
Abstract
In this study, we have assessed the ability of two TAT-fused peptides PYC36
Introduction
Stroke is a leading cause of death and disability worldwide. Current treatments for ischemic stroke such as thrombolysis (tissue plasminogen activator), decompressive hemicraniectomy, and aspirin, are aimed at either restoring perfusion to the compromised tissue or preventing recurrence of the ischemic event (Donnan et al, 2008). Targeted neuroprotective therapies that are able to limit the damage to potentially salvageable brain tissue remain unresolved (Campbell et al, 2008). Therefore, agents that block specific cell death pathways, which are activated after cerebral ischemia, are of potential significance in terms of reducing ischemic infarction and improving motor and sensory function.
One well-characterized cell death pathway activated after cerebral ischemia is the mitogen-activated protein kinase pathway, with most attention being focused on the blockade of the c-Jun N-terminal kinase (JNK) protein of this pathway. Less focus has been on the activator protein-1 complex, namely the c-Jun protein, which is downstream of JNK. The activator protein-1 complex comprising homodimerized c-Jun protein (or heterodimerized with other activator protein-1 proteins; c-fos), which is activated after phosphorylation by JNK, promoting the expression of pro-cell death proteins, such as Fas, Fas-L, DP5, Bax, and c-Jun itself (Besirli et al, 2005; Gao et al, 2005). Meanwhile, JNK is also able to phosphorylate other proteins involved in cell death including p53 and Bcl-2 family members Bad, Bax, Bid, and Bim (Cao et al, 2002; Plesnila et al, 2001; Tsuruta et al, 2004; Wang et al, 2007). After cerebral ischemia, besides being phosphorylated by JNK, c-Jun can also be phosphorylated by other kinases such as VRK1, PLK3, p300, and p38δ, and thereby bypass JNK-mediated activation (Besirli et al, 2005; Bessero et al, 2010; Raivich, 2008; Sevilla et al, 2004). Therefore, repression of both JNK and c-Jun represents attractive targets to inhibit cell death after cerebral ischemia.
With respect to c-Jun, our laboratory has identified 19 peptides (hereafter referred to as c-Jun inhibitory peptides) that downregulate activator protein-1 transcription (Meade et al, 2010a). Furthermore, we have shown that several of these peptides when used synthesized to the cell penetrating peptide TAT, have in vitro neuroprotective activity in glutamate, kainic acid, and oxygen-glucose deprivation neuronal injury models (Craig et al, 2011; Meade et al, 2010a, 2010b). One of these c-Jun inhibitory peptides is PYC36
Materials and methods
Rat Transient Focal Cerebral Ischemia Model
This study was approved by the Animal Ethics Committee of the University of Western Australia and conducted according to the guidelines established for the use of animals in experimental research as outlined by the Australian National Health and Medical Research Council. Male Spontaneously Hypertensive (SH) rats weighing 255 to 305 g were kept under controlled housing conditions with 12 hours light–dark cycle with free access to food and water. Experimental animals were fasted overnight and subjected to 90 minutes of MCAO as follows.
Anesthesia was induced with 4% isoflurane and a 2:1 mix of N2O and O2 via mask. Anesthesia was maintained at 1.8% to 2% isoflurane. Cerebral blood flow was monitored continuously using laser Doppler flowmetry (Blood FlowMeter, AD Instruments, Sydney, NSW, Australia). The probe was located 1 mm posterior to the bregma and 4 mm from the midline of the hemisphere. A cannula was inserted in the right femoral artery to continuously monitor blood pressure and to provide samples for blood glucose and blood gas readings. Blood glucose was measured using a glucometer (MediSense Products, Abbott Laboratories, Bedford, MA, USA) and blood gases were measured using a blood gas analyser (ABL5, Radiometer, Copenhagen, Denmark). Blood pressure was maintained at >90 mm Hg. During surgery, rectal temperature was maintained at 37.5±0.5°C and temporalis muscle temperature as measured with a needle muscle probe (Physitemp Instruments, Clifton, NJ, USA), was maintained at 37.0±0.5°C. A heating fan was used when necessary.
