
Abstract
c-Jun N-terminal kinase 3 (JNK3) is a member of the stress-activated group of mitogen-activated protein kinases. c-Jun N-terminal kinase 3 is a potent mediator of apoptosis and the use of JNK inhibitors or jnk3 gene deletion each protect against brain injury in adults. However, little is known about the role of JNK3 or its mechanism of action in neonatal brain injury. The aim of the present study was to compare the vulnerability of neonatal JNK3 knockout (JNK3 KO) mice and wild-type (WT) mice to cerebral hypoxic—ischaemic injury (HII) using unilateral-carotid occlusion combined with transient hypoxia. The degree of neural tissue loss in JNK3 KO mice was substantially reduced compared with WT mice (JNK3 KO 27.8% ± 2.8% versus WT 48.3% ± 2.0%, P ≤ 0.0001) after HII. Significant attenuation of injury was observed in the cerebral cortex, hippocampus, striatum, and thalamus of JNK3 KO compared with WT mice. Hypoxic—ischaemic injury increased JNK phosphorylation and activity, with JNK3 as the major isoform. Significantly, in JNK3 KO animals there was no difference in the activation of the upstream kinases mitogen-activated protein kinase kinase (MKK4) or MKK7. Downstream of JNK3, HII lead to increased phosphorylation of the transcription factors c-Jun and adenovirus transcription factor-2 (ATF-2), which was attenuated in JNK3 KO mice. c-Jun N-terminal kinase 3 deletion also decrease caspase-3 cleavage and Bim/PUMA expression, coupled with a upregulation of AKT/FOXO3a levels, linking JNK3 to apoptosis. These findings implicate JNK3 involvement in neural cell loss resulting from cerebral HII in the developing brain.
Introduction
Cerebral hypoxic—ischaemic injury (HII) at or around the time of birth is a major contributor to neonatal morbidity and mortality, with many survivors suffering from cerebral palsy (Volpe, 1981). Although the precise mechanisms of neural cell loss are not fully understood, recent neuropathologic studies in animal models and in human infants have indicated that a significant proportion of brain cells die by apoptosis (Edwards and Mehmet, 1996). Among the molecular events leading to the apoptotic death of neural cells is the induction and activation of a number of immediate early genes, many of which act as transcription factors. Among these are c-Jun and ATF-2, which are activated after phosphorylation by stress-activated protein kinases such as c-Jun N-terminal kinase (JNK) and p38, respectively (Irving and Bamford, 2002).
c-Jun N-terminal kinase represents one subgroup of mitogen-activated protein kinase that is activated in response to environmental stress (Wagey and Krieger, 1998). c-Jun N-terminal kinase is activated by the dual phosphorylation of conserved tripeptide motifs by upstream kinases that include MKK4 and MKK7 (Sanchez et al, 1994). One of the converging points downstream in the JNK signalling pathway is the activation (by phosphorylation) of c-Jun (Derijard et al, 1994), although JNKs can also phosphorylate other transcription factors, including ATF-2 and Elk-1 (Davis, 2000). c-Jun N-terminal kinase activation precedes cell death by apoptosis in many cell types, including neurons (Xia et al, 1995). Indeed, high basal levels of JNK activity in the brain are further increased in response to ischaemic stress (Xu et al, 1997). Concomitantly, c-Jun is phosphorylated in neurons subjected to ischaemia, further implicating JNK activation in neuronal cell death (Herdegen et al, 1998).
Mammalian JNK proteins are encoded by three genes, jnk1, jnk2 and jnk3, which are spliced alternatively to create at least 10 JNK isoforms (Dreskin et al, 2001). The jnk1 and jnk2 genes are expressed ubiquitously, while jnk3 is expressed mainly in the brain (Kyriakis et al, 1994). Gene ablation studies in vivo suggest that different JNK isoforms perform distinct roles in cell survival. For example, disruption of the jnk1/jnk2 genes together results in embryonic lethality, while jnk3 null are normal (Kuan et al, 1999). However, targeted disruption of the jnk3 gene, but not jnk1 or jnk2, protects mice from excitotoxic injury (Yang et al, 1997). Furthermore, adult c-Jun N-terminal kinase 3 knockout (JNK3 KO) mice are partially resistant to experimental HII compared with wild-type (WT) mice (Kuan et al, 2003). Similarly, a peptide inhibitor of JNK activation was reported to reduce ischaemic brain injury in both adult and juvenile rats (Kuan et al, 2003; Borsello et al, 2003).
To further investigate the precise relationship between JNK3 signalling and HII-induced apoptosis, we applied the Rice—Vanucci model of neonatal HII (Rice et al, 1981) to WT and JNK3 KO mice. The experimental rationale was designed to investigate the effects of jnk3 deletion on the dynamics of JNK3 signalling and neural cell loss after cerebral HII to the developing brain.
Materials and methods
Reagents
The mouse monoclonal antibody to α-tubulin was purchased from Sigma (Gillingham, UK). Polyvinylidene fluoride membranes were from Milipore Corporation (Bedford, MA, USA) and the LumiGlo chemiluminescence kit was from Cell Signalling Technologies (NEB, Hitchin, UK). Enhanced chemiluminescence Hyperfilm, protein-G-sepharose beads, and the rabbit anti-mouse horseradish peroxidase-conjugated secondary antibody were purchased from Amersham Biosciences (Little Chalfont, UK). Antibodies specific for phospho-JNK, MKK4, phospho-MKK4, MKK7, phospho-MKK7, phospho-c-Jun (Ser73), phospho-p38, p38, phospho-ATF-2(Thr71), ATF-2, phospho-FOXO, phospho-Akt(Ser473), Akt, cleaved caspase-3, Bim, Puma, anti-rabbit immunoglobulins, and the stress-activated protein kinase/JNK assay kit were all obtained from Cell Signalling Technology (NEB, Hitchin, UK). Anti-JNK1/2 and JNK1 antibodies were purchased from Pharmingen (BD Biosciences, Oxford, UK), and the anti-JNK2 antibody was from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). Anti-JNK3 antibody was purchased from Upstate (Lake Placid, NY, USA).
Animal Model
Unilateral HII was induced in neonatal mice using a modification of a model that has been described previously (Rice et al, 1981; Hedtjärn et al, 2002). Briefly, C57/BL6 WT mice at postnatal day 9 (obtained from B & K Universal AB, Uppsala, Sweden) or C57/BL6 mice lacking the gene for JNK3 (prepared in Professor Flavell's laboratory, as described by Yang et al (1997)) were bred at the University of Göteborg, Sweden, hypoxic—ischaemic injury was induced at postnatal day 9 (n = 26 JNK3–/–, n = 20 wt). Mice were anaesthetized with enflurane (3.5% for induction and 1.5% for maintenance) in a mixture of nitrous oxide and oxygen and the left common carotid artery was ligated. After anaesthesia and surgery, the animals were allowed to recover for 60 mins and then exposed to 60 mins of hypoxia in a humidified chamber at 36°C with 10% oxygen in nitrogen. After hypoxia, the pups were returned to and kept with their dams until they were killed.
