
Abstract
Pro-inflammatory cytokine-induced activation of nuclear factor, NF-κB has an important role in leukocyte adhesion to, and subsequent migration across, brain endothelial cells (BECs), which is crucial for the development of neuroinflammatory disorders such as multiple sclerosis (MS). In contrast, microRNA-146a (miR-146a) has emerged as an anti-inflammatory molecule by inhibiting NF-κB activity in various cell types, but its effect in BECs during neuroinflammation remains to be evaluated. Here, we show that miR-146a was upregulated in microvessels of MS-active lesions and the spinal cord of mice with experimental autoimmune encephalomyelitis.
Introduction
In neuroinflammation such as multiple sclerosis (MS), the blood—brain barrier (BBB) becomes compromised with intensive migration of leukocytes into the central nervous system (CNS), contributing to disease pathogenesis. 1 To migrate into the CNS, leukocytes follow a finely regulated sequence of events starting from tethering and rolling along the vessel, then firmly adhering to and migrating out of the vasculature. Firm adhesion is triggered by chemokines and mediated by interactions between integrins on leukocytes and adhesion molecules on brain endothelial cells (BECs). 2
Overwhelming evidence highlights that the nuclear factor, NF-κB, drives transcription of cell adhesion molecules, chemokines, and pro-inflammatory cytokines. 3 The NF-κB complex consists of a family of dimeric transcription factors comprising RelA (p65), RelB, c-Rel, p50, and p52. The classic pathway involves p50/p65 heterodimers complexed to IκB in unstimulated cells. Upon stimulation, IKKβ mediates the phosphorylation-induced ubiquitination of IκB, which frees p50/p65 to translocate to the nucleus to initiate gene transcription. 3 NF-κB also coordinates with other signaling pathways, such as RhoA and transforming growth factor-β to modulate the inflammatory response.4, 5 NF-κB has been proposed as a therapeutic target for the treatment of MS, 3 thereby highlighting endogenous inhibitors of NF-κB as potential novel therapies.
MicroRNAs (miRNAs) are endogenous noncoding RNAs ∼22 nucleotides long that pair to the 3′-untranslated region of messenger RNAs and incorporate them into RNA-induced silencing complexes to repress gene expression. Accumulating evidence implicates certain miRNAs in the pathogenesis of MS and experimental autoimmune encephalomyelitis (EAE): miR-155 contributes to the pathogenesis of EAE by modulating T-cell differentiation and promoting endothelial barrier dysfunction,6, 7 whereas brain endothelial miR-125a-5p tightens BBB and prevents leukocyte migration during inflammation. 8
NF-κB not only initiates the transcription of protein-coding genes but also drives the expression of miRNA precursors, which act as feedback modulators of NF-κB. 9 Among them, miR-146a is an NF-κB-dependent gene and downregulates NF-κB activities by repressing two signal transducers in human monocytes: TNF receptor-associated factor 6 (TRAF6) and IL-1 receptor-associated kinase 1 (IRAK1). 10 Several studies suggest miR-146a as a molecular brake on inflammation, myeloid cell proliferation, and oncogenic transformation. 11 The anti-inflammatory role of miR-146a has been demonstrated in several cell types including monocytes, 10 T cells, 12 astrocytes, 13 human umbilical vein endothelial cells (HUVECs), 14 and human BECs. 15 However, its function in BECs remains to be elucidated. Here we show that miR-146a is upregulated in cytokine-activated BECs and decreases leukocyte adhesion by inhibiting NF-κB through repressing not only TRAF6 and IRAK1 but also RhoA and nuclear factor of activated T cells 5 (NFAT5).
Materials and methods
All animal procedures conform to the Animals (Scientific Procedures) Act 1986 of the UK government and the Animal Research: Reporting of
Human brain tissue and primary human T cells
Human brain tissues of MS patients (four females and two males, 34 to 88 years old) and control patients (six males, 35 to 84 years old) without neurologic diseases were obtained from The UK Multiple Sclerosis Tissue Bank (Imperial College London, London, UK) according to local human ethical guidelines (For clinical information and pathological characterization of MS and control patients see ref. 7). Primary human T cells were obtained from buffy coats (Sanquin, Amersterdam, The Netherlands) of healthy volunteers (after informed consent, following the Netherlands human ethical guidelines) through Ficoll gradient centrifugation, followed by negative selection of the primary T cells using MACS magnetic cell sorting kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. 16
Cell lines
The immortalized human brain microvascular endothelial cell line (hCMEC/D3) was cultured on collagen-coated culture flasks or multi-well plates in EGM-2 MV medium supplemented with 2.5% fetal bovine serum and growth factors (Lonza, Slough Wokingham, UK). 7 The Jurkat T lymphocyte cell line was a kind gift from Dr V Male (Cambridge University, Cambridge, UK) and cultured in suspension in RPMI1640 (Life Technologies, Paisley, UK) with 10% fetal bovine serum.
Induction of experimental autoimmune encephalomyelitis
Adult female and male (10 to 12 weeks) Biozzi ABH mice were purchased from Harlan UK (Bicester, UK). Animals were grouped randomly and maintained on a 12 hours: 12 hours light: dark cycle and received food and water
Laser capture microdissection and isolation of microvessels
Laser capture microdissection of MS brain microvessels was used to collect enriched brain endothelium RNA as previously described.
7
Approximately 200 blood vessels were isolated from each case using PixCell II laser capture microdissection system (Arcturus BioScience, Mountain View, CA, USA) and laser capture microdissection-caps (Applied Biosystems, Warrington, UK). For mouse spinal cords, microvessels were isolated as previously described.