The external carotid artery was ligated and cauterized to create a stump, whereby a 4–0-nylon filament with a 0.39-mm diameter silicone tip (Doccol, Redlands, CA, USA) was inserted into the common carotid artery. The pterygopalatine artery was ligated before filament insertion. The filament was advanced into the left internal carotid artery until laser Doppler flowmetry recorded a drop in cerebral blood flow. The filament was withdrawn after 90 minutes. The procedure was considered successful with a >30% decrease from baseline of cerebral blood flow after insertion of filament and a blood flow increase of >30% of baseline after filament withdrawal. Animals were given postoperative analgesia consisting of pethidine (3 mg/kg intramuscular) and bupivacaine (1.5 mg/kg subcutaneously) at head and leg incision sites. Animals were allowed to recover in a quiet holding room controlled at 27°C. Neurological deficit was confirmed in each animal 1 hour after completion of surgery, by observing flexion of the contralateral forelimb (right side) when suspended by the tail. The rectal temperatures of all experimental animals were monitored for 3 hours postoperatively at 20 minutes intervals and were maintained at 37.5±0.5°C with a heating pad and, when necessary, a heating fan. Occasionally, a cooling water spray was also required if animals became hyperthermic (>38.1°C).
A total of 42 animals were used in the PYC36-TAT dose response experiment. Three animals were euthanased due to subarachnoid hemorrhage, one animal was excluded due to insufficient increase in laser Doppler flowmetry at reperfusion and one animal was excluded due to hyperthermia postsurgery. In the delayed JNKI-1
PYC36d -TAT, JNKI-1d -TAT, and d -TAT Peptides
The peptides were synthesized fused to the TAT cell penetrating peptide (HIV-1 TAT(48–57)), in the protease-resistant
PYC36d -TAT Dose Response Experiment
Treatment groups (N=5) consisted of vehicle (saline), PYC36
Delayed JNKI-1d -TAT Treatment Experiment
Treatment groups (N=7 to 8) consisted of vehicle (saline) and JNKI-1
Tissue Processing and Infarct Volume Measurement
Animals were killed 48 hours after reperfusion with intraperitoneal injections of sodium pentobarbitone (900 mg/kg). After euthanasia, the brain was removed and placed in a sterile container of 0.9% NaCl and then placed in an −80°C freezer for 7 minutes. The brain was then coronally sliced from the junction of the cerebellum and cerebrum to 12 mm rostral to this point in 2 mm thick slices.
Slices were immediately stained with 2% 2,3,5 triphenyltetrazolium chloride (Sigma, St Louis, MO, USA) at 37°C for 15 minutes, followed by fixation in 4% formalin at room temperature for at least 18 to 24 hours before infarct volume measurement. Slices were scanned and images were analyzed by an operator blind to treatment status using ImageJ 3rd edition (NIH, USA). The total infarct volume was determined by measuring the areas of infarcted tissue on both sides of the 2 mm sections. These measured areas were multiplied by half slice thickness (1 mm), and corrected for cerebral edema by multiplying the ratio of affected to normal hemisphere areas (Campbell et al, 2008).
Adhesive Removal Test
This test measured the detection of sensory parameter and reaction to motor component small pieces of adhesive tape placed on the forelimbs (Esneault et al, 2008). Animals had adhesive removal tests performed before surgery for MCAO and at 48 hours after reperfusion. Animals were placed in a transparent enclosure for 2 minutes before starting the tests in order to adapt to placement in a new enclosure (habituation). Adhesive tape (Diversified Biotech, Dedham, MA, USA) was placed on the palmar surface of the paw and the time from first contact (detection) of adhesive tape to time of removal of adhesive was measured and recorded for each forelimb (maximum of 180 seconds).
