
Abstract
The authors studied whether cyclic AMP (cAMP), a widespread regulator of inflammation, modulates the cytokine-mediated expression of the intercellular adhesion molecule, intercellular adhesion molecule-1 (ICAM-1), and the inflammatory nitric oxide synthase 2 (NOS-2), in primary and immortalized brain endothelial cell cultures (GP8.3 cell line). When measured by enzyme-linked immunosorbent assay (ELISA), ICAM-1 was constitutively expressed and was upregulated twofold by interleukin-1β, with no effect of interferon-γ. The NOS-2 activity, assessed by nitrite accumulation, was absent from untreated cultures but was induced by interleukin-1β and interferon-γ acting synergistically. Stimulation of cAMP-dependent pathways with forskolin or dibutyryl cAMP decreased ICAM-1 protein expression, whereas it increased NOS-2 protein expression. For both ICAM-1 and NOS-2, mRNA expression correlated with protein expression. Blockade of NOS activity with L-N-monomethylargiuine (L-NMMA) did not alter ICAM-1 expression, indicating that the nitric oxide released by NOS-2 did not cause the down-regulation of ICAM-1. Analysis of NFκB activation indicated that cAMP acted through a mechanism other than inhibition of nuclear translocation of NFκB. The authors conclude that cAMP modulates the expression of proinflammatory molecules in brain endothelium. This suggests that inflammatory processes at the blood-brain barrier in vivo may be regulated by perivascular neurotransmitters via cAMP.
Keywords
Brain endothelial cells (BEC) have unique anatomic features that restrict the passage of hematogenous cells from the circulation to the brain. However, in the course of the inflammatory reactions that occur during brain tissue damage, infections, or autoimmune diseases, BEC mediate the recruitment and infiltration of leukocytes (Schroeter et al., 1995; Iadecola et al., 1995, 1996; Galea et al., 1998a). This process is initiated by adhesion molecules like the intercellular adhesion molecule-1 (ICAM-1), a cell surface glycoprotein of the immunoglobulin superfamily that binds to β-integrins present in leukocytes (Diamond et al., 1990). Increased ICAM-1 expression has been detected in ischemia (Schroeter et al., 1995), experimental allergic encephalomyelitis (Steffen et al., 1994), multiple sclerosis (Cannella and Raine, 1995), and acquired immunodeficiency syndrome-related dementia (Nottet et al., 1996). Along with adhesion molecules, BEC can express the calcium-independent type of nitric oxide synthase (NOS-2) in the course of brain inflammatory processes (Dorheim et al., 1994; Iadecola et al., 1996; Galea et al., 1998b). The appearance of both ICAM-1 and NOS-2 in BEC probably is induced by proinflammatory cytokines like interleukin-1β (IL-1β), tumor necrosis factor (TNF), or interferon-γ (IFN-γ) produced by either brain cells or infiltrated leukocytes (Buttini, 1994; Benveniste, 1994).
Blockade of ICAM-1 lessens the severity of experimental allergic encephalomyelitis (Archelos et al., 1993; Morrisey et al., 1996) and decreases the size of the infarctions caused by cerebral ischemia (Chopp et al., 1996; Soriano et al., 1996), demonstrating that leukocyte infiltration exacerbates these diseases. By contrast, the nitric oxide (NO) derived from endothelial NOS-2 can have both detrimental and beneficial effects. On the one hand, it could contribute to the damage by reacting with superoxide molecules, thereby generating peroxynitrites, highly cytotoxic agents (Lipton et al., 1998). On the other hand, NO could provide protection by inducing vasodilation (Loscalzo et al., 1995) and inhibiting leukocyte adhesion and platelet aggregation (Radomski et al., 1987; Kubes et al., 1991; Lefer et al., 1999). Local regulation of ICAM-1 and NOS-2 expression thus may alter the outcome of brain inflammatory diseases.
Cyclic adenosine monophosphate (cAMP) is a ubiquitous regulator of inflammatory and immune reactions. Thus, cAMP can block lymphocyte activation (Birch and Polmar, 1982; Jegasothy et al., 1978), inhibit proinflammatory cytokine release from macrophages (Knudsen et al., 1986; Kunkel et al., 1988), and reduce expression of adhesion molecules in peripheral endothelial cells (De Luca et al., 1994; Ghersa et al., 1994) and astrocytes (Ballestas and Benveniste, 1997). By contrast, cAMP can either stimulate (Sowa and Przewlocki, 1994; Eberhardt et al., 1998) or inhibit (Feinstein et al., 1993; Andersen et al., 1996) NOS-2 expression, depending on the cell type (for review see Galea and Feinstein, 1999).
Brain endothelium in culture has adenylate cyclase-coupled receptors (Bacic et al., 1991, Durie-Trautmann et al., 1991) and, in vivo, is innervated by noradrenergic, serotonin, and VIP-ergic fibers (Cohen et al., 1997; Paspalas and Papadopoulos, 1998), indicating the existence of local exposure to cAMP-elevating neurotransmitters. It is thus conceivable that cAMP may regulate the expression of inflammatory molecules in BEC, as it does in other cell types. To test this idea, we determined whether cAMP-dependent signaling affects the cytokine-mediated expression of ICAM-1 and NOS-2 mRNA and proteins in cultured BEC. Studies were performed in both primary cultures and in the immortalized BEC cell line GP8.3 (Greenwood et al., 1996). We found that the increase in intracellular cAMP potentiated the cytokine-induced expression of NOS-2 mRNA and protein, whereas it reduced that of ICAM-1. These observations suggest that cAMP may be a biological modulator of inflammatory processes at the blood-brain barrier.
MATERIALS AND METHODS
Brain endothelial cell cultures
Primary cultures. Cells were obtained following a previously described procedure (Abbott et al., 1992) with modifications. Two-month-old male Sprague Dawley rats were lightly anesthetized with halothane and decapitated. Brains were dissected out of the skulls and placed in buffer A (DMEM, 20 mmol/L Hepes buffer pH 7.6, 100 IU/mL penicillin, and 100 μg/mL streptomycin). The meninges and the choroid plexus were peeled off, and the cerebral cortices were isolated, finely chopped with scalpels, and incubated for 1 hour at 37°C in buffer A containing 0.5% bovine serum albumin, 1 mg/mL collagenase/dispase (Sigma, St. Louis, MO, U.S.A.), 20 U/mL DNAse I (type II, Sigma), and 0.15 μg/mL protease inhibitor TCLK (Sigma). The suspension was briefly centrifuged, the pellet resuspended in buffer A containing 13% dextran, and centrifuged for 10 minutes at 5,800 × g at 4°C in a swinging bucket rotor. The resulting pellet containing brain parenchymal microvessels was saved, and the remaining tissue was resuspended and centrifuged twice to increase the yield. The microvessel pellets were pooled, resuspended in buffer A containing collagenase/dispase, and incubated for 2 hours at 37°C with gentle shaking. The incubation was halted by addition of buffer A, and the vessels were collected by brief centrifugation and seeded on plates coated with 0.1% bovine collagen (Sigma). Culture media consisted of high-glucose (25 mmol/L) DMEM (GIBCO, Gaithersburg, MD, U.S.A.) containing 20 mmol/L Hepes, pH 7.4, and supplemented with 100 μg/mL heparin (Sigma), 100 μg/mL human endothelial cell growth supplement (ECGS), 10% heat-inactivated fetal calf serum (Atlanta Biologicals, Atlanta, GA, U.S.A.), and 10% rat serum. The autologous serum significantly increases the proliferation rate of the cells (Balyasnikova et al., in preparation). Rat serum was routinely prepared from the rats used for microvessel preparations. Blood was extracted, centrifuged at 6000 × g for 30 minutes at 4°C, and the serum sterilized with a 0.1-μM filter. The cells were grown up to confluency (7 to 10 days), the media being changed every other day. Brief exposure to Ca++-and Mg++-free Hanks buffered saline on day 2 or 3 was carried out to remove contaminant cells, which were large cells with multiple processes, perhaps fibroblasts. Examination of confluent cultures under a phase contrast microscope revealed mostly uniform monolayers of intensely refringent cells with the spindle shapes characteristic of endothelial cells. The endothelial nature of the cultures was confirmed by immunohistochemical analysis of the presence of the endothelial marker factor VIII (antibody purchased from DAKO, Carpinteria, CA, U.S.A.). Occasionally, contaminating cells appeared either on top of the endothelial monolayers or forming colonies, although they never amounted to more than 10% of the total number of cells.