Immunohistochemistry
At postnatal day 16, animals were deeply anaesthetized by intraperitoneal injection of 150 μL thiopental (50 mg/mL) and were subjected to intracardial perfusion with 0.9% NaCl followed by 5% buffered formaldehyde (Histofix, Histolab, Göteborg, Sweden). Brains were removed from the skull and immersion-fixed at 4°C for 24 h, dehydrated, embedded in paraffin, and cut into 5 μm coronal sections at 14 evenly distributed anteroposterior levels of the brain. Sections were stained for microtubule-associated protein (MAP-2) (1:1,000, 1 h, mouse-anti-MAP-2, clone HM-2; Sigma). Before immunohistochemical staining, sections were deparaffinized and boiled in citric acid buffer (0.01 mol/L, pH 6.0, 10 mins) and treated with proteinase-K (10 μg/mL; Boehringer Mannheim, Mannheim, Germany). Nonspecific binding was blocked by incubation with horse serum (4%). After incubation with the primary antibody, sections were washed in phosphate-buffered saline and incubated with biotinylated secondary antibodies (horse-anti-mouse 1:250; Vector Laboratories, Peterborough, UK) for 1 h followed by inhibition of endogenous peroxidase (0.6% H2O2 in methanol, 10 mins) and incubation with avidin—biotin enzyme complex (20 μL/mL, 1 h, ABC-Elite; Vector Laboratories). Immunoreactivity was visualized using diaminobenzidine (0.5 mg/mL) enhanced with nickel sulphate (15 mg/mL). Adjacent sections at the level of the hippocampus and striatum were stained with thionin/acid fuschin.
Evaluation of Brain Damage
Intact neurons (dendrites and soma) express MAP-2 and infarction in grey matter is associated with a distinct loss of MAP-2 immunoreactivity. Microtubule-associated protein-2-positive areas in the ipsi- and contralateral hemispheres were outlined by an observer masked to the study groups and were then calculated using the Olympus Micro Image, analysis software version 4.0 (Olympus Optical, Tokyo, Japan). The proportion of the infarction was calculated by subtracting the MAP-2-positive area of the ipsilateral hemisphere from the contralateral hemisphere and was expressed as the percentage of the contralateral hemisphere as described previously (Hedtjärn et al, 2002). Regional brain injury was also evaluated by an observer masked to the study groups using a neuropathologic scoring system where injury in the cortex was graded from 0 to 4, with 0 being no observable injury and 4 being confluent infarction encompassing most of the hemisphere. The damage in the hippocampus, thalamus, and striatum was assessed regarding both hypotrophy (0 to 3) and observable cell injury/infarction (0 to 3), resulting in a scoring for each region ranging between 0 and 6. The total score was the sum score for all the four regions (Hedtjärn et al, 2002).
In Vitro Kinase Assay for c-Jun N-terminal Kinase
c-Jun N-terminal kinase activity was measured using a specific kit (Cell Signalling Technology) following the manufacturer's instructions and using glutathione S-transferase-Jun (1 to 79) fusion peptide as the specific substrate for JNK. In brief, tissue lysates (100 μg protein) were incubated overnight at 4°C with glutathione S-transferase-Jun fusion protein beads. After washing, the beads were resuspended in kinase buffer containing ATP and kinase reaction was allowed to continue for 30 mins at 30°C. Reactions were stopped by the addition of polyacrylamide gel electrophoresis sample loading buffer. Proteins were separated by electrophoresis on a 10% polyacrylamide gel electrophoresis gel, transferred on polyvinylidene fluoride membrane, and finally incubated with phospho-c-Jun(Ser63) antibody. Finally, blots were subjected to enhanced chemiluminescence and kinase activity determined by densitometric analysis.
Specific Analysis of c-Jun N-Terminal Kinase 3
Currently, specific non-cross-reactive antibodies for the major JNK isoforms 1, 2, and 3 are not available. We have adapted a previously published method (Harrington et al, 2002) and for capturing JNK3 after immunodepletion of JNK1 and JNK2 isoforms. To determine specifically the presence of the active JNK3 isoform, cell lysates were first immunoprecipitated with a mixture of JNK1 (crossreacts with JNK1 and 2 but not with JNK3) and JNK2 (crossreacts with JNK2 and 1, but not with JNK3) antibodies already prebound to Protein-G beads to remove both JNK1 and JNK2 from the lysates. This process was repeated twice to ensure that JNK1 and JNK2 are completely removed from the supernatant and was confirmed by Western blot analysis cell lysates before and after immunodepletion using JNK1/2 antibody that recognize both JNK1 and JNK2 isoforms. The residual active JNK3 isoform in the supernatant was subjected to in vitro kinase assay and immunoblot analysis to measure JNK3 activity and protein expression, as described above.
Immunoblot Analysis of Protein Expression and Phosphorylation
Changes in the protein expression and phosphorylation of MKK4, MKK7, JNK, p38, Akt, ATF-2, c-Jun, FOXO3a, Bim, Puma, and cleaved caspase-3 were investigated by Western blotting using non-phospho- and phospho-specific antibodies. Tissues were lysed by sonication in a phosphate lysis buffer (20 mmol/L sodium phosphate, 137 mmol/L NaCl, 25 mmol/L sodium β-glycerophosphate, 2 mmol/L sodium pyrophosphate, 2 mmol/L ethylenediamine-N,N,N′,N′-tetraacetic acid, 10% glycerol, 1% Triton X-100, and a commercial protease inhibitor cocktail). Protein concentration was determined by the bicinchoninic acid method using a commercial assay kit from Pierce (Rockford, IL, USA), with bovine serum albumin as a standard. Extracted cell proteins (50 μg) were separated on a 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis gel and transferred to polyvinylidene fluoride membranes and blocked using Tris-buffered saline, Tris-buffered saline/0.1% Tween-20, and 5% skimmed milk for 1 h at room temperature. The blots were incubated overnight at 4°C with each primary antibody of interest (1:1,000 dilution). After washing in Tris-buffered saline/0.1% Tween-20, the blots were incubated with peroxidase-conjugated goat anti-rabbit antibody or rabbit anti-mouse antibody as appropriate at room temperature for 1 h. After the final wash, blots were subjected to enhanced chemiluminescence and the signal detected with Amersham Hyperfilm. Densitometric analysis was performed using 1D Kodak Digital Science software (Kodak, Oxford, UK).
Statistical Analysis
The data were expressed as means ± s.e.m. The tests used were Mann—Whitney U-test, two-way analysis of variance with Fisher's post hoc test, the χ2 test, and random effects linear regression model. Values of P < 0.05 were considered to be significant. In all cases, n corresponds to the number of animals. Where indicated, data sets were analysed by the Student's t-test, with P-values less than 0.05 considered as statistically significant.
Results
Brain Injury in c-Jun N-Terminal Kinase 3 Knockout and Wild-Type Mice after Hypoxic—Ischaemic Injury
Cerebral HII caused less neural damage in JNK3 KO mice compared with the WT mice. Area measurements comparing the left hemisphere subjected to HI to the right undamaged hemisphere (hypoxia only, H) showed a reduced cell loss in JNK3 KO mice compared with the WT mice throughout the brain (Figure 1A). Overall, this amounted to a 42% reduction in mean tissue loss in JNK3 KO mice (P < 0.0001; Figure 1B). The reduction in brain injury was apparent in all the regions studied: cerebral cortex (JNK3 KO: 1.6 ± 0.3 versus WT: 3.0 ± 0.2, P < 0.01), hippocampus (JNK3 KO: 4.5 ± 0.4 versus WT: 6.0 ± 0.05, P < 0.05), striatum (JNK3 KO 2.2 ± 0.3 versus WT: 4.2 ± 0.2, P < 0.01), and thalamus (JNK3 KO 1.8 ± 0.3 versus WT: 3.6 ± 0.23, P < 0.01) (Figure 1D). These data strongly suggest that JNK3 KO animals are significantly protected from HII.