Statistical Analysis
For infarct volume measurements, each treatment group was compared with its respective vehicle control group by analysis of variance followed by post hoc Bonferroni/Dunn test. The data obtained are presented as mean±standard deviation. Analysis of variance was employed to compare physiological parameters between groups. For the adhesive tape test, data were analyzed using both univariate and multivariate linear regression with the statistical package R (version 2.11.1; http://www.R-project.org). A value of P<0.05 was considered significant for all data sets.
Results
Physiological Measurements
Preischemia, during ischemia, and after reperfusion, there were no significant differences between the treatment groups in arterial blood pressure, blood gases (PaCO2 and PaO2), and blood pH in the PYC36
Physiological data for PYC36
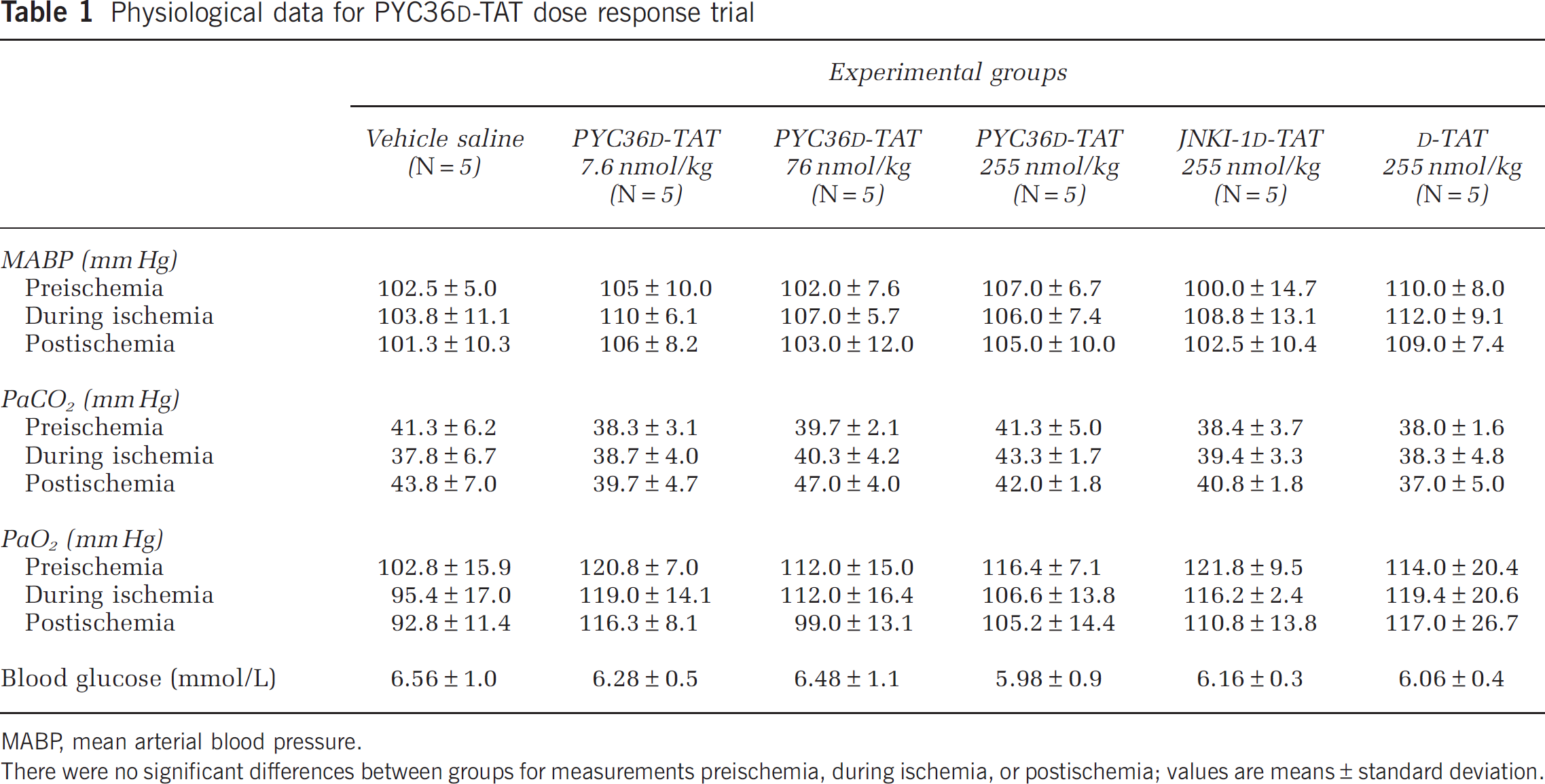
MABP, mean arterial blood pressure.