Immortalized endothelial cells. The generation and characterization of these cells, clone GP8.3, have been described else-where (Greenwood et al., 1996). The cells were plated in 0.1% bovine collagen and grown in high-glucose DMEM supplemented with 20% heat-inactivated fetal calf serum, 50 μg/mL human brain ECGS, and 100 μg/mL heparin. Cells were passaged by a brief digestion of 0.05 mmol/L trypsin and used between passages 10 and 12.
Quantification of NOS-2 and ICAM-1 proteins
The expression of the NOS-2 protein was estimated by measuring the accumulation of nitrites in the culture media using the Griess reagent (Green et al., 1982). The expression of cell surface ICAM-1 was assessed by ELISA. Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS), pH 7.0, for 20 minutes at room temperature. They then were incubated with PBS containing 0.5% casein for 30 minutes at 4°C and were incubated with a monoclonal antibody against rat ICAM (clone 1A29, PharMingen, San Diego, CA, U.S.A.) in PBS/0.5% casein overnight at 4°C. The concentration of the antibody used, 3 μg/mL, was found in pilot experiments to detect cell surface ICAM-1 within the linear range. Cells were incubated in parallel with nonimmune mouse IgG (Sigma) as a negative control. Cells then were incubated for 1 hour with 1:1,000 goat anti-mouse antibody conjugated to alkaline-phosphatase (Sigma) and diluted in PBS/0.5% casein. The alkaline phosphatase substrate (Sigma) was added, and the cells were incubated at room temperature until the reaction developed. Then, the OD405 was measured in a 96-well plate reader. ICAM-1 contents are expressed as relative to the constitutive expression detected by the immunocytochemical reaction.
RNA isolation and reverse transcriptase polymerase chain reaction analysis
We followed procedures that have been previously described (Gilliland et al., 1990; Galea et al., 1998a). Total cytoplasmic RNA was isolated from cells by homogenization in hyperosmotic Tris-HCl buffer, digestion in proteinase K, extraction in phenol-chloroform, and isopropanol precipitation. Synthesis of cDNA was carried out in the presence of random primers using Superscript reverse transcriptase (GIBCO). Samples were amplified by polymerase chain reaction (PCR) using 35 cycles consisting of denaturation at 93°C for 30 seconds, annealing at 60°C for 45 seconds, and synthesis at 72°C for 45 seconds. The PCR products were separated by electrophoresis through a 2% agarose gel with ethidium bromide. All PCR reactions were carried out in a Hybaid Thermal Reactor (Denville Scientific, Denville, NJ, U.S.A.) controlled by tube temperature. For semiquantification, PCR reactions contained a single amount of internal standards for NOS-2 (0.1 fg), ICAM-1 (1 fg). The PCR products were separated by electrophoresis in 2% agarose and quantitated using a computer image analysis system (Biorad).
Electrophoretic mobility assays
To determine whether NFκB may play a role in the regulatory actions of cAMP, we studied by electrophoretic mobility assay (EMSA) the effect of cAMP on the cytokine-stimulated translocation of NFκB to the nucleus.
Preparation of nuclear extracts. The cells were washed with ice-cold PBS, scraped out of the plates, collected by brief centrifugation, resuspended in buffer A (which contained 10 mmol/L Hepes buffer pH 7.9, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.5 mmol/L dithiothreitol, and 0.5 mmol/L phenylmethylsulfonylfluoride), and placed on ice for 10 minutes. Then, nonidet P-40 was added (final concentration 0.6%), and cells were vortexed for 10 seconds. The nuclei were collected by brief centrifugation in a microcentrifuge, the supernatants discarded, and the nuclei were washed once with buffer A and collected again by centrifugation. To obtain nuclear protein extracts, the nuclei were incubated in buffer B (20 mmol/L Hepes buffer, 25% glycerol, 420 mmol/L NaCl, 0.2 mmol/L ethylenediamine tetraacetic acid [EDTA], 0.5 mmol/L dithiothreitol, and 0.5 mmol/L phenylmethylsulfonylfluoride) at 4°C for 30 minutes with gentle rocking. The suspensions were centrifuged at 15,000 × g for 15 minutes at 4°C, and the supernatants stored at −80°C. Protein concentrations were measured by the Bradford method.
EMSA procedure. The NFκB oligonucleotides (5 pmol) corresponding to the palindromic consensus sequences were end-labeled with 10 μCi [γ32P]-ATP (New England Nuclear, Boston, MA, U.S.A.) and 10 U T4 polynucleotide kinase (Promega), and purified in G-50 Sephadex columns (Pharmacia, Piscataway, NJ, U.S.A.). Nuclear extracts (5 to 10 μg protein) were incubated with the radiolabeled oligonucleotide (20,000 to 100,000 cpm) in a final volume of 20 μL in the presence of 1 μg poly dIdC, 20 mmol/L Hepes buffer pH 7.9, 1 mmol/L EDTA, 5% glycerol, 100 mmol/L NaCl, and 0.5 mmol/L dithiothreitol. Incubations were carried out for 20 minutes at room temperature. DNA-protein complexes were resolved by electrophoresis (150 V) in 4% nondenaturing polyacrylamide gels using 0.5× Tris-Borate-EDTA as the running buffer. Control for nonspecific binding was carried out using mutant NFκB oligonucleotides instead of consensus ones. Sequences of oligonucleotides (double-stranded) were as follows: consensus NFκB, 5′ AGT TGA GGG GAC TTT CCC AGG C-3; and mutant NFκB, 5′ AGT TGA GGC GTT TTT CCC AGG C-3′ (Santa Cruz Biotechnology).
RESULTS
Effect of IL-1β and IFN-γ on the expression of NOS-2 and ICAM-1 proteins
Primary cultures. Cells were exposed to IL-1β (1 to 100 ng), IFN-γ (1 to 100 U/mL), or combinations of the two cytokines. Protein expression for ICAM-1 and NOS-2 was measured 24 hours later. No significant basal production of nitrites was detected, indicating lack of constitutive expression of the NOS-2 protein. The result also indicates that the amount of nitrite accumulation from the NOS-3 constitutively present in the cells (data not shown) is undetectable. IL-1β alone increased nitrite accumulation to detectable levels, although not statistically significant compared with the basal expression. Addition of IFN-γ significantly potentiated the effect of IL-1β by eightfold (Fig. 1). By itself, IFN-γ had no effect, suggesting that IFN-γ can “prime” the effect of other cytokines on NOS-2 expression, an effect well documented in several cell types (Feinstein et al., 1994; Martin et al., 1994).