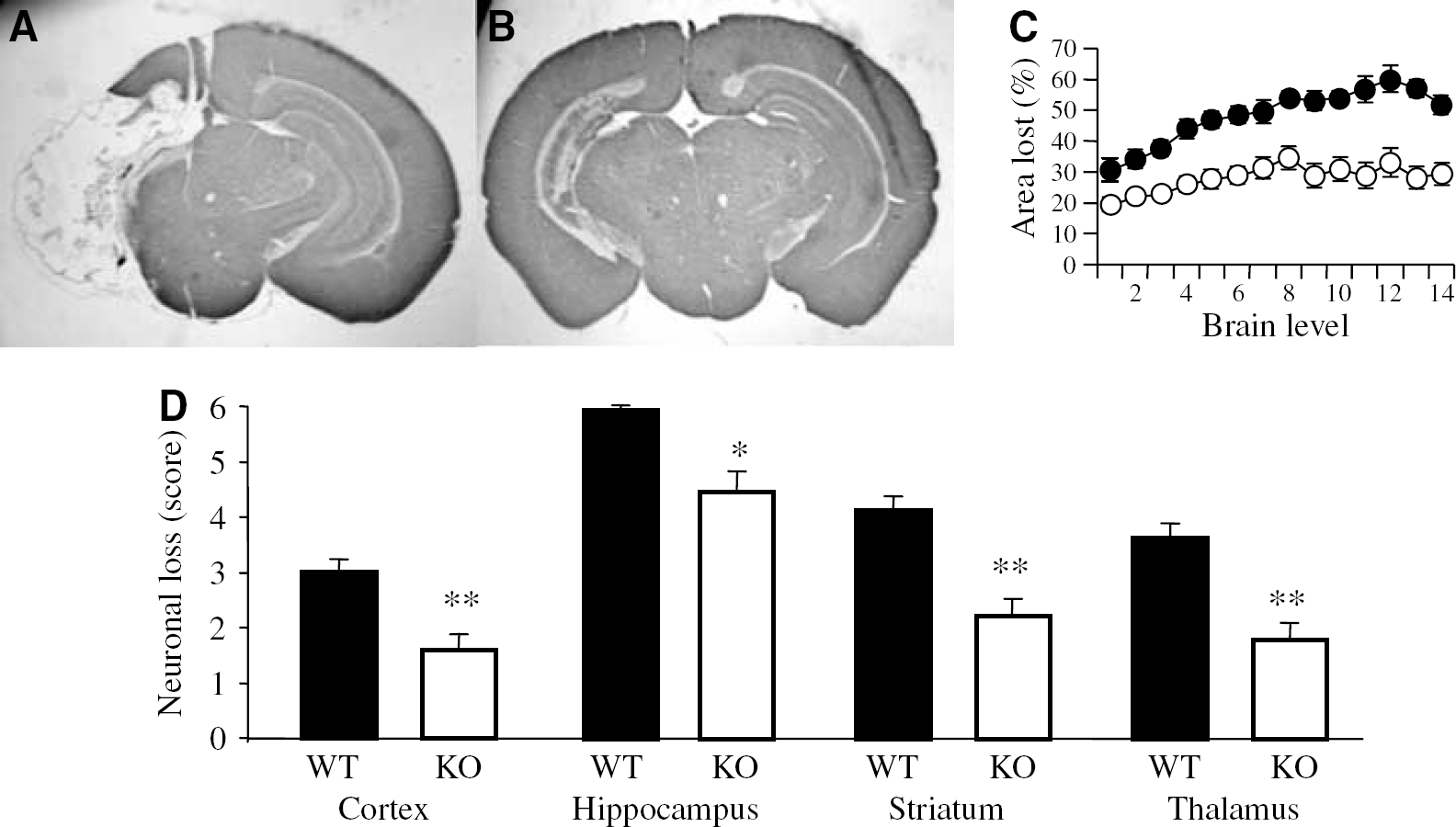
JNK3 contributes to neural cell loss after HII. (
Hypoxic—Ischaemic Injury Induces c-Jun N-Terminal Kinase Phosphorylation and c-Jun N-Terminal Kinase 3 Activation
Immunoblot analysis indicated that basal levels of JNK phosphorylation were high in all three areas of the brain (hippocampus, cortex, and cerebellum). The absence of significant differences between WT and JNK3 KO mice suggested that the high basal expression of phospho-JNK is largely because of JNKs 1 and 2. Immediately after HII, JNK phosphorylation was markedly reduced in the injured hemisphere of the hippocampus (Figures 2A and 2C), but only slightly in the contralateral hemisphere (Figures 2B and 2D). Similar results were observed in the cortex, while in the cerebellum there was no initial change in the expression of phospho-JNK (not shown). After resuscitation from HI, JNK phosphorylation increased in a time-dependent manner in both hemispheres and all three regions of the brain in response to injury (Figures 2 and 3), although the degree of phosphorylation was greatly reduced in JNK3 KO mice. Moreover, the reduction in JNK phosphorylation was more apparent in the injured hemisphere compared with the control in the same brain. In particular, p54 JNK phosphorylation was almost completely absent in JNK3 KO animals, suggesting that this isoform of JNK3 is highly phosphorylated after HII (Figures 2A and 2B). Importantly, the level of JNK1 and JNK2 protein expression was comparable in WT and JNK3 KO animals and was not significantly altered in response to HII (Figures 2A and 2B).