7
Briefly, EAE or normal ABH mice were perfused with 0.5% bovine serum albumin in Hank's balanced solution to rinse out the blood, then the spinal cords were flushed out of vertebral columns. For each replicate, spinal cords of three mice in the same group were pooled together to get sufficient amount of RNA and counted as one (total animals used, nine mice per group), digested with collagenase and dispase (1 mg/mL) at 37 °C for 1 hour, and, finally, homogenized and centrifuged through 25% bovine serum albumin. The pellet of microvessels underwent a second digestion for 30 minutes and microvessel fragments were purified and separated from single contaminating cells by passing through a 70-μm-size mesh filter. Enrichment of endothelium within the microvessel fraction was examined by quantitative RT-PCR for endothelial markers (
RNA extraction and quantitative reverse transcriptase-polymerase chain reaction
Total RNA of hCMEC/D3 cells and microvessels isolated from human brain or mouse spinal cords were extracted using TRIzol reagent (Life Technologies). For each sample, 10 ng of total RNA was used for reverse transcription. Expression of miR-146a and U6B small nuclear RNA were measured with TaqMan MicroRNA Assays Kit (Life Technologies). U6B was used as internal control. QuantiTect SYBR Green PCR kit (Qiagen, Manchester, UK) was used to determine the relative levels of two NF-κB target genes vascular cell adhesion molecule-1 (
Immunohistochemistry and in situ hybridization
Cryostat sections of lumbar spinal cords from 4% paraformaldehyde-perfused EAE mice at day 10 after EAE induction (D10,
Cell transfection
A total of 30 nM of Pre-miR-146a were transfected into hCMEC/D3 cells using siPORT Amine transfection agent (Life Technologies), whereas 60 nM of miR-146a inhibitor (Anti-miR-146a) and siGENOME SMARTpool siRNAs for human RelA, IRAK1, TRAF6, STAT1, ROCK1, RhoA, and NFAT5 (ThermoFisher Scientific, Epsom, UK) were transfected into cells using Lipofectamine 2000 (Life Technologies). Non-targeting scrambled Pre-miR, Anti-miR, or siRNA control pools was used as negative controls, respectively. Cy3 Dye-Labeled Pre-miR or Anti-miR Negative Control #1 (Life Technologies), siGLO Red Transfection Indicator (ThermoFisher Scientific) was used for analyzing transfection efficiency.
Leukocyte adhesion assay under shear flow
Jurkat T-cell and primary human T-cell adhesion assay was performed as previously described with modification.17, 18 Briefly, hCMEC/D3 cells were grown on collagen-coated ibidi μ-Slide VI0.4 six-channel slides (Ibidi GmbH, Martinsried, Germany) until confluence. Primary human T cells were activated by 48-hour stimulation with IL-2 (10 ng/mL) and phytohemagglutinin (1 μg/mL). Then Jurkat T cells or activated primary human T cells (2 × 106/mL) were labeled with 5 mM CMFDA (5-chloromethylfluorescein diacetate, Life Technologies) and were allowed to flow through the channel and accumulate for 5 minutes at low shear stress (0.5 dyn/cm2). Shear stress (τ) was calculated according to τ=176.1
Immunocytochemistry
hCMEC/D3 cells grown on glass coverslips coated with collagen were fixed with 4% paraformaldehyde and permeabilized with methanol and acetone at −20 °C for 10 and 1 minute, respectively, then incubated with rabbit anti-NF-κB p65 (1:100, Cell Signaling Technology, Danvers, MA, USA) followed by goat anti-rabbit IgG Alexa Fluor 488 (1:400, Life Technologies). Cell nuclei were stained using Dapi Fluoromount-G (SouthernBiotech, Birmingham, Alabama, USA). Images were captured with a Zeiss Axiophot fluorescent microscope. Number of cells with nuclear staining of NF-κB p65 was expressed as percentage of total cell numbers demonstrated by DAPI (4',6-diamidino-2-phenylindole). Experiments were performed at three time points (0.5, 6, and 24 hours after cytokine stimulation), each performed in three experimental samples with duplicates each counting over 100 cells in three to six randomly selected fields.
Western blot analysis
Cell lysates were separated by 10% (20% for RhoA) sodium dodecyl sulfate—polyacrylamide gel electrophoresis gels and transferred onto nitrocellulose membranes (Amersham, Buckinghamshire, UK), probed with rabbit anti-IRAK1 or TRAF6 or RelA or ROCK1 (1:1,000, Cell Signaling Technology), or rabbit anti-NFAT5 (1:1,000, Thermo Scientific, Northumberland, UK), or STAT1 (1:500, Santa Cruz Biotechnology, Dallas, TX, USA), or mouse anti-RhoA (1:100, Cytoskeleton, Denver, CO, USA), or mouse anti-VCAM1 (1:100, R&D Systems Europe, Abingdon, UK), or mouse anti-β-actin (1:5,000 Sigma, Dorset, UK), followed with horseradish peroxidise-conjugated goat anti-rabbit IgG (1:3,000; Life Technologies) or anti-mouse IgG (1:3,000 for RhoA, 1:14,000 for β-actin; Pierce Biotechnology, Cheshire, UK). Immunoblots were then developed by enhanced chemiluminescence detection (ECL, Amersham, Buckinghamshire, UK).
Lentiviral transduction of 3′-untranslated region reporter vectors and luciferase assay
The luciferase reporter lentiviral vector constructs containing the puromycin resistance gene and the luciferase gene from the firefly Photinus pyralis with the 3′-untranslated region (UTR) of RhoA (1061 bases), NFAT5 (5′-2100 bases) (Applied Biological Materials, British Columbia, VIC, Canada) were used to transduce hCMEC/D3 cells with a multiplicity of infection of 3. Mutated 3′-UTR versions of putative target genes were obtained by replacing the corresponding miR-146a target sites with AAGA using site-directed mutagenesis. Twenty-four hours after transduction, cells were transfected with Pre-miR-146a or scrambled Pre-miR. Seventy-two hours later, luciferase activity was quantified using the Steady-Glo Luciferase assay system (Promega, Madison, WI, USA) and luminescence was determined using the FLUOstar OPTIMA plate reader (BMG LABTECH GmbH, Ortenberg, Germany).
