
Abstract
The potential role of the chemokine Fractalkine (CX3CL1) in the pathophysiology of traumatic brain injury (TBI) was investigated in patients with head trauma and in mice after experimental cortical contusion. In control individuals, soluble (s)Fractalkine was present at low concentrations in cerebrospinal fluid (CSF) (12.6 to 57.3 pg/mL) but at much higher levels in serum (21,288 to 74,548 pg/mL). Elevation of sFractalkine in CSF of TBI patients was observed during the whole study period (means: 29.92 to 535.33 pg/mL), whereas serum levels remained within normal ranges (means: 3,100 to 59,159 pg/mL). Based on these differences, a possible passage of sFractalkine from blood to CSF was supported by the strong correlation between blood–brain barrier dysfunction (according to the CSF-/serum-albumin quotient) and sFractalkine concentrations in CSF (R = 0.706; P < 0.01). In the brain of mice subjected to closed head injury, neither Fractalkine protein nor mRNA were found to be augmented; however, Fractalkine receptor (CX3CR1) mRNA steadily increased peaking at 1 week postinjury (P < 0.05, one-way analysis of variance). This possibly implies the receptor to be the key factor determining the action of constitutively expressed Fractalkine. Altogether, these data suggest that the Fractalkine-CX3CR1 protein system may be involved in the inflammatory response to TBI, particularly for the accumulation of leukocytes in the injured parenchyma.
Traumatic brain injury (TBI) triggers a cascade of immunological events within the intrathecal compartment, which may either induce beneficial effects such as promoting neuroreparative mechanisms or be detrimental by contributing to the onset of secondary brain damage (Morganti-Kossmann et al., 2001). Cytokines, chemokines, and adhesion molecules orchestrate the complex sequence of cell activation, migration, adhesion, and extravasation into the injured brain tissue. In particular, after focal cortical contusion, the accumulation of blood-derived leukocytes such as neutrophils and macrophages has been demonstrated, whereby the increased permeability of the blood–brain barrier (BBB) plays a determinant role (Stahel et al., 2000b).
Among the large group of chemokines characterized to date (Anthony et al., 2001; Zlotnik and Yoshie, 2000), Fractalkine (CX3CL1) is the only member of the CX3C-chemokine subfamily and has the ability to attract monocytes, T cells, and neutrophils (Bazan et al., 1997; Pan et al., 1997). The uniqueness of this molecule is conferred by its structure, which contains a chemokine domain, a functional adhesion molecule domain (mucine stalk), and a transmembrane region. Fractalkine is membrane-bound and can be shed as a soluble chemotactic form (sFractalkine) by proteolytic cleavage (Tsou et al., 2001). The tissue expression of Fractalkine and its receptor (CX3CR1) is unique because both molecules are also expressed in tissue of nonhemopoietic origin. They are most abundant in the human heart and brain, where they are constitutively expressed (Bazan et al., 1997; Pan et al., 1997). In the central nervous system (CNS), Fractalkine is primarily localized on neurons and endothelial cells, whereas CX3CR1 is expressed by microglia, astrocytes, and neurons (Beck et al., 1999; Harrison et al., 1998; Meucci et al., 2000; Nishiyori et al., 1998; Pan et al., 1997). The role of Fractalkine in the physiology and pathophysiology of the CNS is largely unexplored. However, because of its characteristic structure, multiple properties, and cellular expression pattern, it can be hypothesized that Fractalkine may be involved in the intercellular communication among neurons, microglia and endothelial cells, as well as in extravasation of leukocytes after TBI and other CNS injuries. To date, there are few studies showing the involvement of Fractalkine and its receptor in inflammatory diseases of the CNS (Hughes et al., 2002; Kastenbauer et al., 2003); however, the potential role of Fractalkine in TBI is still unexplored.