There were no significant differences between groups for measurements preischemia, during ischemia, or postischemia; values are means±standard deviation.
Physiological data for delayed JNKI-1
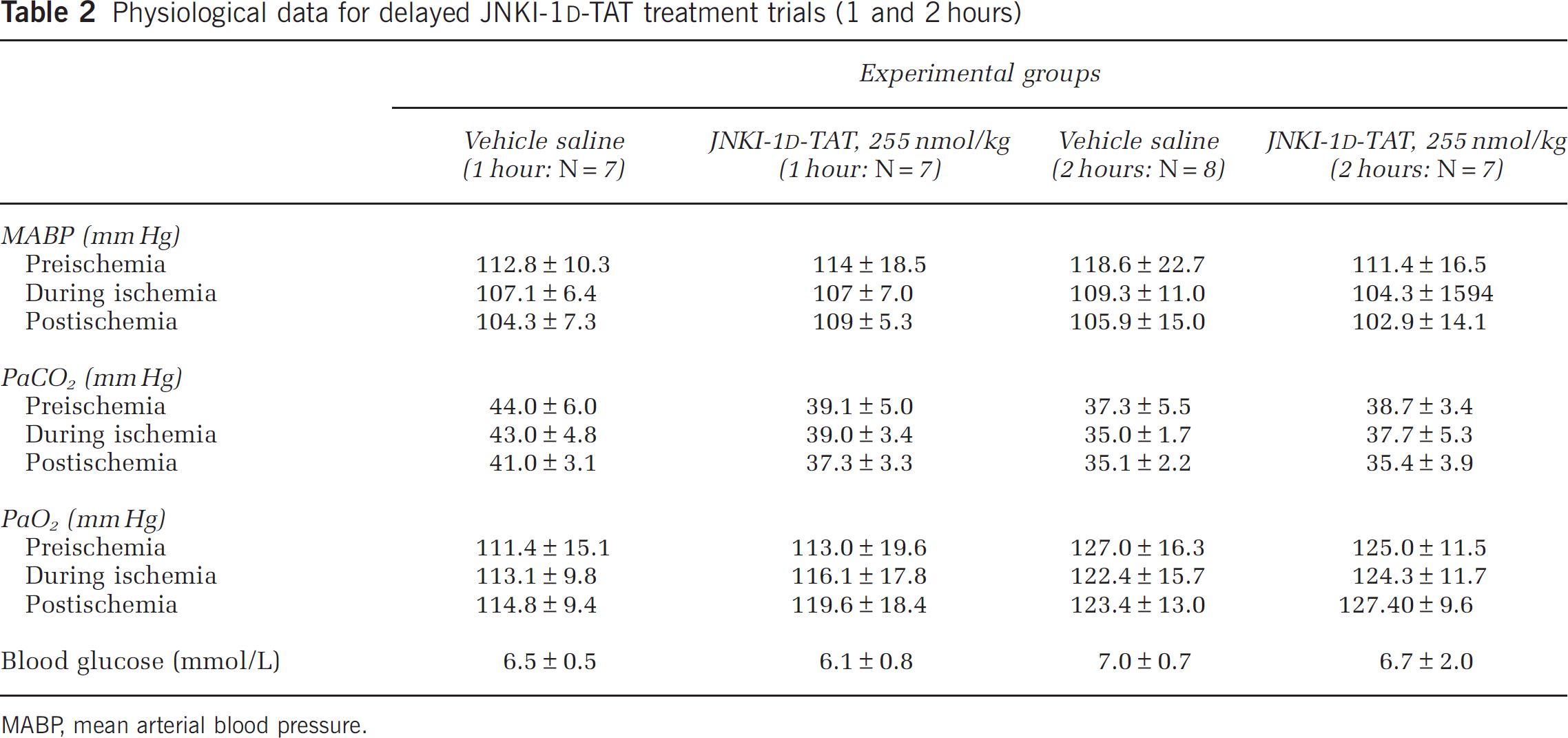
MABP, mean arterial blood pressure.