Statistical analysis
All values are presented as means±s.e.m. Student's paired
Results
Neuroinflammation induces upregulation of central nervous system endothelial miR-146a
To elucidate the role of endothelial miR-146a in BBB dysfunction in neuroinflammatory diseases such as MS, we first characterized its expression in human brain microvessels of MS lesions, isolated by laser capture microdissection. In comparison with normal-appearing white matter, cerebral microvascular miR-146a increased ∼1.5-fold in the active lesions of MS (Figure 1A). We then examined the temporal expression pattern of miR-146a in the spinal cord microvasculature of EAE mice, isolated by enzyme digestion. The enrichment of endothelium was confirmed, as previously reported by our lab,
7
by high gene expression levels of endothelial specific markers (
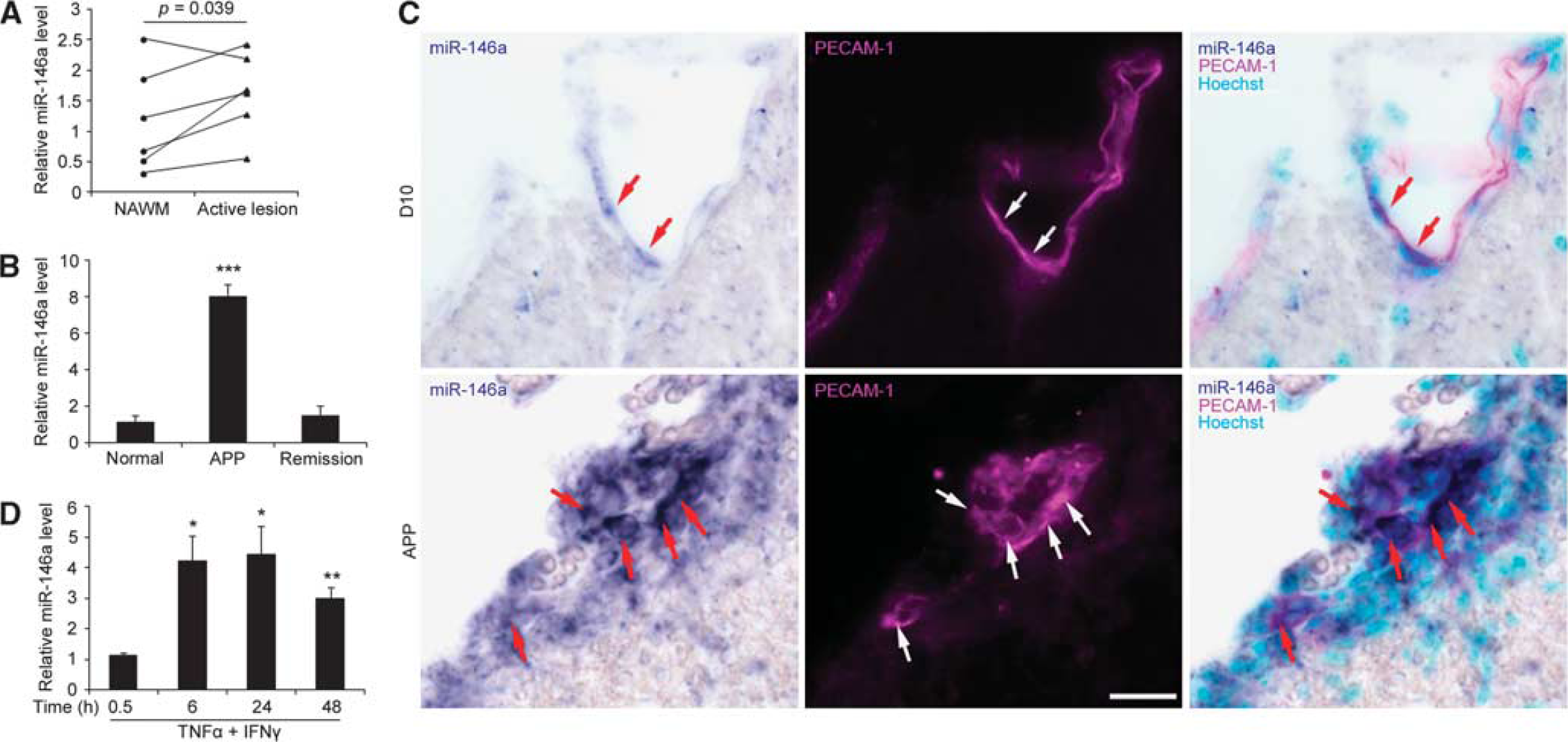
miR-146a is increased in CNS microvessels and cultured brain endothelium during inflammation. (
Brain endothelial miR-146a partially prevents cytokine-stimulated leukocyte adhesion
As miR-146a levels increase in the brain endothelium during neuroinflammation, we then further investigated the role of brain endothelial miR-146a in BBB dysfunction, particularly on leukocyte adhesion to brain endothelium
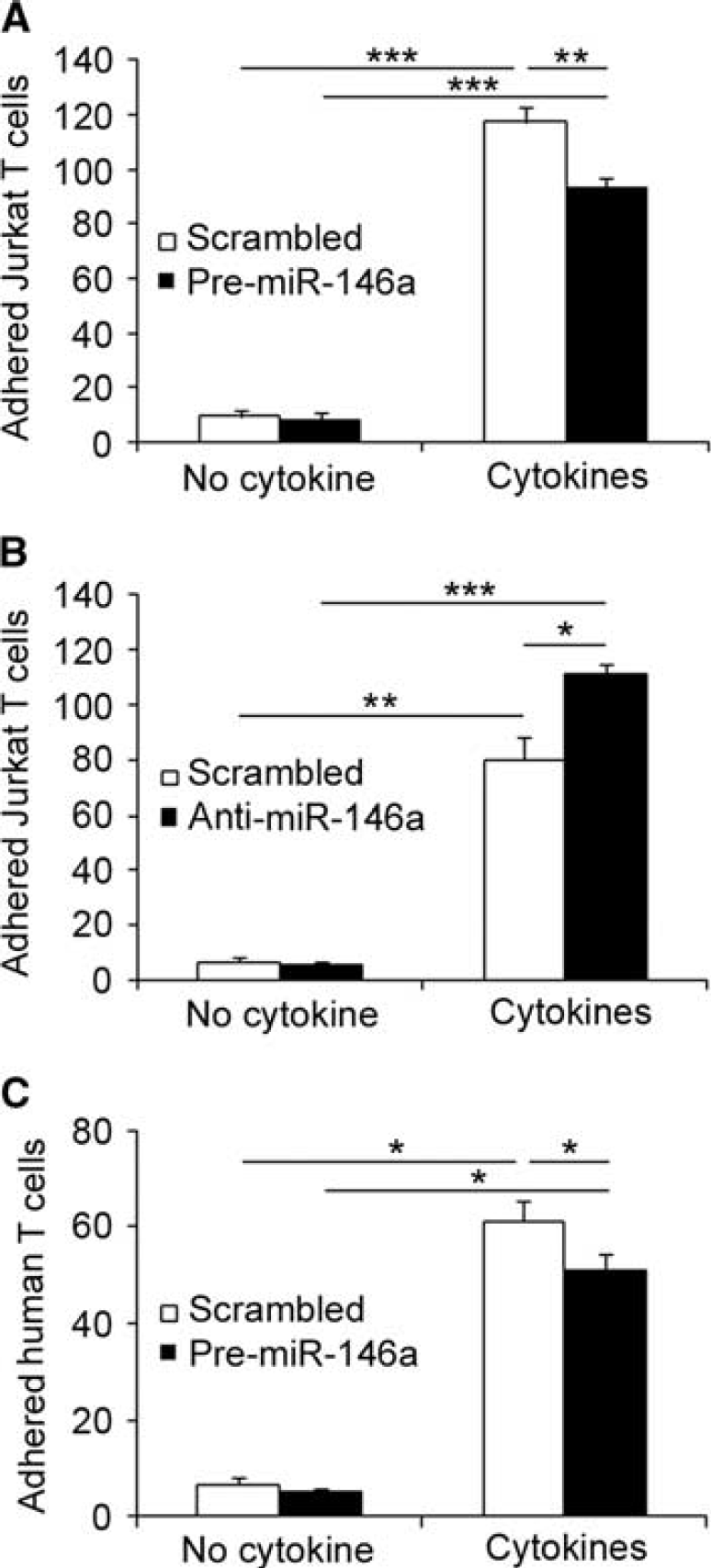
miR-146a modulates Jurkat T-cell adhesion to cytokine-stimulated hCMEC/D3 cells. hCMEC/D3 cells in six-channel slides were transfected with Pre-miR-146a or scrambled Pre-miR, and left untreated or treated with 1 ng/mL TNFα and IFNγ for 24 hours. hCMEC/D3 cells were then exposed to CMFDA-labeled Jurkat or primary human T cells for 5 minutes at 0.5 dyn/cm2. (
miR-146a modulates brain endothelial NF-κB activation
The critical role of NF-κB in leukocyte adhesion to and migration across endothelium is well established, so we next investigated whether miR-146a modulated NF-κB activity in brain endothelium. Following immunostaining of NF-κB p65, our results show that 1 ng/mL of TNFα and IFNγ induced nuclear translocation of NF-κB p65 in hCMEC/D3 cells, which peaked at 0.5 hours and was largely sustained from 6 to 24 hours (Figures 3A–3C). Overexpression of miR-146a decreased cytokine-induced NF-κB p65 nuclear translocation at all three time points, by 8%, 20%, and 10% at 0.5, 6, and 24 hours, respectively, compared with their scrambled controls (Figures 3A and 3B). By contrast, miR-146a knockdown enhanced cytokine-induced NF-κB p65 nuclear translocation by 3%, 7%, and 15% over that of scrambled control at 0.5, 6, and 24 hours, respectively (Figure 3C). In line with a previous report on the role of NF-κB on monocyte adhesion to umbilical vein endothelium, 18 we confirmed that knockdown of RelA (NF-κB p65) via small interference RNA (Figure 3D) decreased Jurkat T-cell adhesion to human cytokine-activated brain endothelium by 67% in comparison with its scrambled control (Figure 3E). These results suggest that cytokine-induced miR-146a upregulation in brain endothelium negatively regulated upstream activators of NF-κB signaling.