The past research of our laboratory has focused on the inflammatory response after TBI. The production of cytokines, chemokines, adhesion molecules, and complement proteins has been investigated in patients with severe TBI (Kossmann et al., 1997; Pleines et al., 1998; Stahel et al., 2001) as well as in animal models of brain injury and cultured cells (Hans et al., 1999; Otto et al., 2000; Otto et al., 2001; Rancan et al., 2001; Rancan et al., 2003; Stahel et al., 2000a). In order to determine whether immune mediators are produced intrathecally or in the periphery, the function of the BBB was assessed in TBI patients in parallel to the measurement of immune parameters in both cerebrospinal fluid (CSF) and serum (Kossmann et al., 1997; Morganti-Kossmann et al., 1999; Pleines et al., 1998).
The objective of the present work was to determine whether Fractalkine and its receptor CX3CR1 might play a role in the inflammatory response after traumatic injury to the brain. In the clinical section of the study, we analyzed the concentrations of sFractalkine in ventricular CSF and serum as well as the BBB dysfunction over 14 days after injury. To extend the results gained through the analysis of human samples, the possible involvement of Fractalkine and its receptor in the cerebral accumulation of leukocytes was investigated in a standardized mouse model of closed head injury (CHI) that produces a cortical contusion with massive accumulation of neutrophils and macrophages (Stahel et al., 2000b).
MATERIALS AND METHODS
Patients
The patients included in this study (n = 12; mean age 36 years; range 17 to 57 years; 2 females, 10 males) were admitted to the University Hospital Zurich, Department of Surgery, Division of Trauma Surgery with a Glasgow Coma Score ≤8 and morphological abnormalities in the primary computed tomography of the brain (Marshall et al., 1991) (Table 1). All patients received intraventricular catheters for continuous intracranial pressure monitoring. Intracranial hematomas were surgically evacuated if necessary. After surgery, the patients were transferred to the intensive care unit and treated according to a standardized protocol (Stocker et al., 1995). No patient was treated with steroids. CSF was drained via intraventricular catheters when the intracranial pressure exceeded 15 mm Hg. On a routine basis, CSF samples were