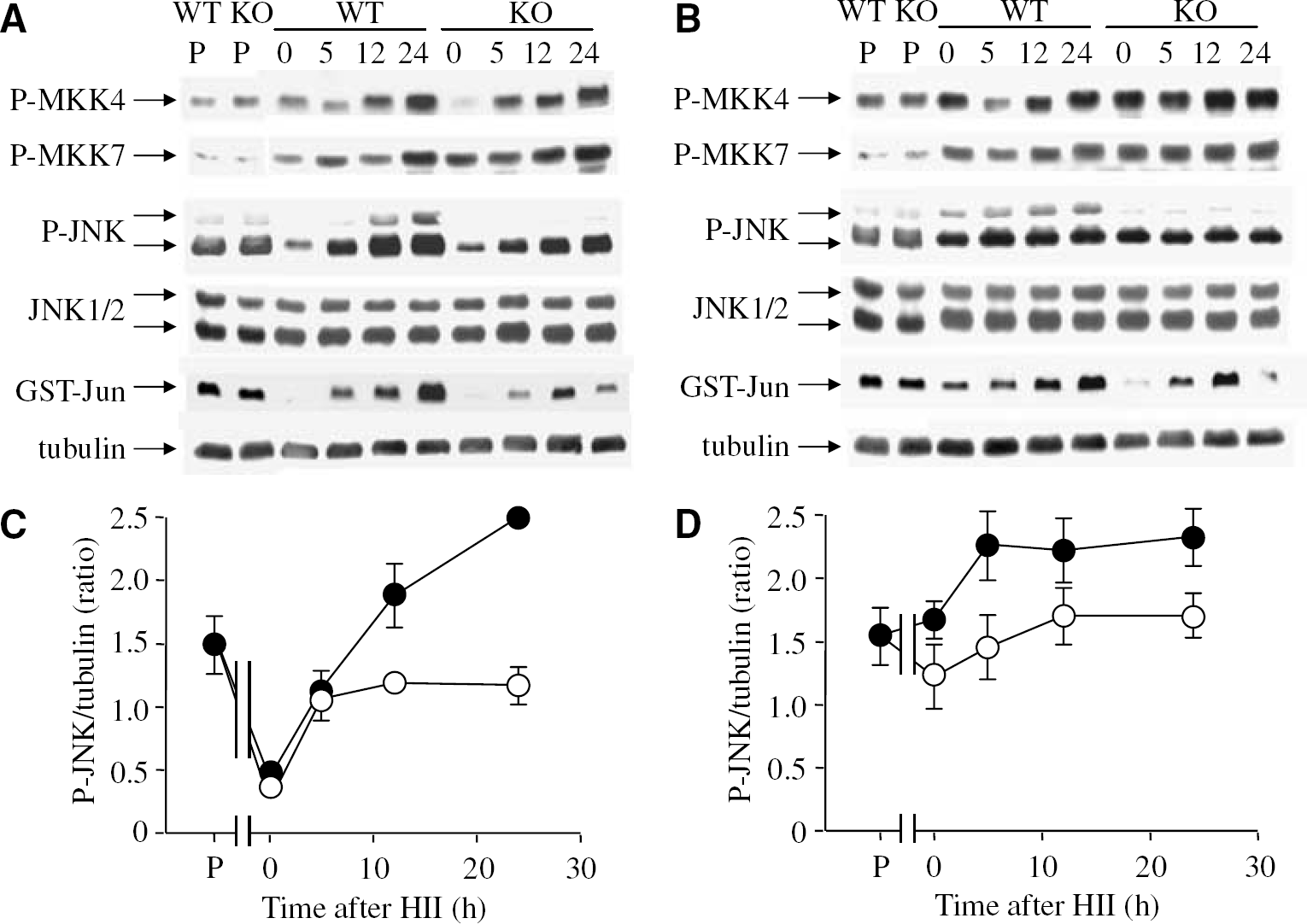
Changes in JNK activation following deletion of the jnk3 gene. Comparison of JNK signalling in WT and JNK3-null (KO) mice was investigated by immune blotting analysis of protein and phosphoprotein levels in the injured, ipsilateral (
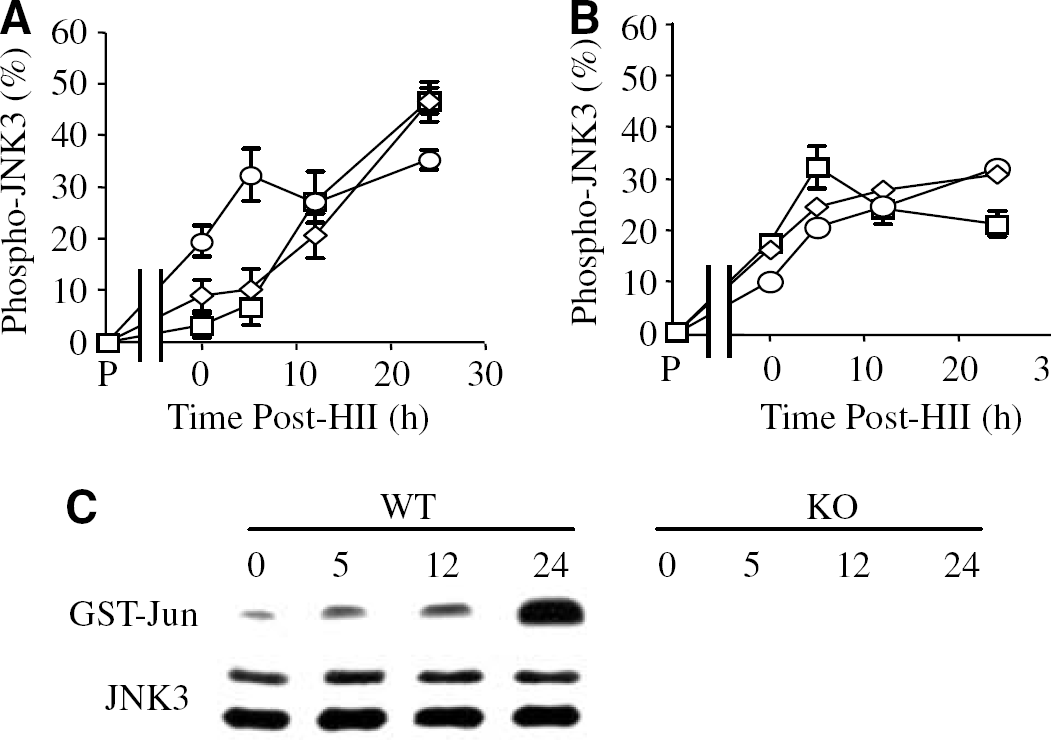
Kinetics of specific JNK3 phosphorylation (
Activation of MKK4/7
To investigate the possibility that the reduced JNK phosphorylation in JNK3 KO mice was the result of attenuated activation of one of the upstream kinases, we next determined MKK4/MKK7 phosphorylation in the hippocampus, cortex, and cerebellum at defined times after HII. In both healthy JNK3 KO and WT mice, the basal phosphorylation of MKK4 was high, while for MKK7 it was low. (See Figures 2A and 2B for the hippocampus. Data for the cortex and cerebellum are not shown.) MKK4 and MKK7 phosphorylation were increased in both hemispheres of the brain after HII, with no significant differences between WT and JNK3 KO mice (Figures 2A and 2B). These results show that jnk3 deletion does not affect MKK4/MKK7 activation after HII.
Kinetics of c-Jun N-Terminal Kinase Activation After Hypoxic—Ischaemic Injury
Having determined the pattern of neonatal HII and overall JNK phosphorylation in JNK3 KO mice, we are able to evaluate both the dynamics of JNK3 activation and the specific contribution of JNK3 to neural cell loss. c-Jun N-terminal kinase 3 phosphorylation was determined by subtracting phospho-JNK levels after HII in the JNK3 KO mice (representing phospho-JNK1 and -JNK2) from the total phospho-JNK in WT mice (representing phospho-JNK1, -JNK2. and -JNK3) and then normalizing to a loading (α-tubulin) control. The results showed that while JNK3 did not contribute to the basal level of phospho-JNK in the uninjured brain (Figure 3), JNK3 phosphorylation was increased immediately after HII in both the injured (HI) nonischaemic (H) hemispheres in all three regions of the brain (Figure 3A). However, we also observed significant differences in the dynamics of JNK3 phosphorylation. For example, phospho-JNK3 increased gradually after HII in the injured, ipsilateral areas of the hippocampus and cortex and comprised between 35% and 45% of total phospho-JNK at 24 h after HII (Figure 3A). In contrast, in the contralateral, nonischaemic hemisphere, phospho-JNK3 peaked earlier (5 h) after injury and comprised only between 20% and 30% of total phospho-JNK (Figure 3B). The dynamics of JNK phosphorylation in the cerebellum was similar in both hemispheres of the brain and this coincided with comparable neural cell death in this region (not shown). In addition, we used an immunodepletion method to determine the specific contribution of JNK3 in the response of WT mice to cerebral HII. Analysis of hippocampus lysates isolated from WT mice after HII indicated that JNK3 activity was absent from control, but increased gradually after HII to a peak at 24 h, although JNK3 protein levels remained similar; as expected, neither JNK3 activity nor expression of the protein were detectable in the JNK3-deficient mice (Figure 3C). Overall, these data indicate that JNK3 is significantly phosphorylated and activated in response to HII and that this correlates with neural cell loss.