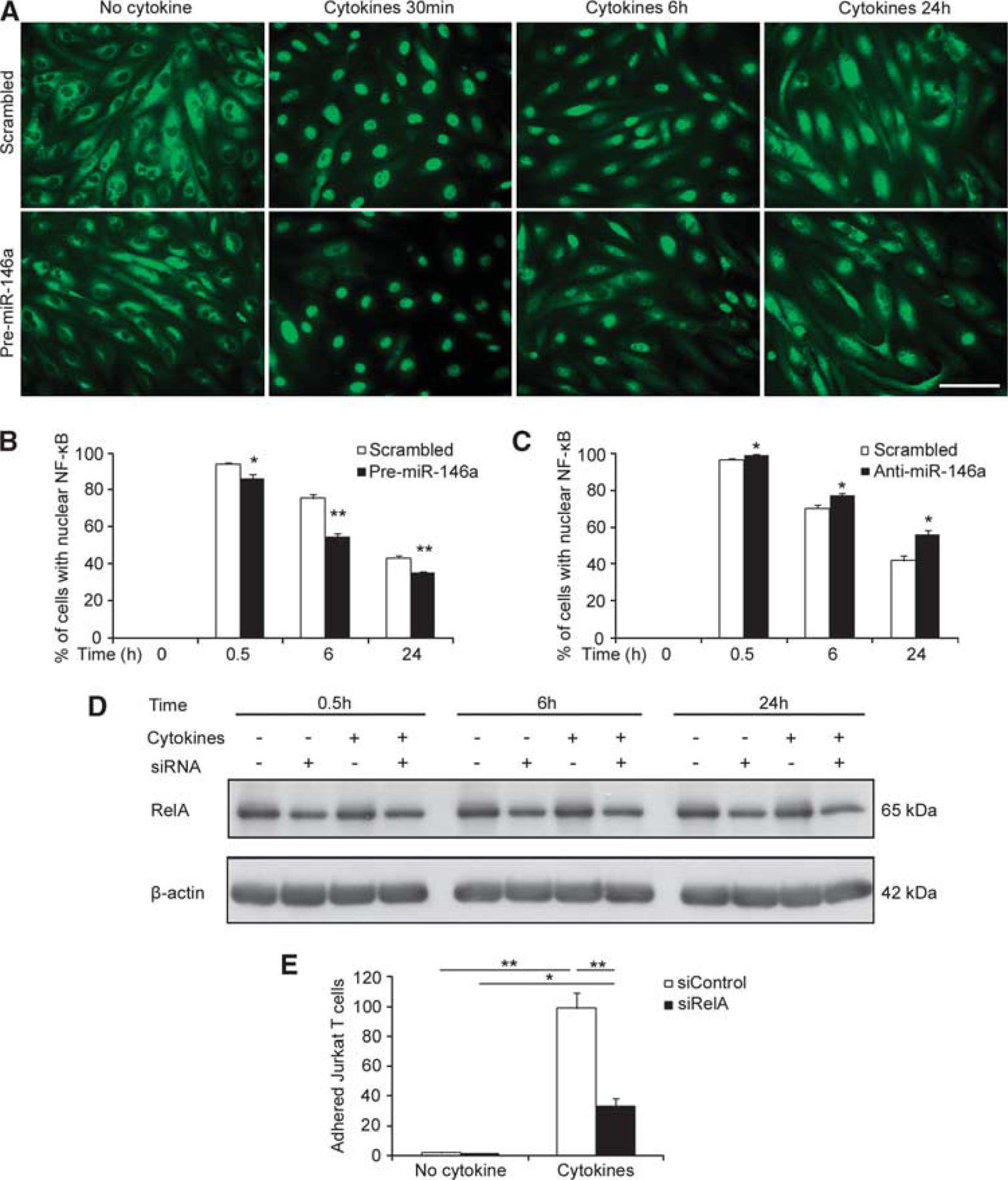
miR-146a in hCMEC/D3 cells negatively modulates nuclear NF-κB translocation. (
miR-146a targets IRAK1 and TRAF6 in brain endothelium
As microRNAs regulate cellular processes and cell signaling through silencing target molecules, using different online databases (microRNA.org, MiRanda, and TargetScan), we identified four potential target genes for miR-146a with known NF-κB regulatory activity, STAT1, ROCK1, RhoA, and NFAT5, in addition to the well-known miR-146a targets, IRAK1 and TRAF6. Indeed, these two genes have already been validated as targets of miR-146a in several cell types including monocytes, T cells and astrocytes,10, 12, 13 and are involved in the regulation of NF-κB activity and expression of NF-κB target genes such as
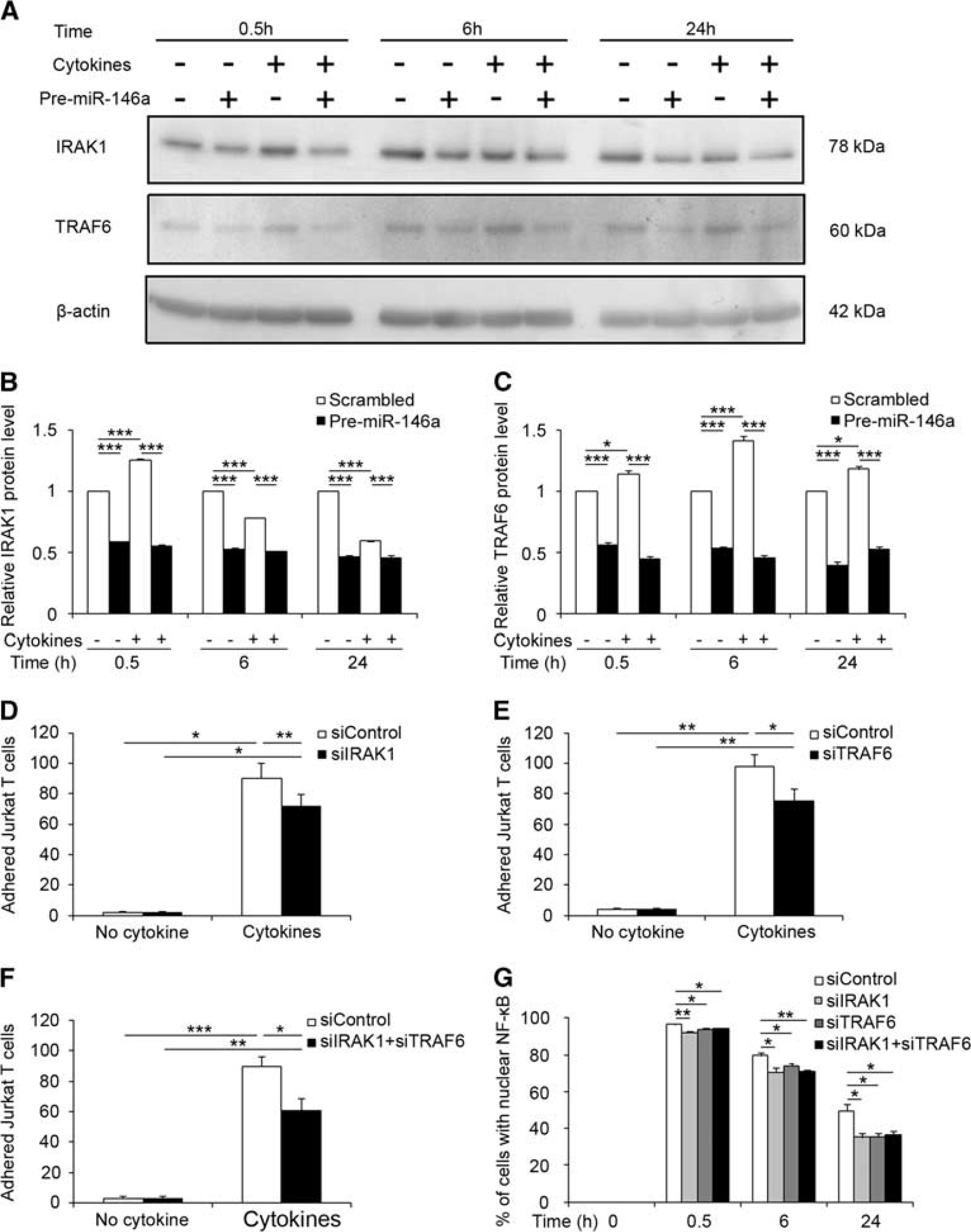
miR-146a targets IRAK1 and TRAF6 to modulate NF-κB activation and Jurkat T-cell adhesion. (
Two additional potential miR-146a targets, STAT1, and ROCK1, have been, respectively, validated in prostate carcinoma cells 19 and Treg cells. 20 However, in brain endothelium, miR-146a suppressed neither STAT1 nor ROCK1 protein expression (Supplementary Figure 2).
miR-146a targets NFAT5 and RhoA in brain endothelium
We further investigated two novel potential targets for miR-146a in brain endothelium that have not been previously validated: RhoA and NFAT5. We transfected hCMEC/D3 cells with Pre-miR-146a, then treated them with 1 ng/mL TNFα and IFNγ for 0.5, 6, and 24 hours. In controls, cytokines