screened every 3 days for bacterial infections and no ventriculostomy-related infections were observed. Catheters were removed when the intracranial pressure remained below 15 mm Hg for 24 hours. Clinical recovery of TBI patients was evaluated using the Glasgow Outcome Score (Jennett et al., 1976). Patients with significant systemic injuries, pelvic or long bone fractures, and spinal cord injuries were excluded from the study. The study did not interfere with clinical management of head trauma patients and received approval by the University Hospital Ethics Board, Zurich, Switzerland.
Clinical and experimental data of the 12 patients with severe traumatic brain injury included in the study
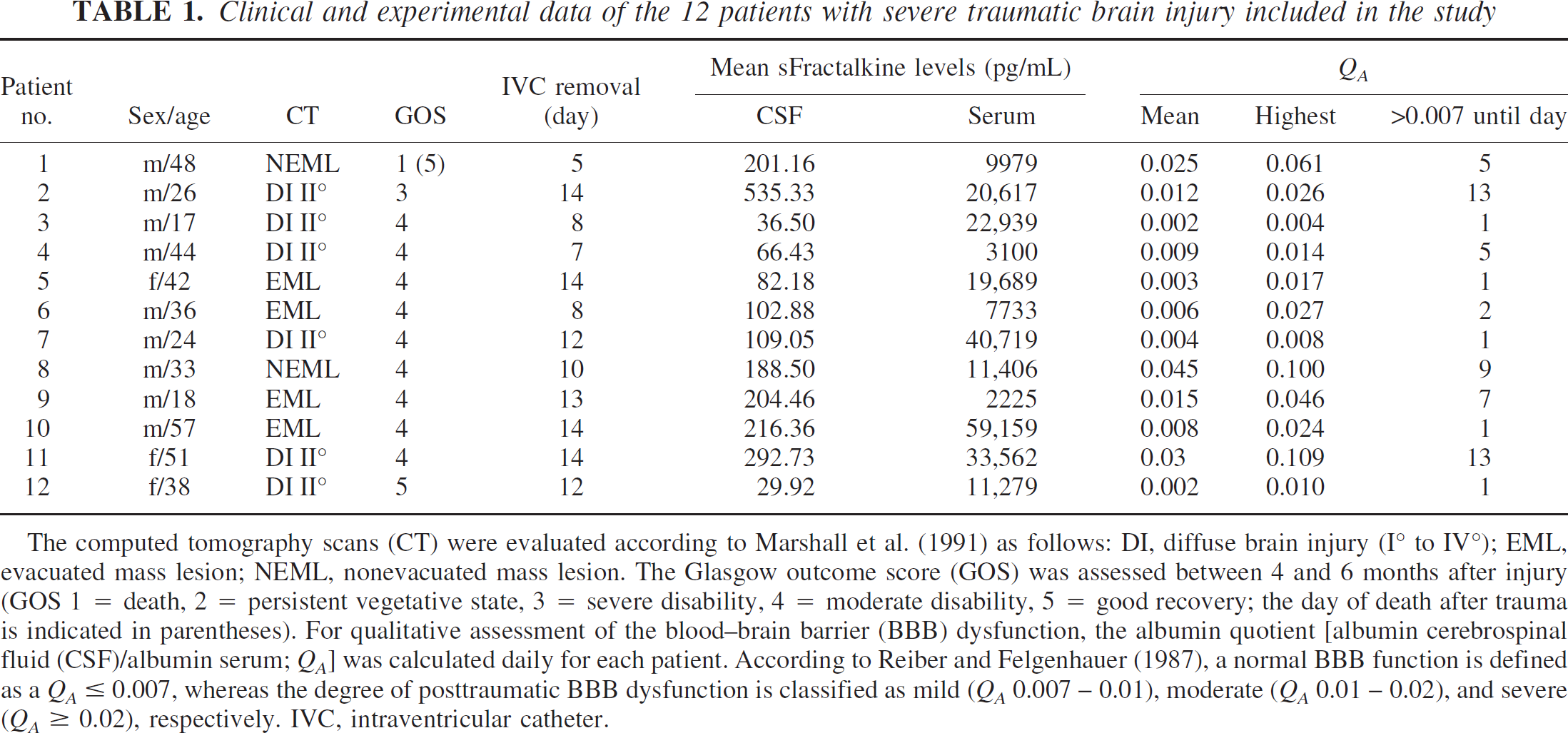
The computed tomography scans (CT) were evaluated according to Marshall et al. (1991) as follows: DI, diffuse brain injury (I° to IV°); EML, evacuated mass lesion; NEML, nonevacuated mass lesion. The Glasgow outcome score (GOS) was assessed between 4 and 6 months after injury (GOS 1 = death, 2 = persistent vegetative state, 3 = severe disability, 4 = moderate disability, 5 = good recovery; the day of death after trauma is indicated in parentheses). For qualitative assessment of the blood–brain barrier (BBB) dysfunction, the albumin quotient [albumin cerebrospinal fluid (CSF)/albumin serum; QA] was calculated daily for each patient. According to Reiber and Felgenhauer (1987), a normal BBB function is defined as a QA ≤ 0.007, whereas the degree of posttraumatic BBB dysfunction is classified as mild (QA 0.007–0.01), moderate (QA 0.01–0.02), and severe (QA ≥ 0.02), respectively. IVC, intraventricular catheter.
Serum and cerebrospinal fluid sampling
Cerebrospinal fluid (total n = 134) and serum (total n = 180) samples were collected daily. After sampling, drained CSF was immediately centrifuged at 1,000 rpm for 10 minutes at 4°C, aliquoted, and frozen at −70°C until analysis. Collection was performed for up to 14 days after trauma or until removal of the intraventricular catheters. Paired blood samples were taken and processed at the same time points as CSF samples. Cerebrospinal fluid from patients (n = 5) without neuropathology or trauma utilized as controls had been kindly provided by Prof. Hess, Clinic of Neurology, University Hospital Zurich. Control serum was taken from eight healthy volunteers.