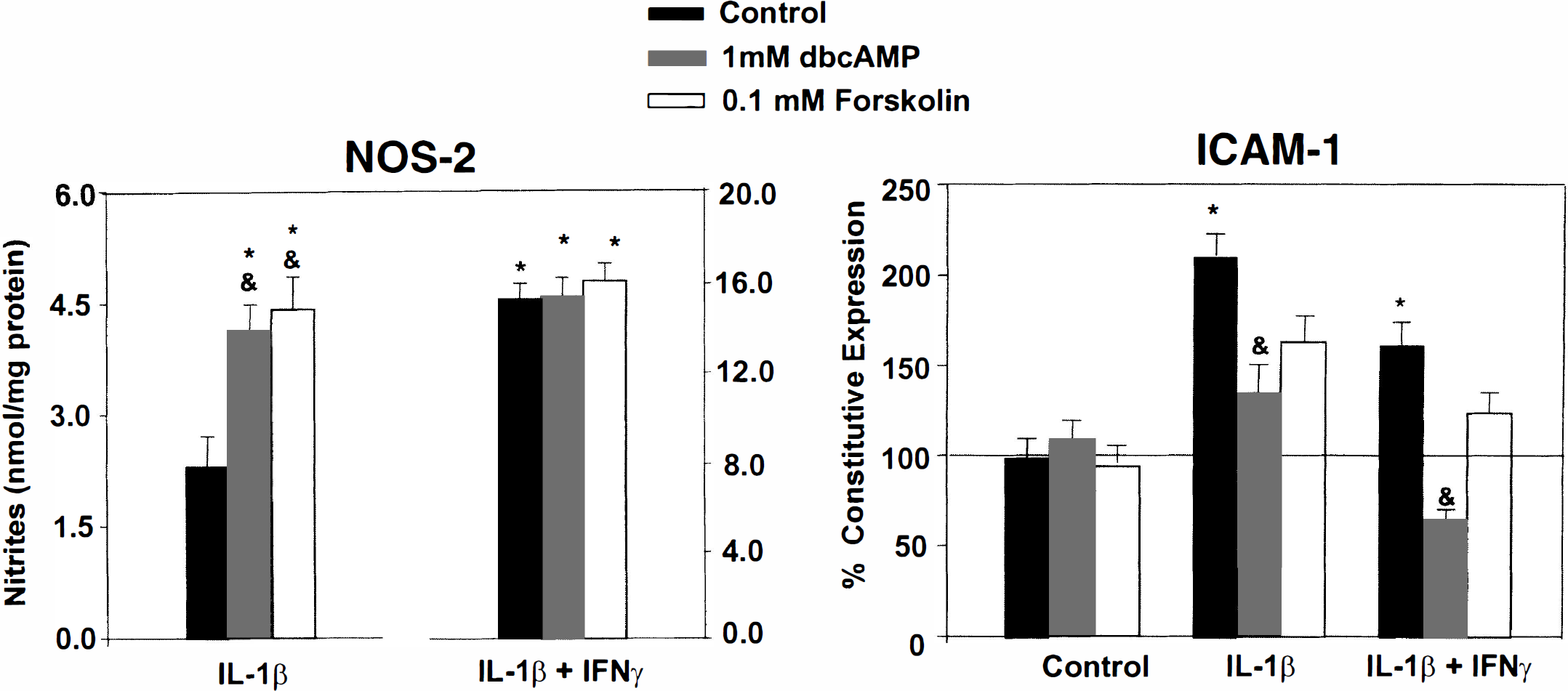
Effect of cytokines and cAMP-elevating agents on the expression of NOS-2 and ICAM-1 proteins in primary endothelial cell cultures. Cells were exposed to four different treatments: (1) DMEM alone; (2) IL-1β; 100 ng/mL alone or together with IFN-γ; 100 U/mL; (3) cytokines plus forskolin or dbcAMP; or (4) forskolin or dbcAMP alone. NOS-2 and ICAM-1 protein expression was assessed 24 hours later by the release of nitrites and ELISA, respectively. Notice that responses to IL-1β and IL-1β/IFN-γ are displayed at different scales. The “DMEM” group was used as the blank for “cytokines,” whereas “forskolin or dbcAMP alone” was the blank of “cytokines plus forskolin or dbcAMP.” Interleukin-1β and IFN-γ synergized to induce NOS-2 enzyme expression, whereas only IL-1β induced an increase in the content of cell surface ICAM-1 protein. Stimulation of cAMP-dependent pathways stimulated IL-1β-mediated NOS-2 expression, but it inhibited the expression of ICAM-1 protein. Forskolin or dbcAMP alone did not change the expression of NOS-2 or ICAM with respect to the incubation with DMEM alone. Values are the means ± SD of three independent experiments. *P < 0.05, with respect to basal expression. (&) P < 0.05, with respect to expression in the absence of cAMP-elevating agents (analysis of variance [ANOVA] followed by Fisher's post hoc analysis).
Unlike NOS-2, the ICAM-1 protein was constitutively present in the endothelial cell membrane (Fig. 1, black bars). IL-1β (100 ng/mL) increased ICAM-1 content by twofold. Alone, IFN-γ (100 U/mL) did not induce ICAM-1 expression, nor did it potentiate the effect of IL-1β. Instead, it caused a small but statistically significant decrease of the IL-1β-mediated effect.
Immortalized brain endothelial cells. No constitutive NOS-2 expression was detected in the immortalized BEC. Although neither IL-1β (1 ng/mL) nor IFN-γ (10 and 100 U/mL) induced nitrite release on their own, together they synergized to induce NOS-2 expression (Fig. 2). In contrast, ICAM-1 protein was constitutively present in the cell surface, and its expression was increased by approximately 70% by 1 ng/mL IL-1β. No further increase was observed by greater concentrations of IL-1β. The IFN-γ alone (10 and 100 U/mL) did not induce ICAM-1 expression, nor did it potentiate the effect of IL-1β.