increased NFAT5 expression (by ∼20% over untreated control) from 0.5 to 6 hours, after which levels decreased slightly (Figures 5A and 5B). In contrast, RhoA levels decreased by 20% at 6 hours, then increased by 20% at 24 hours (Figures 5A and 5C). We show that ectopic expression of miR-146a suppressed levels of both NFAT5 and RhoA (Figures 5A–5C), in either untreated or cytokine-treated hCMEC/D3 cells at all three time points by western blot analysis. Moreover, knockdown of NFAT5 or RhoA expression using small interference RNAs (siNFAT5, siRhoA; Supplementary Figure 3C), significantly decreased leukocyte adhesion to cytokine-activated brain endothelium by 33% or 37%, respectively (Figures 5D and 5E). siNFAT5 downregulated nuclear translocation of NF-κB by 15%, 14%, and 11% at 0.5, 6, and 24 hours, respectively, compared with their scrambled controls, while siRhoA decreased nuclear translocation of NF-κB by 7% at all three time points (Figure 5F). Furthermore, in hCMEC/D3 cells transduced with lentiviral luciferase reporter vectors containing the 3’-UTR of RhoA or NFAT5, overexpression of miR-146a significantly decreased luciferase activity by 50% and 30%, respectively, in comparison with scrambled controls, but no decrease was detected in cells transduced with lentiviral luciferase reporter vector containing mutated 3’-UTR versions of either RhoA or NFAT5 (Figure 5G). These results demonstrated that miR-146a have a key role in modulating NF-κB activity by repressing multiple cytokine-effectors.