Involvement of c-Jun N-Terminal Kinase and ATF-2 in c-Jun N-Terminal Kinase 3 Signalling
Next, we investigated the involvement of c-Jun and ATF-2 in JNK3 signalling after HII. Immunoblotting analysis using a phospho-Ser73-specific antibody revealed rapid changes in c-Jun phosphorylation after cerebral HII (Figure 4). The kinetics of c-Jun phosphorylation closely followed JNK phosphorylation in all areas of the brain. As was observed for phospho-JNK, the basal levels of phospho-c-Jun also did not differ between WT and JNK3 KO mice, again suggesting that JNK3 is not involved in maintaining basal c-Jun activity.
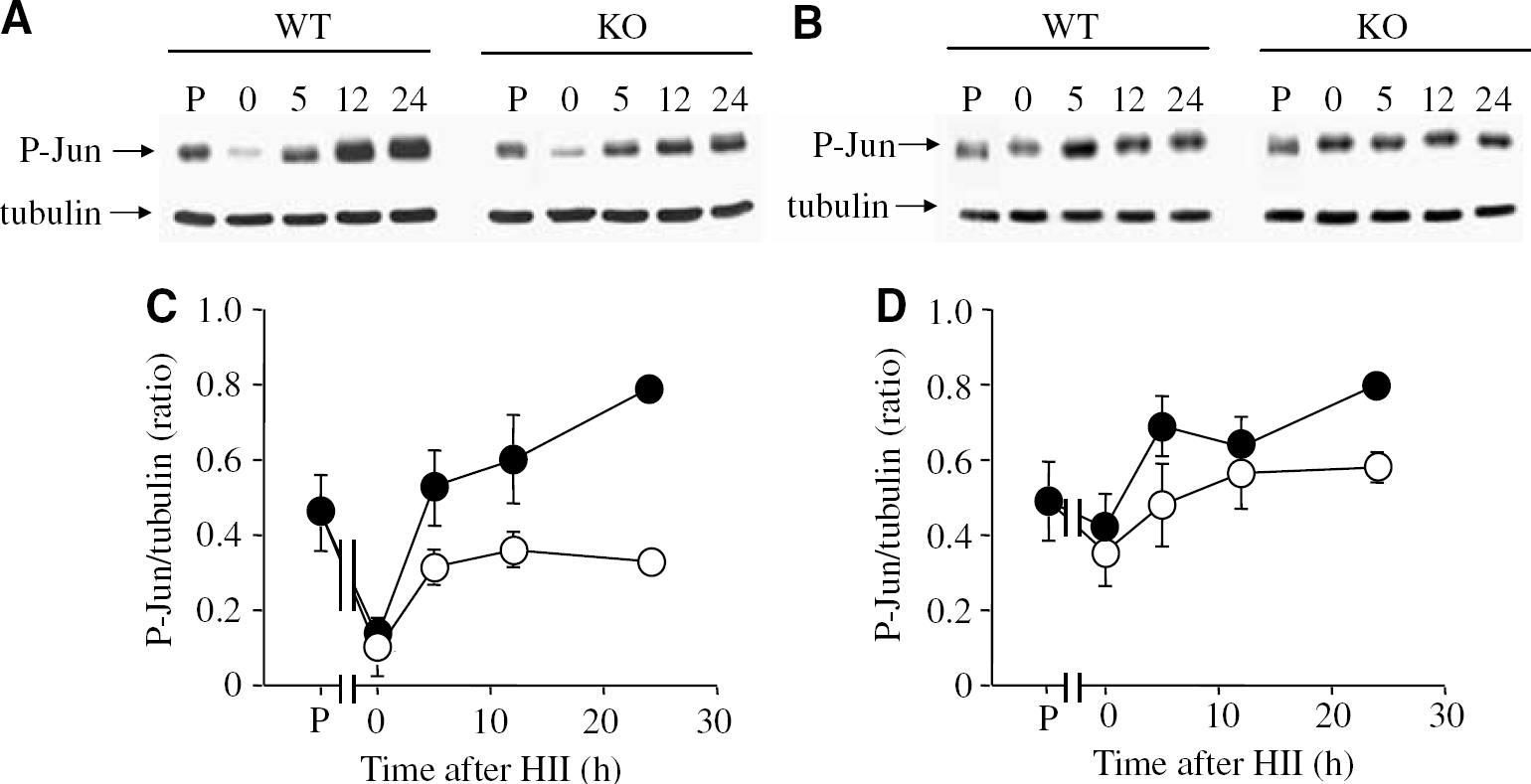
Effects of cerebral HII on downstream substrates of active JNK. (
c-Jun phosphorylation was significantly reduced immediately after HII in the injured hemisphere of the hippocampus (Figures 4A and 4C) and cortex (not shown) and slightly reduced in the contralateral hemisphere of the hippocampus (Figures 4B and 4D) and cortex and both hemispheres of the cerebellum (not shown). c-Jun phosphorylation in the ipsilateral hippocampus increased in a time-dependent manner after HII and to a lesser extent in the contralateral hemisphere. Significantly, our data indicated reduced levels of c-Jun phosphorylation in JNK3 KO mice, thus implicating JNK3 in c-Jun phosphorylation after HII.
Since ATF-2 is both a potential substrate for JNK and a dimerization partner for c-Jun, we next investigated the possible contribution of JNK3 to ATF-2 phosphorylation. As shown in Figure 5, ATF-2 phosphorylation was dramatically reduced in the hippocampus immediately after HII and, subsequently, increased reaching a maximum at 12 h after HII. In sharp contrast, ATF-2 phosphorylation was downregulated in the injured, but not in the contralateral, hemisphere of JNK3 KO mice. Because ATF-2 is a substrate for p38 mitogen-activated protein kinase, we additionally analysed the kinetics of p38 phosphorylation after HII. The levels of expression and phosphorylation of p38 did not differ between WT and JNK3-deficient mice (Figures 5A and 5B), consistent with a direct role for JNK3 in ATF-2 phosphorylation. The kinetics of ATF-2 phosphorylation are consistent with the involvement of this transcription factor downstream of JNK3 signalling, but upstream of neural loss after HII.