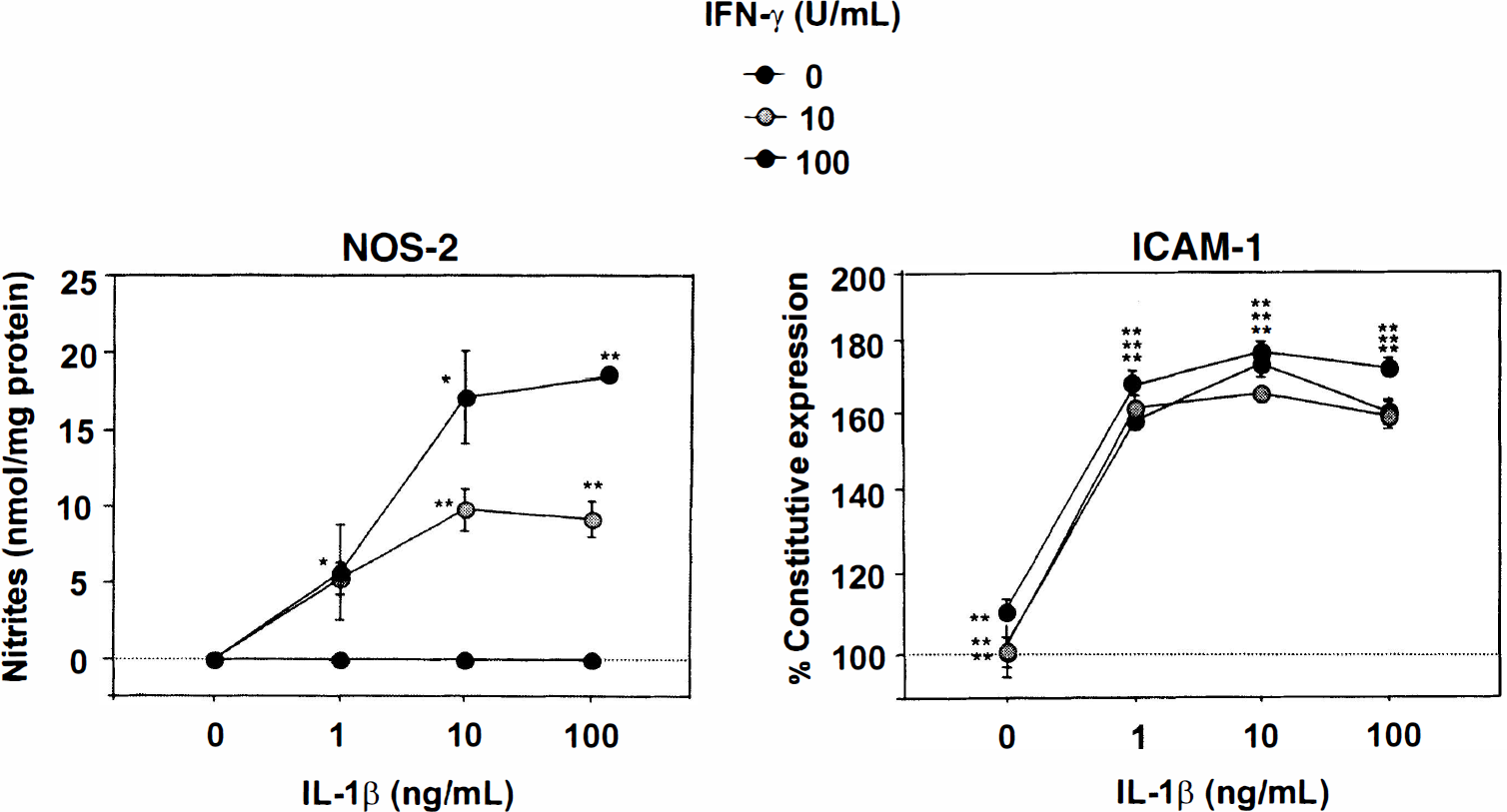
Effect of IL-1β and IFN-γ on the expression of NOS-2 and ICAM-1 proteins in immortalized RBEC (clone GP8.3). Cells were incubated at the indicated concentration of cytokines, and 24 hours later, NOS-2 and ICAM-1 protein expression was assessed by the release of nitrites and ELISA, respectively. Both IL-1β and IFN-γ induced NOS-2 expression in a dose-dependent and synergistic fashion, whereas only IL-1β induced expression of ICAM-1 protein. Data are representative of three separate experiments. Values are the means ± SD of triplicate measurements. *P < 0.05 and **P < 0.01 compared with basal expression (ANOVA followed by Fisher's post hoc analysis).
Effect of cAMP on the expression of ICAM-1 and NOS-2 proteins
Effect on NOS-2 expression.
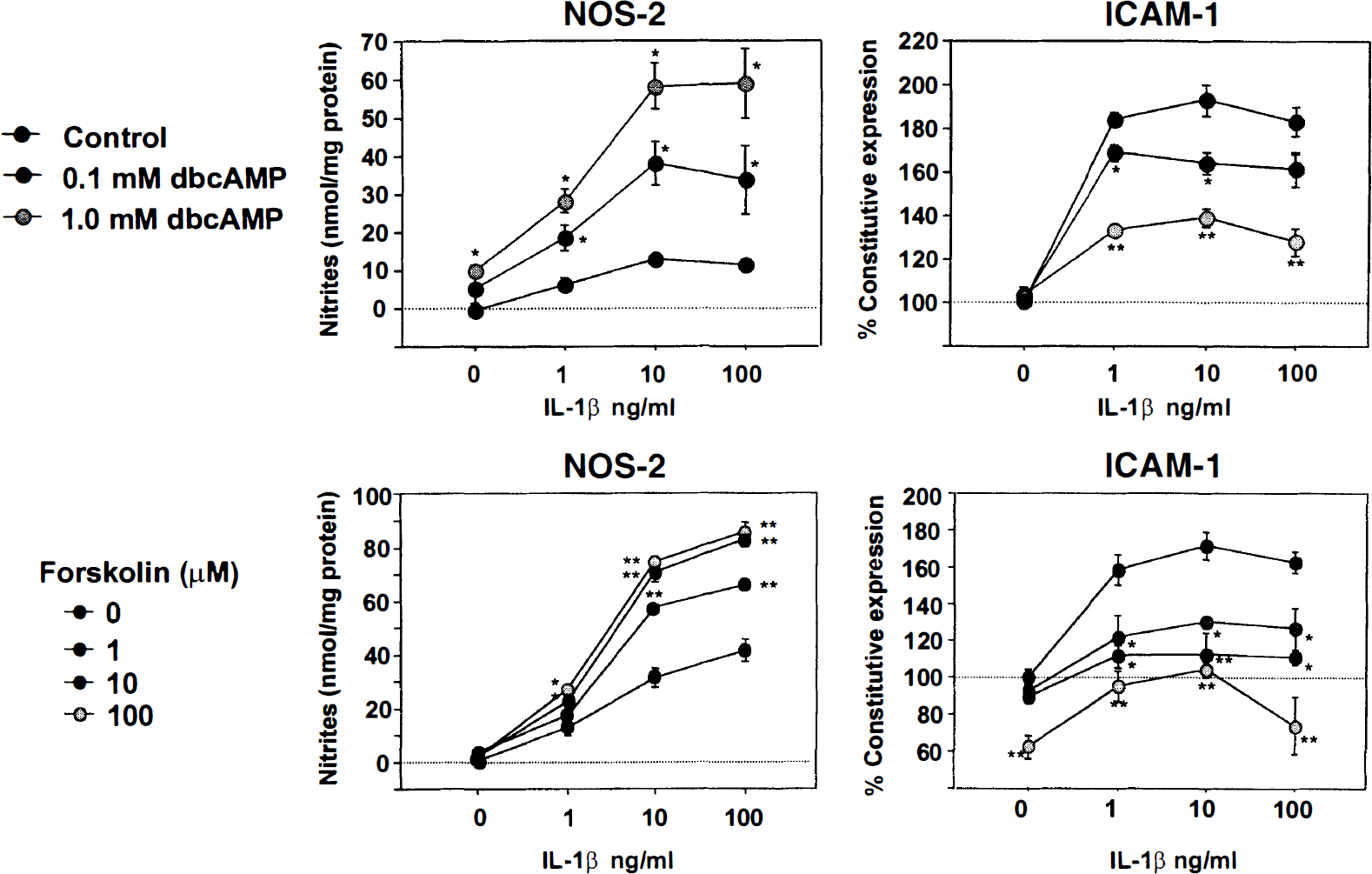
Effect of elevations of intracellular cAMP on ICAM-1 and NOS-2 protein expression in immortalized BEC (clone GP8.3). Cells were incubated for 24 hours with DMEM alone, IL-1β (100 ng/mL) alone or together with IFN-γ (100 U/mL), cytokines plus forskolin or dbcAMP, or forskolin or dbcAMP alone. The “DMEM” group was used as the blank for “cytokines,” whereas “forskolin or dbcAMP alone” was the blank of “cytokines plus forskolin or dbcAMP.” Activation of cAMP-dependent pathways decreased ICAM-1 but increased NOS-2 protein expression. Exposure to dbcAMP or forskolin had no effect. Data are representative of three separate experiments. Values are the means ± SD of triplicate measurements. *P < 0.05 and **P < 0.005 with respect to expression in the absence of dbcAMP or forskolin (ANOVA and Fisher's post hoc analysis).