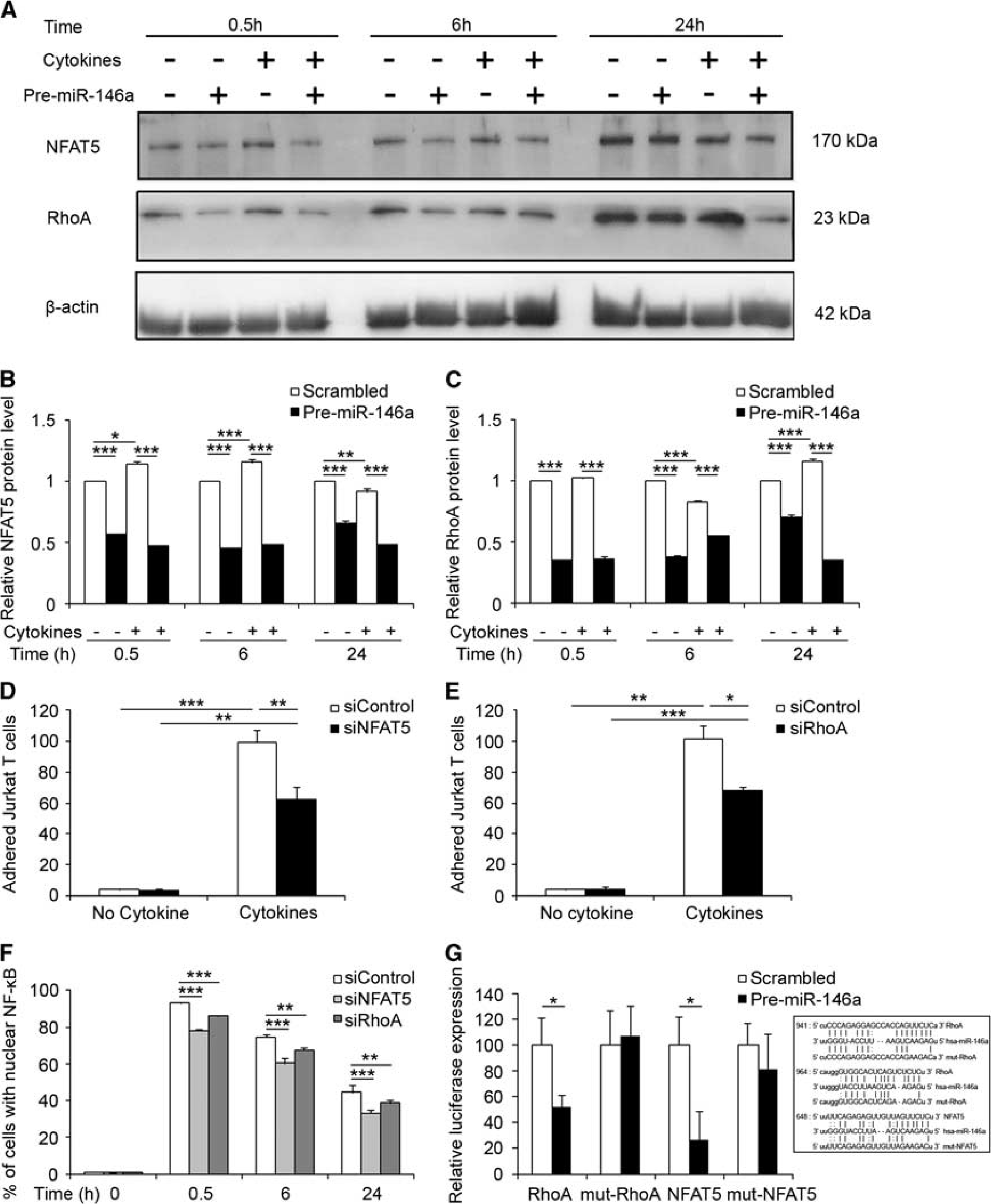
miR-146a targets NFAT5 and RhoA to modulate NF-κB activation and Jurkat T-cell adhesion. (
miR-146a inhibits expression of NF-κB target genes in brain endothelium
As miR-146a modulates NF-κB activation by repressing multiple targets, we then examined its effect on the mRNA and protein expression of two NF-κB target genes:
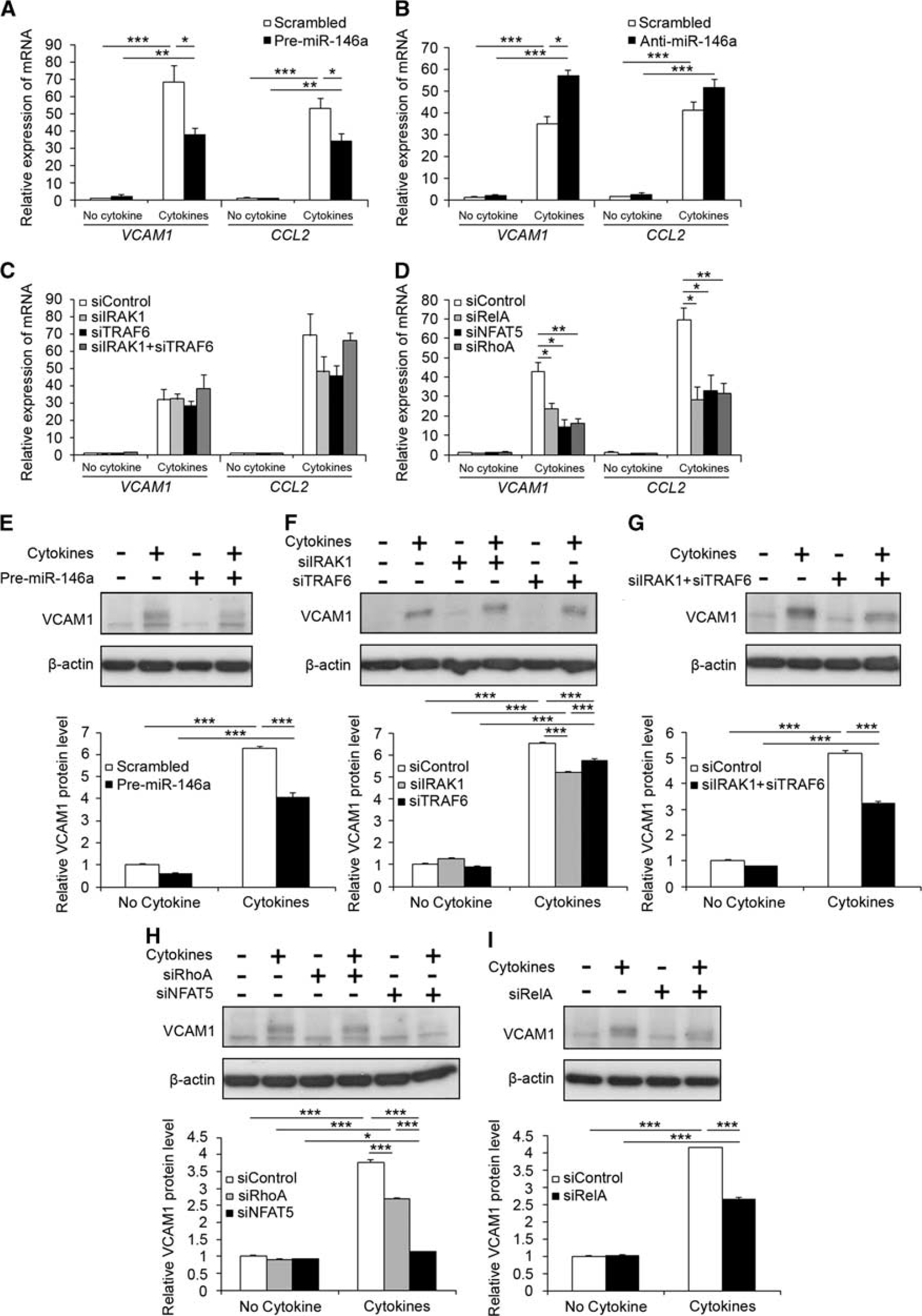
miR-146a modulated expression of NF-κB target genes. (
Taken together, these results indicate that cytokine-induced increase in brain endothelial miR-146a negatively regulates NF-κB activation via repression of multiple targets upstream of NF-κB signaling, highlighting its anti-inflammatory role in brain endothelium.