Detection of sFractalkine in human cerebrospinal fluid and serum samples
Soluble Fractalkine was measured by ELISA using capture and detection goat anti-Fractalkine antibodies and human recombinant Fractalkine as a standard (all reagents from R&D Systems Europe, Abingdon, UK). The assay was established and adapted for CSF and serum measurements in our laboratory. Briefly, ELISA plates were coated overnight at 4°C with 2 μg/mL goat polyclonal anti-human-Fractalkine antibodies in a carbonate buffer (NaHCO3/Na2CO3 5 Mmol/L, pH 9.6). The next day the plates were washed with phosphate buffered saline pH 7.4 containing 0.05% Tween-20, and blocked with phosphate buffered saline containing 1% bovine serum albumin and 5% sucrose for 2 hours at room temperature (RT). Before the assay, the CSF samples were concentrated by centrifugation using Centricon tubes and the concentration factor was assessed by weighing the samples before and after concentrating (Amicon Inc., Beverly, MA). Before application to the ELISA plate, CSF samples and the native serum samples were diluted 3-fold and 20-fold, respectively, with TrisHCl pH 7.3 containing 0.1% bovine serum albumin and 0.1% Tween-20. CSF samples were incubated overnight at 4°C, whereas serum samples were incubated for 2 hours at RT. After washing, a secondary biotinylated goat anti-human-Fractalkine antibodies was added at 150 ng/mL in Tris containing 0.1% bovine serum albumin and 0.1% Tween-20 at pH 7.3 and incubated for 2 hours at RT. After repetitive washes, a solution of streptavidin conjugated with horseradish peroxidase (R&D Systems) diluted 1:200 was added in phosphate buffered saline with 1% bovine serum albumin and incubated for 20 minutes at RT. After three washes, the substrate solution (R&D Systems) was added. After incubation for 30 minutes at RT, the reaction was stopped with 1 mol/L H2SO4 and the absorbance was measured at a wavelength of 450 nm with 550 nm as the reference wavelength (microplate reader from Dynatech Laboratories Inc., Chantilly, VA). A standard curve was established using various dilutions of recombinant human Fractalkine ranging from 33 to 8,000 pg/mL.
Assessment of the blood–brain barrier function
To assess the BBB function or dysfunction, the CSF-/serum-albumin quotient (QA) was calculated daily in the TBI patients. QA values below 0.007 are regarded as normal. A QA between 0.007 and 0.01 reflects a mild, between 0.01 and 0.02 a moderate, and above 0.02 a severe BBB dysfunction (Reiber and Felgenhauer, 1987). Albumin levels were measured by automatized laser photometry (BNA Automat, Behring Werke, Marburg a.L., Germany).
Mouse model of closed head injury
A total of 40 mice of the C57BL/6 strain were used in this study. They were 8- to 16-week-old males weighing 28 to 32 g. The animals were bred in a specific pathogen-free environment, kept under standardized conditions of temperature and light, and fed with food and water ad libitum. The animal model received ethic approval by the “Kantonales Veterinaeramt Zurich”, Bew. Nr. 143/2000. Experimental CHI was performed as described previously by our group (Stahel et al., 2000b). Briefly, the mice were ether anesthetized and their skull was exposed by a longitudinal incision of the skin. Focal trauma was induced to the left hemisphere lateral to the midline in the midcoronal plane, using a weight-drop device with a silicon tip of 3-mm diameter fixed at the end of the impacting rod of 333 g falling from a height of 2 cm, resulting in a focal injury to the left cortex. After trauma, the mice received supporting oxygenation with 100% O2 until fully awake, the wound was sutured, and the animals were brought back to their cages with food and water ad libitum. Sham-operated animals underwent anesthesia and scalp incision but were not subjected to head trauma. The mice were killed at various time-points after trauma or after sham surgery. In all animals, the brains were immediately removed after decapitation, snap-frozen in liquid nitrogen, and stored at −70°C until analysis.
Evaluation of neurological impairment
For assessment of posttraumatic neurological impairment and recovery, the group of mice killed at 1 week (n = 4) was used. A 10-point Neurological Severity Score (NSS) was used as described elsewhere (Stahel et al., 2000b). The score consists of 10 individual clinical parameters. A maximal NSS of 10 points indicates severe neurological dysfunction, with failure of all tasks, whereas a score of zero is achieved by healthy uninjured mice. In the present study, the NSS was assessed independently by two investigators who were blinded to the study groups at 1, 24, 48, and 72 hours and 1 week after trauma. The ΔNSS, calculated as the difference between the NSS at 1 hour and the NSS at any later time-point, reflects the degree of spontaneous recovery after TBI, as described earlier (Stahel et al., 2000b).