PYC36d -TAT Dose Response Experiment
Intravenous administration of the PYC36
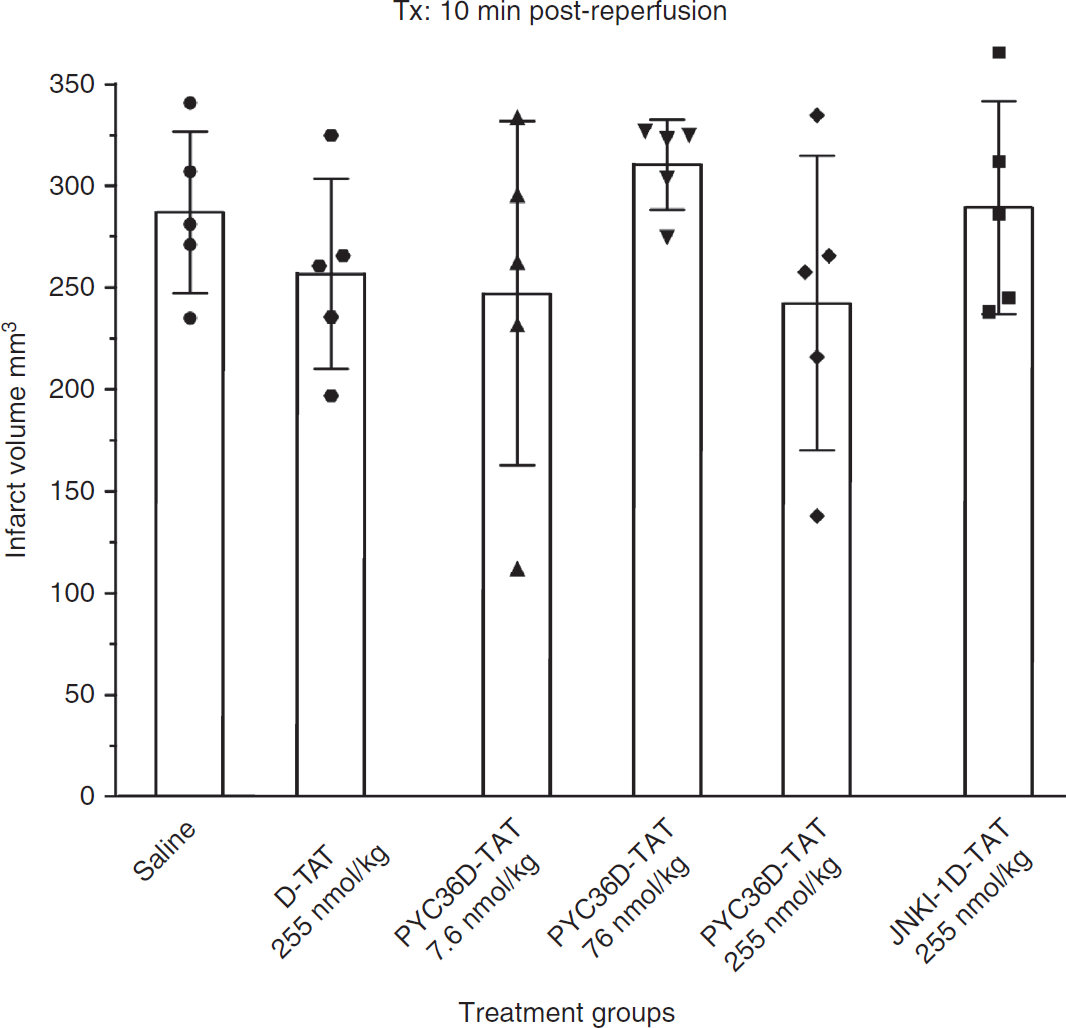
Effect of different doses of PYC36
In the adhesive tape removal test, there were no significant differences between any of the treatment groups in the posttreatment reaction or removal times of both left and right sides (data not shown).