Effect on ICAM-1 expression.
These results indicate that in both primary and immortalized BEC, stimulation of cAMP-dependent pathways respectively potentiate and inhibit the cytokine-induced expression of NOS-2 and ICAM-1.
Effect of cAMP on ICAM-1 and NOS-2 mRNA steady-state contents
To determine whether cAMP alters the expression of NOS-2 and ICAM-1 mRNA, immortalized endothelial cells were incubated with IL-1β (20 ng/mL) plus IFN-γ (100 U/mL) in the presence and absence of 0.1 mmol/L dbcAMP. Five hours later, a time at which maximal expression of NOS-2 occurs after inflammatory activation (Galea et al., 1994), cells were processed to measure contents of ICAM-1 and NOS-2 mRNA by semiquantitative PCR. Consistent with the protein expression, non-stimulated cells showed constitutive expression of ICAM-1 but not NOS-2 mRNA. On exposure to cytokines, both mRNA were upregulated (Fig. 4). The dbcAMP did not have an effect on its own, but it modified the cytokine-mediated expression: NOS-2 mRNA was upregulated approximately three times, whereas ICAM-1 mRNA was decreased by approximately 50%. That is, the changes in mRNA contents induced by cAMP-dependent pathways correlated with the changes in protein expression.
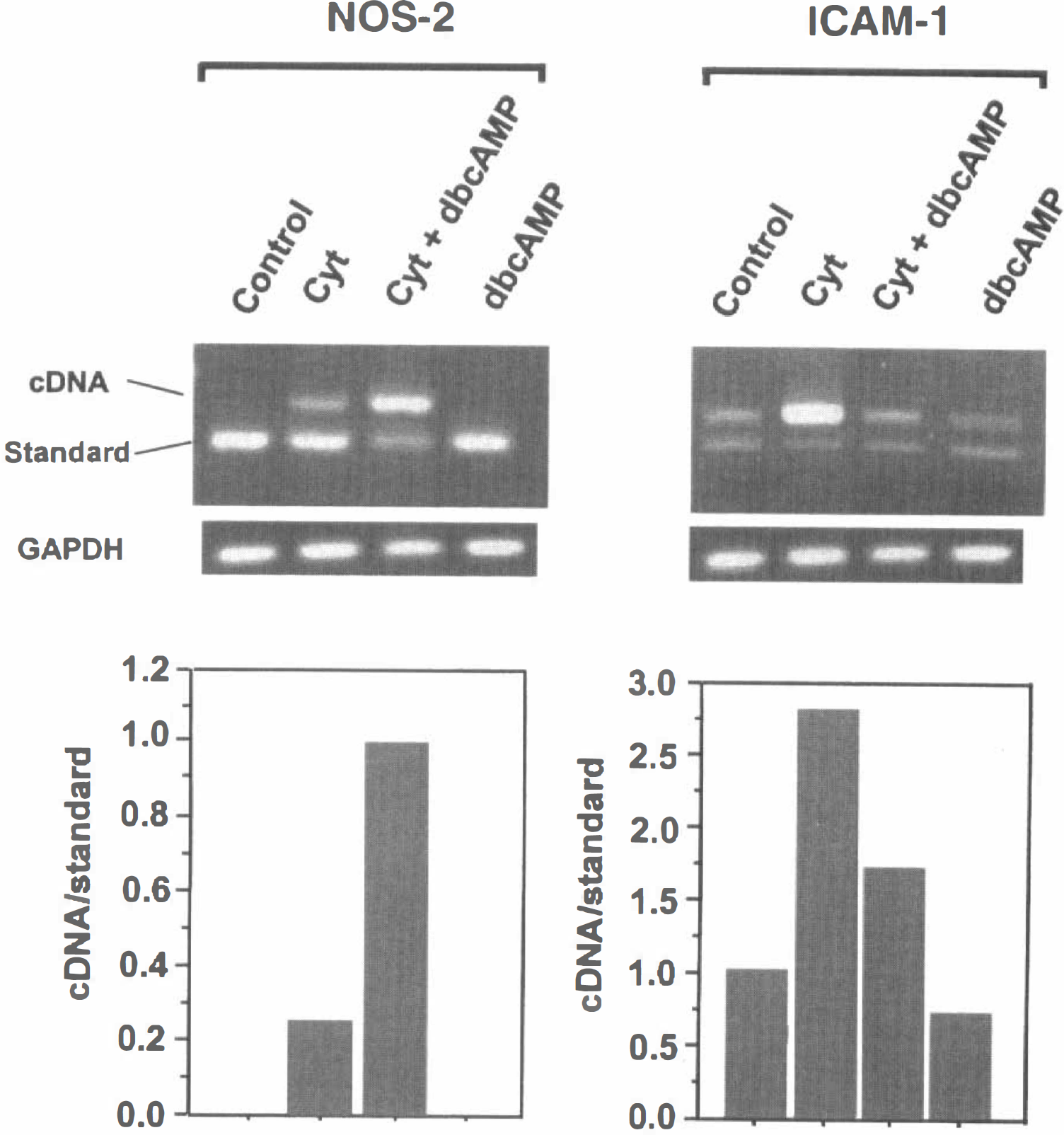
Effect of cAMP on the steady-state expression of NOS-2 and ICAM-1 messenger RNA (mRNA). Immortalized endothelial cells (clone GP8.3) were incubated for 5 hours with IL-1β (20 ng/mL) and IFN-γ (100 U/mL), with or without 0.5 mmol/L dbcAMP, and processed for analysis by RT-polymerase chain reaction (RT-PCR). Semiquantification was performed by amplification of the cDNA with NOS-2 or ICAM-1 internal standards. Parallel analysis of GAPDH in the same samples revealed no significant intersample variations. Increase of intracellular cAMP respectively inhibited and potentiated ICAM-1 and NOS-2 mRNA expression. Data are representative of four independent experiments. Top: Agarose gel showing PCR products corresponding to NOS-2 or ICAM2 cDNA, the internal standards, and GAPDH. Bottom: Relative amounts of cDNA versus internal standards.
Does NOS-2-derived NO mediate the decrease in ICAM-1 expression induced by cAMP?
