphorylation of Trk occured in the membrane fraction, whereas ERKs were phosphorylated in cytosolic and nuclear fractions. There was no obvious change in phosphorylation of MAPK in the striatum, and the level of ERK activation in the neocortex and CA3 was intermediate when compared with the CA1 and DG regions. The apparent increase in TrkB and ERK phosphorylation in postischemic neurons might be indicative of growth factor signal activation in these neurons, which was consistent with the demonstrated cell repair roles of TrkB and ERK in physiologic and pathologic paradigms (Lindvall et al., 1992; Medio, 1993; Ginty et al., 1994, Fukunaga and Miyamoto, 1998; Hetman et al., 1999). The activation of ATF-2 and c-Jun in CA1 neurons after ischemia was also consistent with the fact that ATF-2 and c-Jun are expressed and activated in response to stress (Dragunow et al., 1993; Bogoyevitch et al., 1996; Herdegen et al., 1998). Activation of cell surviving pathways such as TrkB-ERK might contribute DG granule cells resistance to ischemia, whereas activation of JNK-c-Jun and p38-ATF-2 in CA1 dying neurons in the late period of reperfusion might reflect cell damaging signals present in these neurons.
The specificity of the phospho-specific antibodies used in the current study has been extensively characterized by many investigators (Dieckgraefe et al., 1997; von Willebrand et al., 1998). Moreover, dephosphorylation in phospho-ERKs (Fig. 2, S3) and phospho-Trk (Fig. 3) during ischemia observed in the current study further suggested that the antibodies were phospho-specific. Previous studies also demonstrate that phosphorylation of ATF-2 and c-Jun are decreased during ischemia (Hu et al., 1999). We knew that ATP is depleted within approximately 2 to 3 minutes after the onset of ischemia (Siesjö, 1988). Therefore, protein kinases would not have ATP available for phosphorylation of proteins, and active phosphatases would continue to dephosphorylate most phosphoproteins (Hu and Wieloch, 1994).
In surviving neurons of the DG, the Trk—ERK pathway was highly activated. These results agree with previous studies that demonstrated that levels of trkB mRNA and protein are markedly increased in DG neurons after brain injuries (Lindvall et al., 1992). The current study extended this observation to show that phosphorylation of Trk was also highly increased in the membrane fraction from DG tissue after ischemia. An increase in phosphorylation of ERK after forebrain ischemia has been reported previously in several studies (Campos-Gonzalez and Kindy, 1992; Hu and Wieloch, 1994; Saluja et al., 1999; Shamloo et al., 1999). However, results from the current study demonstrated that active 42 kDa ERK not only increased in surviving DG neurons, but was redistributed into the nuclei (Fig. 2). These results could be attributed to the initiation of a signal transduction cascade coinciding with expression of neurotrophins and activation of their receptor. Phosphorylation of ERKs and redistribution of active 42 kDa ERK into the nuclei can activate transcription factors in surviving DG neurons after ischemia. CREB is activated by the Trk—ERK pathway (Xing et al., 1998; Impey et al., 1998; Hansen et al., 1999), and we have shown previously that CREB phosphorylation is increased primarily in surviving DG neurons (Hu et al., 1999). In addition, a reduction in brain-derived neurotrophic factor protein in hippocampal CA1 preceded the delayed neuronal damage after ischemia (Yamasaki et al., 1998). These results supported the idea that the activation of Trk—MAP kinase—CREB pathway in DG neurons might contribute to the resistance of these neurons to ischemia. Furthermore, the lack of activation of this surviving pathway in CA1 neurons might render these neurons vulnerable to ischemia.
In CA1 dying neurons, phospho-ATF-2 and phospho-c-Jun were selectively phosphorylated in the late period of reperfusion after ischemia. This was in accordance with an increase in JNK activity and expression of c-Jun protein during the late period of reperfusion after brain ischemia (Dragunow et al., 1993; Neumann-Haefelin et al., 1994; Ferrer et al., 1997; Herdegen et al., 1998). Because JNK-c-Jun and p38-AFT-2 are activated after stress in many systems (Dragunow et al., 1993; Bogoyevitch et al., 1996; Herdegen et al., 1998), selective activation of these stress signaling pathways may reflect the presence of stress signals in CA1 dying neurons in the late period of reperfusion. The presence of predominantly more stress signals versus surviving signals in CA1 pyramidal neurons than in dentate granule cells may contribute to the selective vulnerability of hippocampal neurons after ischemia.