Detection of Fractalkine in mouse brain homogenates
For the quantification of Fractalkine in mouse brain tissue, 15 animals were subjected to experimental CHI and killed at 4, 24, 48, and 72 hours and 1 week postinjury (n = 3 per time-point). Sham-operated animals (n = 3) and normal mice (n = 3) were used as controls. Mouse brains were homogenized in ice-cold extraction buffer containing TrisHCl 50 nmol/L pH 7.2, NaCl 150 nmol/L, Triton-X 1%, and 1 tablet of protease inhibitor cocktail per 10 mL solution (Boehringer Mannheim) at a ratio of 1:4 (tissue:buffer). Samples were homogenized with a Polytron homogenizer (Kinematica, Kriens, Switzerland) shaken for 90 minutes on ice, and centrifuged at 3,000g for 15 minutes at 4°C, aliquoted, and frozen until analysis. The concentrations of total protein were measured in all brain extracts by Bradford assay (Bio Rad Laboratories, Munich, Germany). Quantification of total (soluble and membrane-bound) mouse Fractalkine by ELISA was performed using the same protocol described for human samples, with slight modifications. Homogenized tissue was diluted 1:10 with diluting buffer. For coating, anti-mouse-Fractalkine antibodies (1 μg/mL) and for detection, biotinylated anti-mouse-Fractalkine antibodies (250 ng/mL) were used. A standard curve based on serial dilutions of recombinant mouse-Fractalkine (15 to 8,000 pg/mL; R&D Systems) was utilized for estimation of Fractalkine concentrations in brain homogenates.
Determination of Fractalkine and CX3CR1 mRNA levels in mouse brains by Northern blot hybridization
For quantification of total Fractalkine and CX3CR1 mRNA expression after CHI, total cellular RNA was isolated from brain hemispheres collected from 19 C57BL/6 mice killed at 24 hours after sham operation (n = 3) or 4, 24, 48, 72 hours and 1 week postinjury (n = 3 per time-point, except n = 4 at 1 week). Ten micrograms of each RNA sample was electrophoresed on a denaturing 1.2% agarose gel containing 2.2 mol/L formaldehyde and transferred to a nylon membrane (Duralon UV; Stratagene, La Jolla, CA) in 5x SSC (1x SSC = 150 mmol/L sodium chloride, 15 mmol/L sodium citrate). [32P-UTP]-labeled cRNA probes were transcribed from linearized pBSII KS+ or pGEM Teasy vectors containing 1.2 or 1.0 kb cDNA fragments complementary to Fractalkine and CX3CR1 mRNAs, respectively. After prehybridization, blots were hybridized overnight at 77°C with labeled probe (3 × 106 cpm/mL) in 5x SSC, 0.5% nonfat dry milk, 1% sodium dodecyl sulfate, 10% dextran sulphate, 25 μg/mL polyadenylic acid, 25 μg/mL polycytidylic acid, and 100 μg/mL salmon sperm DNA. Blots were washed at 75°C to a final stringency of 0.5x SSC, and were exposed to a phosphorimager screen for 24 to 72 hours at RT. The intensity of the radioactive signal representing the relative levels of Fractalkine and CX3CR1 mRNAs was quantified by densitometry using ImageQuant analysis software (Molecular Dynamics, Sunnyvale, CA). To standardize for the amount of total RNA loaded onto each lane, blots were rehybridized with a [32P-UTP]-labeled cRNA probe against mouse GAPDH, transcribed from a 1.1-kb fragment subcloned into pBluescript. (Stratagene, La Jolla, CA, U.S.A.) For each sample, density values for Fractalkine and CX3CR1 were expressed as a percentage of the respective GAPDH value.