In peripheral endothelium, NO inhibits the expression of adhesion molecules by blocking NFκB activation (De Caterina et al., 1995; Lefer et al., 1999). To determine whether the cAMP-induced increase in NO production by NOS-2 could account for the parallel decrease in ICAM-1 expression in BEC we examined the following possibilities: (1) if inhibition of NOS-2 activity by the L-arginine analog L-NMMA reversed the decrease in ICAM-1 expression induced by activation of cAMP-dependent pathways; and (2) whether cAMP could inhibit the activation of NFκB induced by cytokines.
Immortalized cells were stimulated with IL-1β (100 ng/mL) and IFN-γ (100 U/mL) in the presence of 0.1 mmol/L dbcAMP, or 0.01 mmol/L forskolin, and 100 μmol/L L-NMMA. As expected, cytokines induced the expression of both ICAM-1 and NOS-2 protein, and stimulation of cAMP-dependent signaling potentiated and inhibited NOS-2 and ICAM-1 protein expression, respectively (Fig. 5). Incubation with L-NMMA inhibited the cytokine-induced nitrite release, demonstrating that the L-arginine analog had effectively inhibited NOS-2 activity. However, in the same cells, L-NMMA failed to alter the cAMP-induced reduction of ICAM-1 expression. This result indicates that the increase in NO production induced by cAMP did not account for the parallel reduction of ICAM-1 expression.
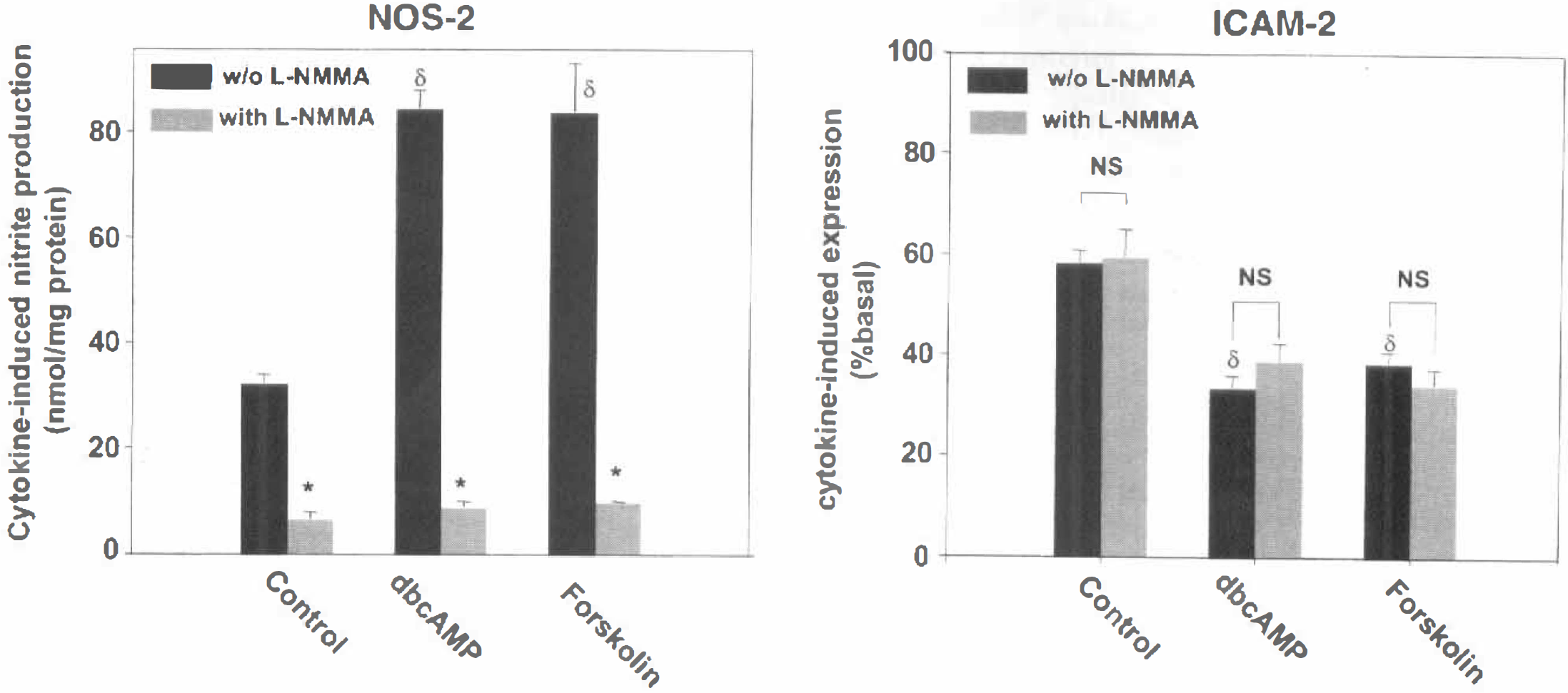
Effect of L-NMMA on the expression of ICAM-1 and NOS-2 proteins. GP8.3 cells were incubated with 100 ng/mL IL-1β and 100 U/mL IFN-γ in the presence of 1 mmol/L dbcAMP, 0.1 mmol/L forskolin, and the NOS inhibitor L-NMMA. Twenty hours later, NOS-2 and ICAM-1 protein expression was assessed. Inhibition of NOS-2 activity did not alter the effect of cAMP on ICAM-1 expression. Values are the means ± SD of three independent experiments. §P < 0.05 with respect to the expression in the absence of dbcAMP or forskolin; *P < 0.05 with respect to the expression in the absence of L-NMMA (ANOVA followed by Fisher's post hoc analysis).
To measure NFκB activation, nuclear extracts were prepared from cells exposed to cytokines or dbcAMP and processed for EMSA using oligonucleotides containing consensus or, for control, mutated NFκB binding sites. Because the NOS-2 enzyme had to be synthesized de novo, we tested short and long incubation times to consider the possibility that the NOS-2-released NO may act on the sustained rather than on the acute NFκB activation.
A highly abundant DNA-protein complex of fast mobility appeared in all samples, both when using consensus and mutated oligonucleotides, indicating that it did not contain NFκB proteins (labeled as nonspecific in Fig. 6). Exposure to cytokines induced the formation of two additional complexes (I and II) at the two times analyzed. Supershift analysis confirmed the presence of the p65 NFκB protein in both complexes (data not shown). The presence of complex II in control cells indicated some constitutive NFκB activation. Addition of dbcAMP did not modify significantly the band pattern detected in the presence of cytokines, nor did it induce NFκB activation on its own. This result indicates that, in BEC, activation of cAMP-dependent signaling does not alter the cytokine-induced movement of NFκB transcription factor to the nucleus.