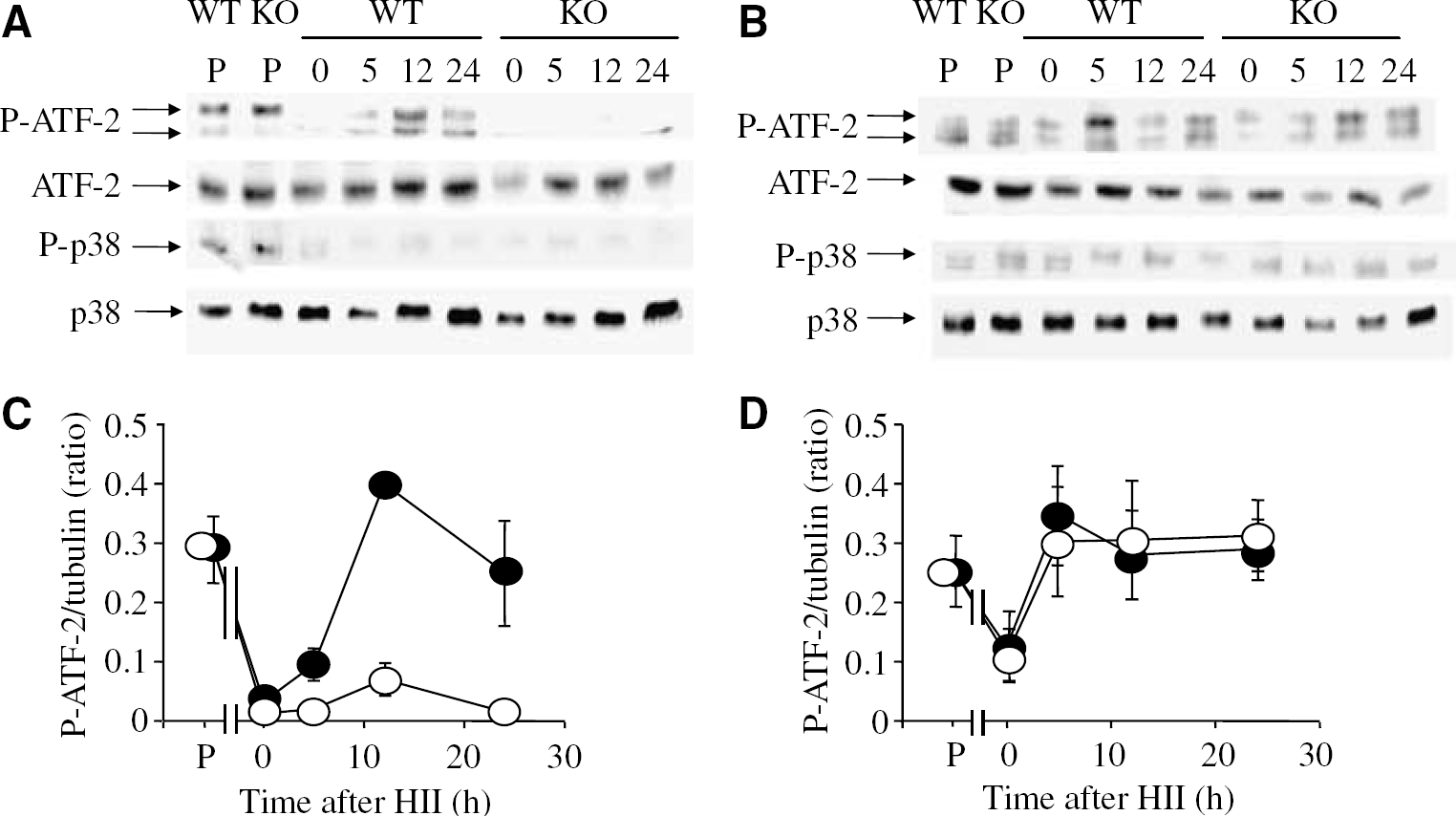
Cerebral HII induces ATF-2 but not p38 phosphorylation in the hippocampus. (
Apoptotic Signalling in Response to Hypoxic—Ischaemic Injury
It has been well documented that HII leads to apoptosis after cerebral HII. To investigate the involvement of JNK3 signalling in this mode of cell death, we monitored the activation of caspase-3 by analysing expression of the active cleavage product (15 to 17 kDa) using a specific antibody. First, we found that caspase-3 was processed after HII but only in the injured hemisphere where neural cell death was significant (Figures 6A and 6B), suggesting that both hypoxia and ischaemia are required for activation of the caspase cascade. After cerebral HII, caspase-3 cleavage occurred in a time-dependent manner and was significantly lower in JNK3 KO mice (Figure 6C), suggesting that caspase-3 cleavage is at least partially mediated by JNK3 activation after HII. However, the cleavage of caspase-3 in the hippocampus and cortex, but not in the cerebellum after HII (not shown) stresses the fact that JNK3 activation is not sufficient to trigger apoptosis.
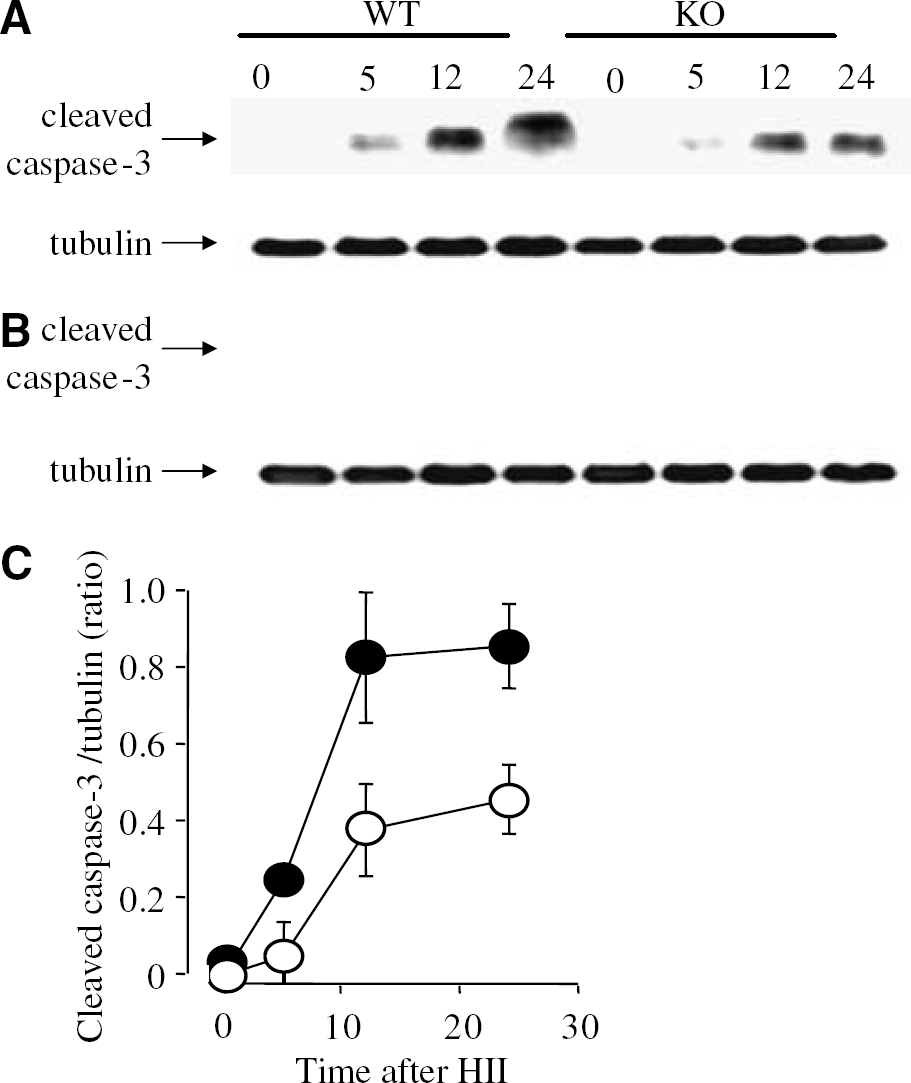
Cerebral HII results in caspase-3 cleavage only in the injured hippocampus. (
As Akt/protein kinase B activation has been shown to be important for neuronal cell survival, we next compared the phosphorylation of Akt and its substrate FOXO3a in JNK3-deficient and WT neonatal mice subjected to cerebral HII. Akt phosphorylation increased at 5 h after HII and was downregulated by 24 h, although in the ipsilateral (HI) hemisphere of JNK3 KO animals the disappearance of the phospho-Akt signal was significantly delayed, suggesting a possible role for JNK3 in switching off Akt activity (Figure 7C). Importantly, total Akt and FOXO3a expression was comparable in WT and JNK3 KO animals and was not significantly altered in response to HII (not shown). Perhaps not surprisingly, the kinetics of FOXO3a phosphorylation were similar to Akt; moreover, no major differences were observed in Akt/FOXO3a phosphorylation between WT and JNK3 KO mice in the contralateral hemisphere (Figure 7). These findings suggest crosstalk between Akt signalling and the activation of JNK3 after HII.