Data analysis
Statistical analysis was performed on commercially available software (SigmaStat, Hearne Scientific Software Pty Ltd, Melbourne, Australia). Soluble Fractalkine levels in human CSF and serum as well as NSS of mice were analyzed using a one-way (time postinjury) repeated-measures analysis of variance. Protein levels of total Fractalkine as well as total Fractalkine and CX3CR1 mRNA levels in mouse brains were analyzed using a one-way (time postinjury) analysis of variance. Student-Newman-Keuls post hoc tests were applied for multiple comparisons. Correlation was assessed with Spearman rank order correlation. All data are presented as mean values with the corresponding standard deviation. A P value less than 0.05 was considered statistically significant.
RESULTS
Soluble Fractalkine levels are increased in CSF but not in serum of traumatic brain injury patients
The concentration of sFractalkine was measured daily in the CSF and serum of 12 patients with severe TBI over a period of 14 days or less, depending on the removal of the intraventricular catheters based on the therapeutic regimen. Table 1 summarizes the clinical, demographic, and biochemical data of the patients included in this study. Based on the Marshall classification (Marshall et al., 1991) of the computed tomography scans, six patients had diffuse brain injury grade II (DI II°) and six had intracerebral hemorrhage that were in part surgically evacuated (evacuated mass lesion; n = 4) or not evacuated (nonevacuated mass lesion; n = 2). With the exception of one, all patients recovered neurologically to various extents after TBI, having Glasgow outcome scores between 3 and 5 within the first 6 months after trauma.
The upper limit of normal sFractalkine concentrations in CSF (95.7 pg/mL) was calculated as the mean + 3 SD (42 + 53.7 pg/mL; dashed line in Fig. 1A) from measurements in healthy control individuals (range 12.6 to 57.3 pg/mL; n = 5). In TBI patients, sFractalkine was already increased on the day of admission. Maximal levels were observed at day 1 after injury, decreasing gradually up to day 11, but remaining over the normal range throughout the entire study period. Statistical analysis revealed a significant overall difference (P = 0.004, one-way repeated measures analysis of variance) as well as significant differences at days 1 to 4 postinjury between TBI patients and controls. No significant difference could be detected at the day of admission; however, only three patients could be included in the study at this early time-point. The means of sFractalkine in CSF calculated for the single patients over the whole time analyzed ranged between 29.92 and 535.33 pg/mL (Table 1).