Delayed JNKI-1d -TAT Experiment
Delayed intravenous administration of the JNKI-1
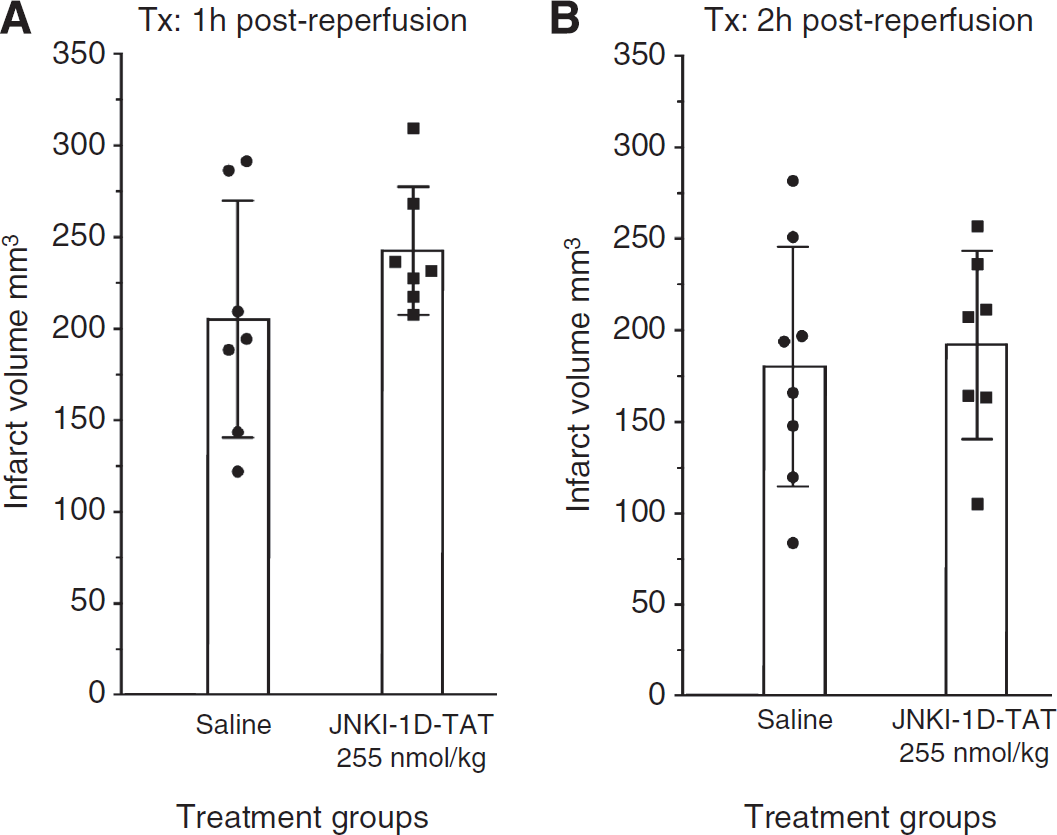
Effect of JNKI-1
In the adhesive tape removal test, there was no statistically significant difference between saline and treatment groups in the 1-hour postreperfusion administration trial (data not shown). In the 2-hour postreperfusion administration trial, there was no difference in tape removal for the left side; however, vehicle-treated animals were significantly better at removing tape on their right side compared with JNKI-1
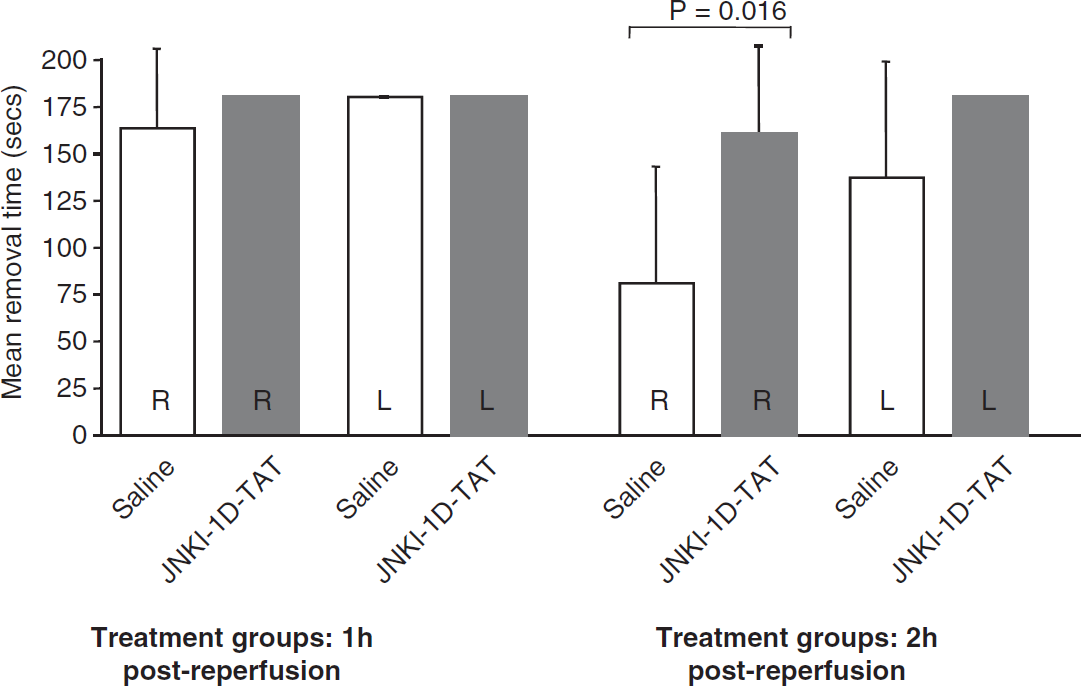
Effect of JNKI-1
Discussion
The initial focus of this study was to assess the neuroprotective potential of the c-Jun inhibitory peptide PYC36
Our experimental trial for the assessment of PCY36
In an attempt to reveal any neuroprotective effects with these downstream mitogen-activated protein kinase pathway inhibitory peptides, we performed two additional trials with JNKI-1
Based on our negative experimental outcomes with both PCY36
It is also possible that the JNKI-1
Despite the use of SH rats, which are considered to produce a consistent ischemic lesion (Howells et al, 2010), an unusual outcome of our study was the different mean infarct volumes obtained for control animals between trials. Mean infarct volume for saline-treated controls in the 10-minutes, 1-, and 2-hour treatment trials was 287, 206, and 180 mm3, respectively. The decreasing infarct volumes in each trial are suggestive that the ischemic severity was less and/or animals were more resistant to ischemia. Since the surgical protocol for each trial was the same and each trial was performed independently, any differences in ischemic severity/animal susceptibility would be expected to apply equally across all treatments within a trial, and are therefore not expected to confound treatment outcomes. One explanation for this may be seasonal fluctuations of brain neuropeptides or hormones. Although these animals are nonphotoperiodic rodents, maintained in rigorously controlled laboratory conditions for generations, studies have shown intrastrain variations of neuropeptides and hormones (Bissette et al, 1995; Vázquez et al, 2004a, 2004b; Wong et al, 1983), which may have a compounding factor on infarct volumes after MCAO. Moreover, the smaller infarct volumes obtained in the delayed JNKI-1
Taken together, our findings reveal that two mitogen-activated protein kinase pathway inhibitory peptides PYC36
Footnotes
Acknowledgements
The authors would like to acknowledge Joanne Chieng for technical assistance.
PMW is an Executive Director for Phylogica Ltd Pty. NM is a Senior Scientist working for Phylogica. BPM is a Phylogica shareholder.