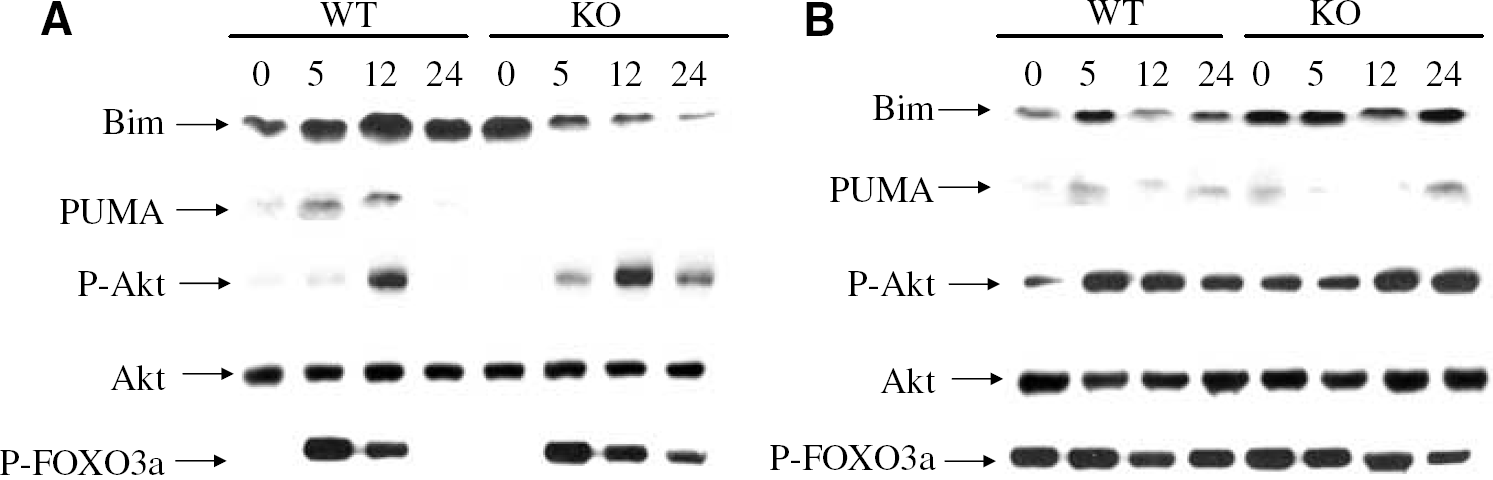
Effects of cerebral HII on the phosphorylation of Akt and FOXO3a or the expression of the proapoptotic proteins, Bim and PUMA. Increased Bim/PUMA expression levels are induced in WT (WT) compared with JNK3-null (KO) mice by cerebral HII. In contrast, reduced phospho-Akt/phospho-FOXO3a levels are present in WT compared with KO mice after HII. Tissue samples were prepared from the ipsilateral (
To investigate the relationship between JNK3 activation and expression of proapoptotic members of the Bcl-2 protein family, we compared the expression of two of these proteins, Bim and PUMA, after cerebral HII. The levels of expression of the two proteins increased in the injured hippocampus after HII, but were dramatically reduced in JNK3-deficient mice (Figure 7A). In the nonischaemic contralateral hippocampus, however, the expression levels of Bim and Puma were comparable in both WT and JNK3 KO mice (Figure 7B). While these data are consistent with a role for JNK3 in the regulation of Bim and Puma expression in the injured hemisphere, they do not exclude the possibility that other proapoptotic signals specifically increase the levels of proapoptotic Bcl-2 proteins in areas of neural damage after cerebral HII.
Discussion
In this study, we report that jnk3-deficient mice are protected against HII in the neonatal period. c-Jun N-terminal kinase inhibition has previously been shown to be neuroprotective against excitotoxicity and ischaemia and specific deletion of jnk3 protects adult mice from HII (Kuan et al, 2003). It was important to elucidate the role of jnk3 during HII in the neonatal setting as several protective strategies in the adult brain are not protective in the immature brain (Gill et al, 2002). Indeed, periods of enhanced vulnerability during brain development, such as the increased susceptibility of oligodendrocyte (OL) precursors to HII compared with mature OLs (Back et al, 1998), may be mediated by changes in JNK3 activity. We previously reported that increased JNK3 activation in OL precursors after exposure to apoptotic stimuli correlated with their high sensitivity to cell death, while the resistance of mature OL was associated with low levels of JNK3 activity (Pirianov et al, 2006). In the present study using a neonatal model of HII, we observed significant neuroprotection in jnk3-deficient mice in different regions of the brain.
It has been suggested that neural cell death in response to HII is preceded by a robust activation of mitogen-activated protein kinase family members (Nozaki et al, 2001), although whether JNK activation is the main event leading to cell death is a topic of heated debate. High basal levels of JNK activity in the healthy, uninjured brain suggests that this mitogen-activated protein kinase also regulates physiologic processes other than apoptosis (Xu et al, 1997). In this respect, Kuan and co-workers have shown that there is differential activation of JNK isoforms under basal versus stress-induced conditions (Kuan et al, 2003). Thus, JNK1 contributes to the high basal level of JNK activity, while JNK3 is the major isoform activated in response to cell stress. We previously found that while JNK3 activity is absent in healthy OL lineage cells, it represents 40% to 60% of total JNK activity in response to apoptotic stimuli (Pirianov et al, 2006). In the present study, we did not find any differences in basal phospho-JNK levels between WT and JNK3-deficient mice, confirming that JNK3 does not contribute to basal JNK activity in the brain. Interestingly, the high basal level of JNK phosphorylation was markedly reduced immediately after HI but only in the ipsilateral (injured) hemisphere of the brain. This may be because of early cell death by necrosis during the initial HII; delayed cell death by apoptosis (a relatively late event in this model) might therefore be specifically associated with JNK3 activation. In support of this notion, continuous JNK activation is required for several hours for neuronal apoptosis induced by HII and even the relatively late inhibition of JNK after injury is still protective (Borsello et al, 2003; Carboni et al, 2005).