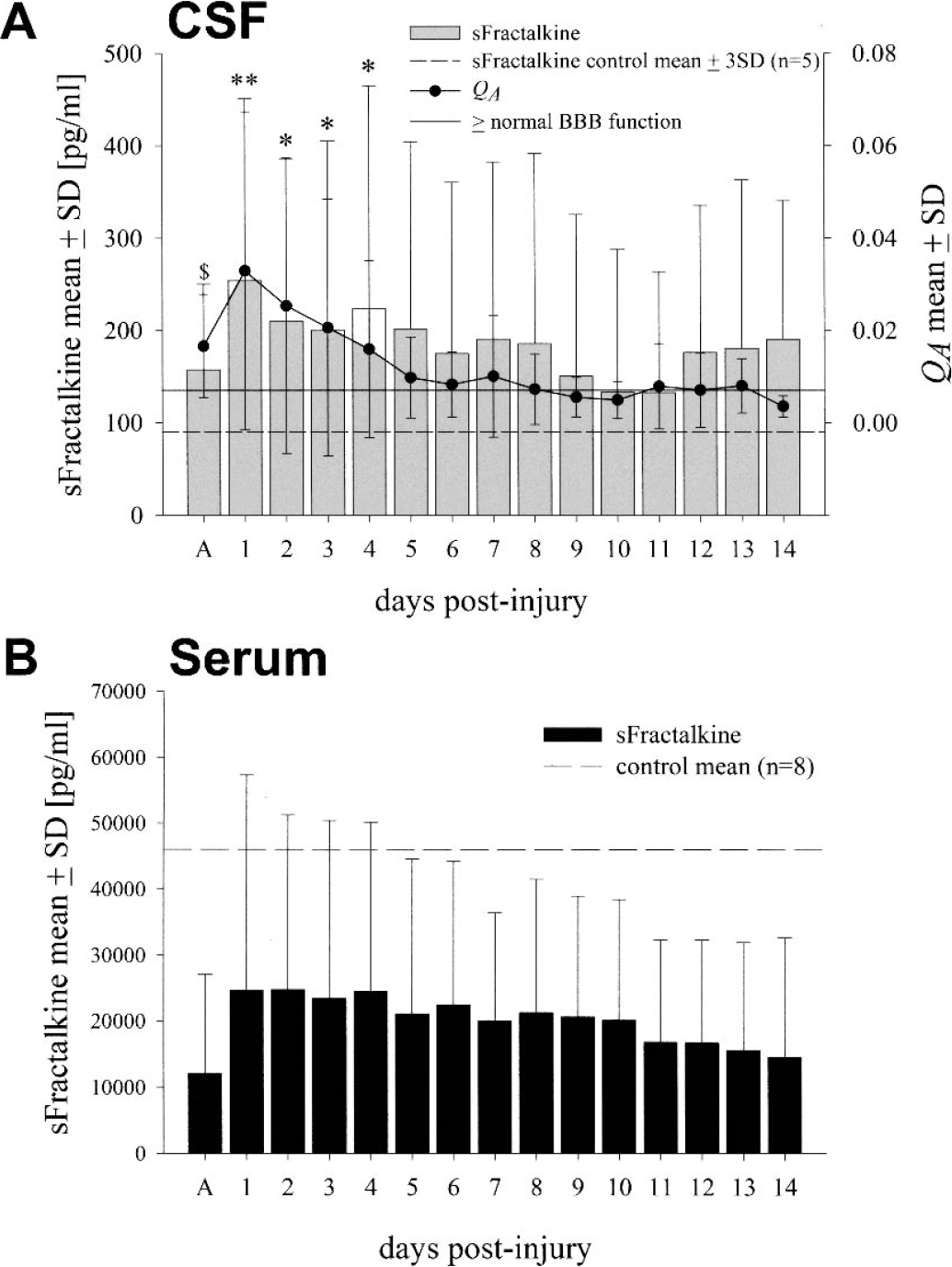
Soluble Fractalkine measurements in cerebrospinal fluid (CSF)
Interestingly, measurements of serum sFractalkine in healthy control individuals revealed very high concentrations (45,968 ± 17,097 pg/mL, range 21,288 to 74,548 pg/mL; n = 8) with cut-off level at 97,259 pg/mL (mean + 3 SD), which were not exceeded in any of the serum samples of the TBI patients (Fig. 1B). The means of sFractalkine measurements in serum of TBI patients ranged between 3,100 and 59,159 pg/mL (Table 1).
Soluble Fractalkine in cerebrospinal fluid correlates with blood–brain barrier dysfunction and neurological outcome
The evidence of greater levels of sFractalkine in serum as compared to CSF suggested that the increased permeability of the BBB caused by head trauma might allow the passage of this protein from serum into the CSF. To test this hypothesis, the function of the BBB was assessed according to the QA as previously used in neurological disease as well as TBI (Pleines et al., 1998). Table 1 gives an overview on the maximal QA values for each patient as well as the duration in days of BBB dysfunction (QA > 0.007) during the study period. Interestingly, the early time profile of the QA closely paralleled the increase of sFractalkine in CSF (Fig. 1A). A moderate BBB dysfunction appeared clearly at the day of admission, having a mean QA > 0.01, whereas the most severe disturbance (QA > 0.02) was present at day 1 after injury and coincided with maximal sFractalkine mean concentrations. Thereafter, the BBB dysfunction persisted up to day 7 (QA > 0.007) and returned to normal values (QA < 0.007) from day 8 onwards. The highest CSF sFractalkine concentrations were found in the patients with long-lasting BBB dysfunction (patients no. 2 and no. 11; Table 1). Statistical analysis using the mean values of sFractalkine in CSF and mean QA for each patient over the entire study period indicated a strong correlation of these two parameters (R = 0.706; P < 0.01).