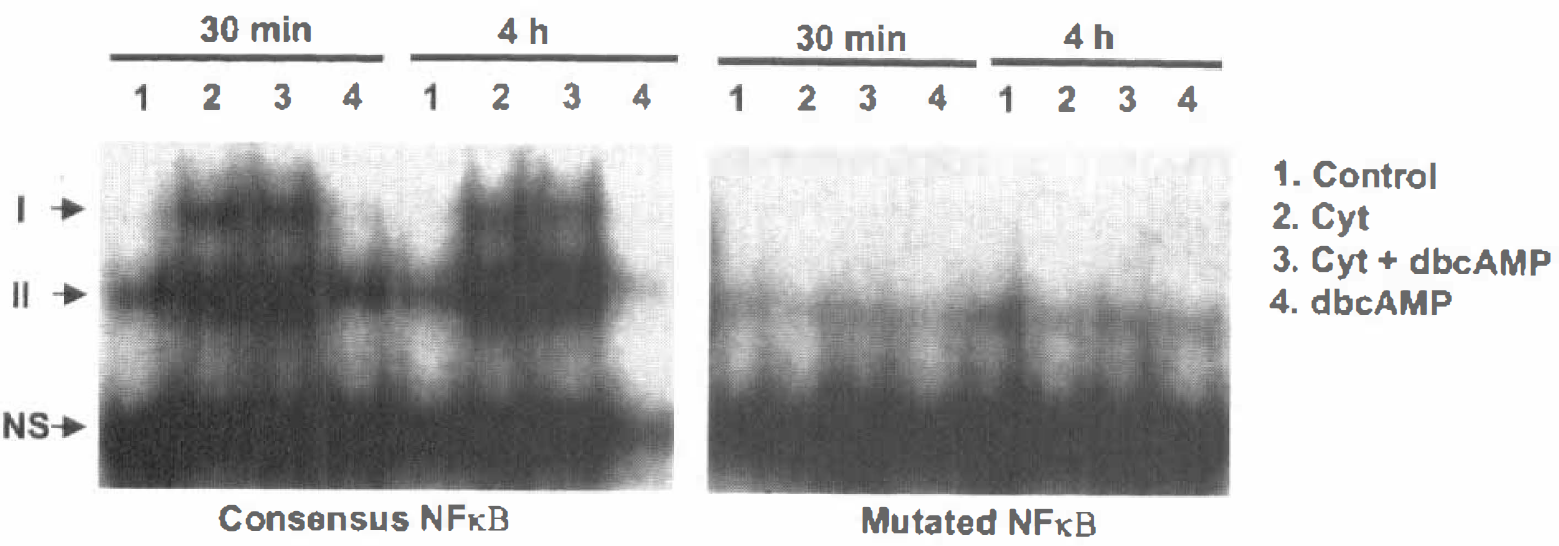
Effect of cAMP on the activation of NFκB. Nuclear extracts were prepared from immortalized endothelial cells (GP 8.3) incubated with cytokines (Cyt), in the presence or absence of dbcAMP, for 30 minutes or 4 hours. Extracts were incubated with radiolabeled oligonucleotides containing a consensus or a mutated NFκB binding site, and separated by electrophoresis. I and II are NFκB-containing DNA-protein complexes. Activation of cAMP-dependent pathways did not modify the cytokine-induced translocation of NFκB to the nucleus. Data are representative of four independent experiments. NS: nonspecific complex.
DISCUSSION
We have shown that proinflammatory cytokines induced the expression of NOS-2 and ICAM-1 proteins and mRNA in cultured BEC, that cAMP regulated this expression, although in an opposite manner, and that the effect of cAMP was independent of NO and NFκB activation.
Both IL-1β and IFN-γ acted synergistically to induce NOS-2 expression, whereas only IL-1β induced an increase in ICAM-1 expression. This result is surprising because IFN-γ, alone or in combination with other cytokines, has been shown to be a potent inducer of ICAM-1 expression on many cell types, including astrocytes (Frohman et al., 1989), smooth muscle cells (Braun et al., 1995), BEC (Fabry et al., 1992; Wong and Dorovini-Zis, 1992; McCarron et al., 1993), and human umbilical endothelial cells (Sancho et al., 1999). The reasons for the discrepancy between these studies and ours are not clear.
Whereas ICAM-1 and NOS-2 have been widely studied individually, this is the first time that the regulation of the two molecules has been analyzed at the same time. Stimulation of cAMP-dependent signaling had a dual effect on the response to cytokines: it potentiated the expression of NOS-2, whereas it inhibited ICAM-1 expression, affecting both the mRNA and protein contents. This finding was unexpected because it is believed that the production of these proinflammatory molecules is regulated in unison, triggered in the early phases of inflammation, and inhibited in later phases by suppressors of inflammation (Vodovotz et al., 1993; Bogdan et al., 1994; Shimizu et al., 1999).
Experiments were conducted in primary endothelial cell cultures, and in the immortalized endothelial cell line GP8.3, to determine whether the latter, much easier to generate, can be used as a model for blood-brain barrier inflammation. Several differences were found in the responses of the two cell types. First, in primary cultures IL-1β alone induced expression of NOS-2 and IFN-γ greatly potentiated this effect, whereas in the GP8.3 cells, NOS-2 expression was elicited only in the presence of both cytokines. Second, cAMP potentiated NOS-2 expression in primary cultures when the inflammatory stimulus was IL-1β but not in the presence of IL-1β and IFN-γ. By contrast, cAMP could potentiate the effect of IL-1β plus IFN-γ in GP8.3. cells. These observations suggest that, in primary cultures but not in GP8.3 cells, the NOS-2 expression elicited by the combination of the two cytokines was maximal, and thus it could not be further increased by stimulation of cAMP-dependent pathways. It thus appears that the sensitivity to cytokines may be reduced in GP8.3 cells with respect to the primary endothelial cell cultures, perhaps because of the loss of cytokine receptors.
Another difference observed was related to the effect of the drugs used to stimulate cAMP-dependent pathways. Thus, dbcAMP was the most potent inhibitor of ICAM-1 expression in primary cultures, whereas forskolin was the most effective in GP8.3 cells. Unlike dbcAMP, which is a nonmetabolizable cAMP analog, forskolin causes a transient increase in the content of intracellular cAMP because of the actions of phosphodiesterases. Conceivably, some phosphodiesterase activities may have been lost in the generation of the GP8.3 cells, thereby explaining the greater effectiveness of forskolin. Although these findings reveal differences between primary and immortalized endothelium regarding cAMP-dependent signaling, the effect of cAMP on NOS-2 and ICAM-1 expression was similar in both cells types. We thus conclude that the GP8.3 cells can be used instead of the primary cultures to gain insight into mechanisms of action of this second messenger.
Stimulation of cAMP-dependent pathways can inhibit adhesion molecule expression in several cell types. Thus, elevation of intracellular cAMP reduces the TNF-α- or IL-1β-mediated upregulation of ICAM-1 and VCAM1 in smooth muscle cells (Panettieri et al., 1995) and astrocytes (Ballestas and Benveniste, 1997). Also, cAMP antagonizes the TNF-α-mediated expression of endothelial leukocyte adhesion molecule, ELAM1, and vascular cell adhesion molecule, VCAM1, in human BEC (Pober et al., 1993; Ghersa et al., 1994). Reports on the effect of cAMP on ICAM-1 expression in endothelial cells are, however, conflicting. Whereas one group has found suppression (Ochi et al., 1995), others report potentiation (Ghersa et al., 1994) or no effect (Deisher et al., 1993; Pober et al., 1993; Morandini et al., 1996). These observations, together with ours, reveal that cAMP-dependent signaling exerts a widespread modulatory-mostly suppressive-action on the expression of adhesion molecules in the course of inflammatory activation, although differences exist, depending on the cell and adhesion molecule being analyzed. Expression of NOS-2 also is widely regulated by cAMP. However, cAMP can play stimulatory or inhibitory roles, depending on the cell type (for review see Galea and Feinstein, 1999). Thus, endotoxin- or cytokine-mediated NOS-2 mRNA and NOS-2 protein expression is inhibited by cAMP in glial (Feinstein et al., 1993), pancreatic, and liver cells (Andersen et al., 1996; Harbrecht et al., 1996). By contrast, increased cAMP stimulates NOS-2 expression in smooth muscle cells (Eberhardt et al., 1998), adipocytes (Nisoli et al., 1997), and, as shown here, BEC.