Discussion
miR-146a has been demonstrated as a NF-κB inhibitor in several cell types other than brain endothelium. In this study, we provide evidence that brain endothelial miR-146a represses multiple targets: NFAT5, RhoA, IRAK1, and TRAF6, to inhibit NF-κB activity, and subsequently negatively modulate leukocyte adhesion (Figure 7).
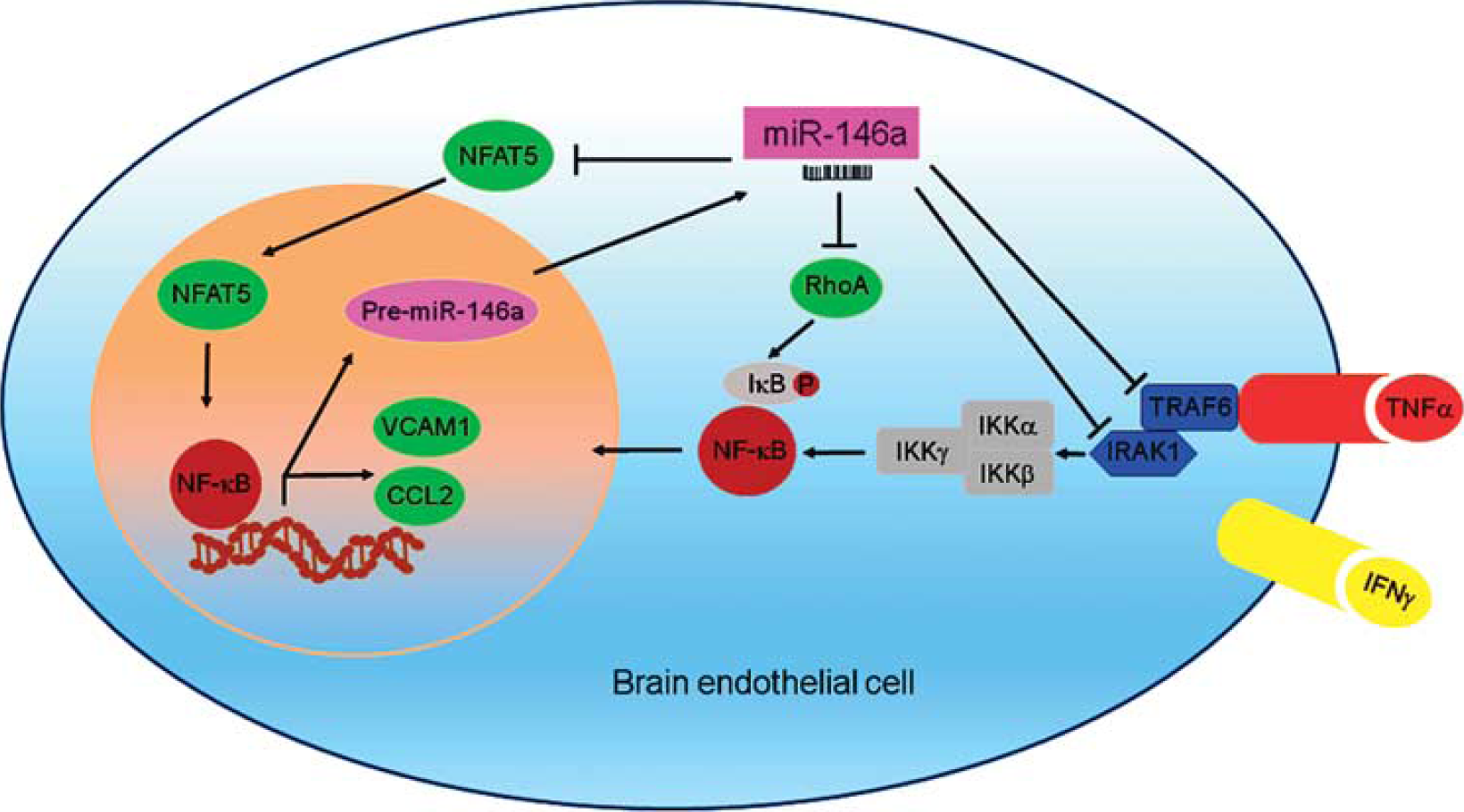
Mechanisms of miR-146a inhibiting NF-κB and T-cell adhesion. Pro-inflammatory cytokines TNFα and IFNγ act through their specific receptors on brain endothelial cells, and activate receptor-associated molecules IRAK1 and TRAF6, which then initiate IKKβ-mediated phosphorylation and ubiquitination of IκB, leading to the activation and nuclear translocation of NF-κB (p50/p65). NF-κB initiates transcription of mRNAs including those for CCL2 and VCAM1. CCL2 acts as chemoattractant for T cells, which are attracted and adhere to the brain endothelium through interacting with VCAM1. NF-κB also initiates negative feedback modulators including primary miR-146a. Primary miR-146a is processed by nuclear RNase III enzyme Drosha into Pre-miR-146. Pre-miR-146a is transported to the cytoplasm where it is further processed by cytoplasmic RNase III enzyme Dicer into mature miR-146a. Both NFAT5 and RhoA positively regulate NF-κB activities. Thus miR-146a inhibits NF-κB activation through repressing IRAK1, TRAF6, NFAT5, and RhoA, leading to decreased expression of CCL2 and VCAM1, which results in decreased T-cell adhesion to brain endothelium. IFNγ, interferon gamma
NF-κB has been shown to play a central role in several autoimmune diseases such as MS.
3
The NF-κB pathway fulfils diverse roles in development, maturation, activation and proliferation of lymphocytes, and in regulating pro-inflammatory and anti-inflammatory function of dendritic cells and macrophages.
3
Brain endothelial transcripts regulated by NF-κB may include not only miR-146a
11
but also nitric oxide synthases, pro-inflammatory cytokines, chemokines and cell adhesion molecules.