After cerebral HII, the combined activation of excitatory amino-acid receptors (Yang et al, 1997) and the increased formation of oxygen free radicals and of intracellular Ca2+ (Takita et al, 2004) are possible triggers for JNK3 activation. Activated JNK3 will, in turn, phosphorylate c-Jun, which has been shown to trigger the transcription of a large number of death genes including the proapoptotic Bcl-2 family member, Bim and the death receptors TNFR (p55) and CD95/Fas (Whitfield et al, 2001; Kuan et al, 2003). Furthermore, JNK3 signalling is implicated in the mitochondrial release of cytochrome c leading to caspase-3 activation either via a Bim-dependent mechanism or through direct targeting of the mitochondria (Whitfield et al, 2001; Kuan et al, 2003; Borsello et al, 2003). This might partly explain the critical role of JNK3 in perinatal brain injury as caspase-3 activation appears to be particularly important in damage to the immature brain (Gill et al, 2002).
In the present study, we found that cerebral HII to the neonatal brain increases JNK phosphorylation, with JNK3 as the major activated isoform. While JNK3 is activated in both hemispheres of the brain, it is more pronounced in the ipsilateral hemisphere. Moreover, both hypoxia and ischaemia are required for JNK activation and infarct formation. Although JNK3 activity largely correlates with infarct formation, JNK3-deficient animals are not entirely protected from HII, we can conclude that other signalling pathways independent of JNK3 activation can also contribute to neural cell loss. To take this argument one step further, we also find that JNK3 activation is not sufficient for the execution of apoptotic cell death, since neither caspase-3 activation nor significant neural cell loss occur in the contralateral (uninjured) hemisphere of the brain where JNK3 activity increases in WT mice after HII.
What are the signals that mediate JNK3-induced cell death? It is well documented that active JNK3 can phosphorylate a number of transcription factors and substrates. In the present study, we investigated the effects of JNK3 deletion on c-Jun and ATF-2 signalling. Our results are consistent with previous findings that c-Jun is an important player in JNK3 signalling in the hippocampus after HII (Kuan et al, 2003) and we found that c-Jun is phosphorylated by JNK3 in all regions of the brain. Our results further implicate ATF-2 downstream of JNK3 signalling in the hippocampus in response to HII. Significantly, while phospho-p38 levels were reduced in the ipsilateral hippocampus after HII in WT mice, phospho-ATF-2 levels were maintained. In contrast, deletion of JNK3 resulted in a marked reduction in ATF-2 phosphorylation, which correlated with the size of the infarct. This result suggests that in the absence of significant p38 activity, JNK3 appears to be the primary kinase that phosphorylates ATF-2. While we cannot exclude the possibility of a very early increase in p38 activation (Hee Han et al, 2002), our data suggest that JNK3 is a major regulator of ATF-2 activity. Consistent with this possibility, ATF-2 is an important dimerization partner of c-Jun and the resulting heterodimer plays a critical role in neuronal apoptosis (Ham et al, 2000). Based on these findings, we suggest that the JNK3/c-Jun/ATF-2 pathway is likely to be pivotal in neural cell death resulting from HII to the developing brain.
Recent reports have implicated JNK activation in the apoptotic cell death driven by the BH3-only members of the Bcl-2 family. Although the precise mechanism by which JNK might activate these proapoptotic proteins is unclear, there is evidence for both a transcriptional and post-translational role. For example, Bim is a potential target of JNK that is transcriptionally upregulated in neurons undergoing JNK-dependent cell death (Kuan et al, 2003). However, JNK can directly phosphorylate Bim or Bmf, which may cause the release of these BH3 proteins from the cytoskeleton, subsequently to engage the mitochondrial death pathway (Lei and Davis, 2003). In fact, JNK phosphorylation of Bim at Ser63 is critical for neuronal apoptosis and JNK can also trigger the translocation of Bax and Bim to the mitochondria after cerebral ischaemia (Becker et al, 2004). In this context, our data are consistent with recent published data demonstrating that JNK3 plays a major role in the upregulation of Bim after HII. However, further investigations are required to study the precise mechanisms by which JNK mediates the proapoptotic actions of Bcl-2 BH3 members in neural cell death after HII. Similarly, the proapoptotic Bcl-2 family protein PUMA has also been implicated in neural cell death (Reimertz et al, 2003). PUMA function is regulated by p53 (Jeffers et al, 2003), which, in turn, is a substrate for JNK (Buschmann et al, 2001). Consistent with this crosstalk, we found that PUMA is upregulated after cerebral HII in WT but not in JNK3 KO mice.
Since the commitment to apoptosis is determined by the balance between death and survival signals, it is not surprising that the expression of antiapoptotic molecules also increase after cerebral HII (Noshita et al, 2001). Akt signalling has been shown to be an important survival signal for many cell types including neuronal cells and promotes survival through its ability to phosphorylate and inactivate several apoptosis-related proteins, including Bad, the forkhead transcription factors (FOXO family), caspase-9, and glycogen synthase kinase-3β (Brunet et al, 1999). Akt activity increases transiently after ischaemia and is subsequently downregulated at the time when apoptotic cells appear (Ouyang et al, 1999). We also find that phosphorylation of Akt and its substrate FOXO3a are initially activated (up to 12 h) after HII, but are then downregulated (after 24 h). Our observation that Akt and FOXO3a phosphorylation are maintained in the injured hemisphere of the hippocampus in JNK3 KO mice further suggests a possible crosstalk between JNK3 and apoptosis. In addition, we recently found that two proteins (insulin growth factor-1 and hexarelin) with strong neuroprotective effects in this neonatal model of HII both upregulated phosphorylation of Akt (Brywe et al, 2005a, b ). Is there a common substrate for the JNK3 and the Akt signalling pathways? One possible candidate is FOXO3a, which can be phosphorylated by JNK at a site that is different from that recognized by Akt, resulting in FOXO3a activation in the nucleus (Essers et al, 2004). In the present studies, it is significant that both hypoxia and ischaemia are required for brain damage. The outcome of cell death or survival might therefore depend on the balance of JNK activity and Akt phosphorylation. Indeed, recent reports document a mechanism by which Akt can regulate JNK phosphorylation, which involves the c-Jun-interactive proteins (Song and Lee, 2005).
In summary, we show that the vulnerability of neonatal mice to cerebral HII is significantly dependent on JNK3 activation, with a more subtle role for JNKs 1 and 2. While our results confirm the role of c-Jun phosphorylation in this process, they also identify ATF-2 as a major potential player. In jnk3-deficient mice, decreased caspase-3 cleavage and Bim/PUMA protein levels, coupled with the maintenance of AKT signalling, suggest that the primary mode of JNK3 action is to promote apoptosis. While more detailed investigations are required to understand the precise crosstalk between JNK3 and apoptotic signalling, our data clearly suggest that JNK3 inhibitors might be promising therapeutic agents for neonatal brain injury.
Footnotes
Acknowledgements
We are grateful to Nigel Kennea in the Institute of Reproductive and Developmental Biology for genotyping the mice by RT-PCR.