For both ICAM-1 and NOS-2, protein and mRNA contents changed in parallel after increasing intracellular cAMP. This suggests that cAMP-dependent pathways target pre-translational events such as regulation of mRNA stability or gene transcription. The 3′ untranslated regions of both the ICAM-1 (Wertheimer et al., 1992) and the NOS-2 (Galea et al., 1994) mRNA contain several AUUUA motifs. These sequences modulate mRNA turnover and are typically abundant in short-lived mRNA (Kruys et al., 1990). However, evidence in other cell types demonstrates that regulation of ICAM-1 (Occhi et al., 1995; Ballestas and Benveniste, 1997) and NOS-2 (Eberhardt et al., 1998) expression by cAMP occurs primarily at the level of gene transcription. We thus speculate that, in brain endothelium, the changes in production of NOS-2 and ICAM-1 protein and mRNA also occur via changes in the transcriptional rates of the corresponding genes.
The question then arises as to how cAMP-dependent pathways exert opposite effects on the transcription of the ICAM-1 and NOS-2 genes. One possibility is that the increase in NOS-2 and the decrease of ICAM-1 were causally related. It is well established that NO can inhibit leukocyte adhesion to the vascular walls under basal conditions or during inflammation. Studies using NO donors or NOS inhibitors have demonstrated that NO acts by suppressing the expression of adhesion molecules such as platelet endothelial adhesion molecule, VCAM1, P-selectin, and ICAM-1 (Luvara et al., 1998; Liu et al., 1998; Scalia and Lefer, 1998; Lefer et al., 1999). The mechanism of action of NO appears to be the reduction of gene transcription by blockade of NFκB activation (De Caterina et al., 1995; Khan et al., 1996; Kupatt et al., 1997), either by scavenging reactive oxygen species (Kupatt et al., 1997) or by upregulation of the endogenous inhibitor of NFκB, IKB-α (Spiecker et al., 1998). Considering this evidence, it is tempting to hypothesize that the NO derived from NOS-2 causes the inhibition of ICAM-1 expression. However, inhibition of NOS-2 activity did not prevent the cAMP-mediated down-regulation of ICAM-1 expression, nor did it alter NFκB activation. These results suggest that, unlike in peripheral endothelium, NO does not play an antiadhesive role in brain endothelium. Alternative interpretations are, nevertheless, possible. First, NO could decrease expression of adhesion molecules other than ICAM-1. Precedents for this exist in saphenous endothelial cells, where NO donors inhibited the expression of VCAM1 and E-selectin but not ICAM-1 (De Caterina et al., 1995), and in human lung microvascular cells, in which increases in intracellular cAMP by phosphodiesterase inhibitors decreased the cytokine-induced expression of E-selectin but did not alter ICAM-1 expression (Blease et al., 1998). Second, down-regulation of ICAM-1 by NO may occur in resting conditions but not during inflammation. This has been observed in vitro in human umbilical endothelial cells (Biffl et al., 1996) and in an in vivo model of acute inflammation (Cockrell et al., 1999).
Alternatively, it has been proposed in peripheral endothelial cells that the inhibitory effect of cAMP on adhesion molecule expression is mediated by cAMP response element binding protein (CREB), the transcription factor activated by protein kinase A (Parry and Mackman, 1997). The evidence suggests that CREB may block the transcriptional activity of the NFκB migrated to the nucleus by quenching essential transcriptional coactivators (Parry and Mackman, 1997). Evidence also indicates a role for CREB in the upregulation of NOS-2 by cAMP. Thus, in smooth muscle-derived cells, analysis of the activity of serial deletion constructs of the NOS-2 promoter, combined with site-directed mutagenesis, reveals that binding of CREB to the site of another transcription factor, C/EBP, accounts for the stimulatory effects of cAMP (Eberhardt et al., 1998). Whether these mechanisms apply to brain endothelium remains to be determined.
Considering that the upregulation of ICAM-1 exacerbates the damage in the course of brain disorders with inflammatory components (e.g., ischemia or demyelinating diseases [Archelos et al., 1993; Morrisey et al., 1996; Chopp et al., 1996; Soriano et al., 1996]), the inhibitory actions of cAMP-dependent signaling should result in protection. Less clear is the role of the parallel upregulation of NOS-2 expression, since blockade of NOS-2 activity in vivo results in both amelioration (Cross et al., 1994; Nagayama et al., 1998) and worsening of brain diseases (Ruuls et al., 1996). The discrepancy may arise from the fact that NOS-2 is expressed in different cell types, namely glial cells, macrophages, and endothelial cells, in association with various brain disorders (Endoh et al., 1994; Cross et al., 1994; Iadecola et al., 1996; Oleszak et al., 1997; Galea et al., 1998b). Because of the highly cytotoxic effects of NO when combined with oxygen radicals, expression of NOS-2 generally is believed to generate further damage. Whereas strong evidence indicates that this may be the case for NOS-2 expressed in macrophages (MacMicking et al., 1997), the NO locally released by endothelial NOS-2 may, by contrast, reduce the brain damage by virtue of its vasodilatory (Loscalzo et al., 1995) and, perhaps, its antiadhesive actions, as discussed earlier. If so, the dual effect of cAMP on the expression of NOS-2 and ICAM-1 in brain endothelium could result in neuroprotection.
It has long been recognized that cAMP-dependent pathways play a role in the regulation of brain microvascular function (Palmer et al., 1986; Cohen et al., 1996). Brain microvessels, including capillaries, arterioles, and venules, are innervated by serotoninergic (Itakura et al., 1985; Cohen et al., 1995), noradrenergic (Cohen et al., 1997), and VIPergic neurons (Paspalas and Papadopoulos, 1998), all of which release neurotransmitters that act via cAMP. Local application of serotonin or noradrenaline, or manipulation of nuclei where perivascular fibers originate, results in changes in blood-brain barrier permeability or local blood flow (Palmer et al., 1986; Sharma et al., 1986; Sarmento et al., 1991). Whereas the vascular smooth muscle cells account for the latter, alterations in permeability are conceivably confined to the endothelial compartment. The ability of endothelial cells to respond directly to the perivascular fibers is underscored by anatomical, biochemical, and functional evidence. First, there is direct apposition of nerve terminals to the endothelial cell basal membrane (Cohen et al., 1997; Paspalas and Papadopoulos, 1998). Second, BEC have receptors for serotonin (Bacic et al., 1991), VIP (Martin et al., 1992), and noradrenaline (Karnushina et al., 1982; Durieu-Trautmann et al., 1991), coupled to cAMP synthesis. Finally, the elevation of cAMP in endothelial cell cultures decreases the transcellular permeability (Borges et al., 1994; Rist et al., 1997; Hurst and Clark, 1998). Our observation that cAMP-dependent pathways in BEC control the cytokine-mediated expression of ICAM-1 and NOS-2 supports the idea, originally developed in a previous study (Galea et al., 1998a), that, in addition to controlling blood flow and permeability, locally released neurotransmitters may regulate inflammatory reactions at the blood-brain barrier.
Footnotes
Abbreviations used
Acknowledgements
The authors thank Drs. Douglas Feinstein and Sergei Danilov for continuous and generous advice on scientific matters.