3
In cultured brain,
23
umbilical vein,
18
or retina
24
endothelial cells, inhibition of NF-κB activity reduces TNFα-induced permeability and/or leukocyte migration.
It has been demonstrated that miR-146a negatively modulates NF-κB through downregulating TRAF6 and IRAK1.10, 11, 12, 13 In BECs, we also demonstrated that miR-146a repressed translation of TRAF6 and IRAK1 in response to TNFα and IFNγ. Knockdown of TRAF6 and IRAK1 either individually or simultaneously show similar effects as miR-146a on Jurkat T-cell adhesion and nuclear translocation of NF-κB, although simultaneous knockdown of both proteins together did not result in synergistic effects. TRAF6 and IRAK1 have been mainly associated with IL-1β signal transduction leading to NF-κB activation,10, 11, 12, 13 but several reports demonstrate a role for these two signaling molecules in TNFα-induced NF-κB activation.26, 27, 28 In addition, an endogenous IL-1 inhibitor, IL1 receptor antagonist (IL-1RA), blocked IL-1β- but not TNFα/INFγ-induced NF-κB activation and Jurkat T-cell adhesion to BECs (Supplementary Figure 4), indicating that TNFα-induced IL-1 expression was not involved in the miR-146a-mediated TRAF6 and/or IRAK1 inhibition on NF-κB activity. This effect is, however, limited, suggesting the involvement of other NF-κB modulators targeted by miR-146a that are specific for TNFα and IFNγ. However, levels of another two known miR-146a targets, ROCK1 (ref. 19) and STAT1 (ref. 20), were not affected by miR-146a overexpression, suggesting tissue-specific actions of miRNAs.
We also identified two other novel miR-146a targets known to modulate NF-κB activity, NFAT5, and RhoA. NFAT5 together with NFAT1–4 belong to the Rel family, sharing 15% to 17% identity with p50 in the Rel homology region. 29 Most reports on NFAT5 refer to its transcriptional activity in the context of hypertonicity. 29 However, its role in inflammation is only beginning to emerge, with evidence that pro-inflammatory cytokines TNFα, IL-1, IL-6, IL-18, and chemokines, such as CXCL8, CCL2, and CCL5 are transcriptional targets of NFAT5 in various cell types and under various inflammatory stimuli.29, 30 Furthermore, there appears to be cross-talk between NFAT5 and NF-κB 30 or Wnt/β-catenin, 31 where the latter is important for BBB development. Accumulating evidence suggests the onset of local or systemic hyperosmolality during the course of various inflammatory disorders, such as diabetic microvascular lesions and inflammatory bowel disease. Recently, it has been reported that a high-salt diet leads to a more severe EAE clinical outcome through an effect relying on p38-MAPK and NFAT5-dependent Th17 development. 32 We provide further evidence that NFAT5 is involved in the activation of cerebral endothelium by cytokines, particularly concerning regulation of NF-κB activation and T-cell adhesion.
RhoA, Cdc42, and Rac1 are members of the Rho family of small GTPases. Endothelial Rho has been shown to have a crucial role in leukocyte migration across BECs either by modulating the cytoskeleton to embrace leukocytes by promoting docking structures 33 or by acting as an intercellular adhesion molecule-1 (ICAM1) signal transducer. 34 In other endothelium (mostly HUVECs), RhoA has been shown to modulate expression of cytokines, adhesion molecules, and chemokines via Rho/ROCK/(actin cytoskeleton)/NF-κB signaling cascade. 4 Rho family members and their exchange factors Dbl, Ost, and Vav potently activate NF-κB, though other transcription factors such as the activating protein-1 (AP-1) family member c-Jun and may also coordinately act together to modulate NF-κB activities. Our results demonstrating a link between miR-146a and RhoA are in agreement with the role of RhoA in NF-κB activation and leukocyte adhesion to endothelium.
We have also examined the levels of adhesion molecules downregulated by miR-146a (Figure 6), because of the well-documented observation that NF-κB initiates adhesion molecule expression and chemokine production in multiple cell types including brain endothelial cells. 3 It seems a reasonable assumption therefore, that cerebral endothelial miR-146a may regulate signal transducers for NF-κB activation posttranscriptionally, thereby leading to decreased expression of NF-κB transcriptional targets. Indeed, our results are in line with those obtained in HUVECs, where manipulating levels of miR-146 had a significant indirect effects on transcription of ICAM1, VCAM1, E-selectin and CCL2, whereas these effects are not only because of targeting of NF-κB activation but also other signaling molecules such as mitogen-activated protein kinase/early growth response pathway and AP-1 signaling. 15 Unexpectedly, we also observed that siIRAK1 and/or siTRAF6 decreased VCAM1 protein levels without affecting its corresponding mRNA levels. It is possible that the role of these two signaling proteins in the inflammatory activation of brain endothelial cells induced by cytokines is a complex process and that, in addition to their role in NF-κB-mediated transcriptional activation, IRAK1, and TRAF6 may regulate inflammatory molecules such as VCAM1 at the posttranslational level. As an example, IL-1 activates a translational control mechanism that modulates expression of a group of genes, 35 including IL-6, via IRAK1. Alternatively, combination of pro-inflammatory cytokines increases the half-life of VCAM1 mRNA, 36 and TRAF6 and IRAK1 actively regulate the ubiquitin-proteasome system. 37 It is thus conceivable that VCAM1 protein turnover is affected by ablation of IRAK1 and/or TRAF6 in the absence of apparent changes of VCAM1 mRNA levels by a mechanism that remains to be determined.
In summary, we have delineated further the underlying mechanisms through which miR-146a affects T-cell adhesion to human brain endothelium by identifying two novel targets NFAT5 and RhoA, and we have demonstrated the role of IRAK1 and TRAF6, two signaling molecules usually associated with IL-1R and Toll-like receptor responses, in brain endothelial activation by TNFα and IFNγ, we have characterized the temporal and spatial expression of miR-146a in CNS endothelium in MS-active lesions, EAE model, and cultured brain endothelium. As the complex signaling networks appear to control inflammation progression,28, 38 it is unlikely that rational therapeutic strategies should focus on modulating the activity of one signaling molecule. Indeed, the emerging role of miRNAs in directly regulating complex signaling networks involved in inflammation, such as cell adhesion molecules39, 40 by targeting the expression of several genes makes them suitable therapeutic targets for neuroinflammatory disorders. The limited increase of miR-146a in MS brain endothelium further stresses the necessity to increase miR-146a through therapeutic manipulation to counteract inflammatory responses, thereby potentially preventing the progression of MS pathogenesis.
Footnotes
Acknowledgements
The authors are grateful to Julia Barkans for general laboratory infrastructure assistance, and Radka Gromnicova for preparation of cerebral endothelium culture for some of our experiments.
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.