With regard to the clinical outcome after severe TBI, besides patient No.1, who died 5 days postinjury, the poorest outcome (Glasgow Outcome Score = 3) was associated with the highest CSF sFractalkine concentrations (patient no. 2; Table 1), whereas patient no. 12, having the lowest sFractalkine concentrations in CSF, showed good recovery (Glasgow Outcome Score = 5; Table 1). This is corroborated by statistical analysis, revealing an inverse relationship of sFractalkine means in CSF with the neurological outcome (Glasgow Outcome Score) (R = −0.587; P < 0.05), suggesting that in this group of head trauma patients, increased levels of intracranial Fractalkine correspond to a poor recovery.
No differences were noticed in terms of sFractalkine concentrations in CSF when comparing patients with focal injuries (EMN, NEML) versus patients with diffuse brain injury (data not shown).
Total Fractalkine is not increased in brain tissue of mice subjected to closed head injury
To extend the results gained through the analysis of human samples, the possible involvement of Fractalkine and its receptor in the cerebral accumulation of leukocytes was investigated in a standardized mouse model of CHI that produces a cortical contusion with massive accumulation of neutrophils and macrophages (Stahel et al., 2000b). After CHI, the NSS was assessed at different time points after injury. At 1 hour, the mice showed the strongest deficit and thereafter, gradually recovered up to 1 week. Neurological improvement is best shown as ΔNSS calculated as the difference between the NSS at 1 hour and the NSS at later time-points (Fig. 2).
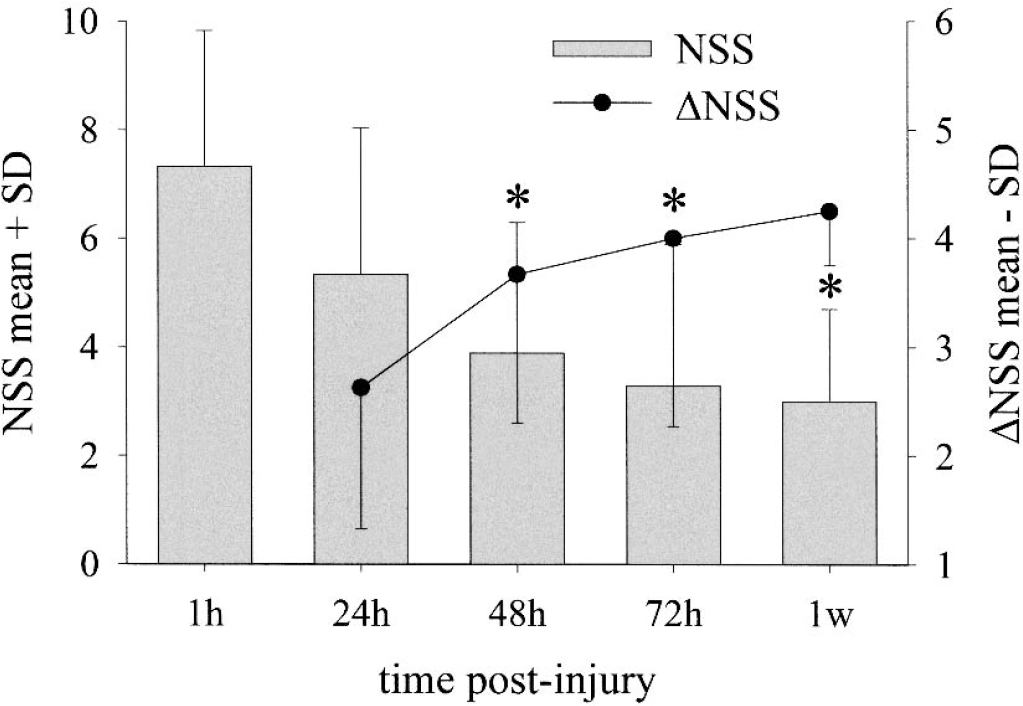
Neurological deficit and recovery in mice after closed head injury. Experimental closed head injury was produced in mice (n = 4) and 10 well-defined tests were applied to monitor the neurological performance of the animals. NSS (Neurological Severity Score) = 0 indicates normal behavior and NSS = 10 indicates worst deficit. Bars represent mean NSS scores and the curve represents the neurological recovery ΔNSS calculated as the difference between the NSS at 1 hour and the NSS at later time-points. (*P < 0.05 NSS vs. NSS at 1 hour, one-way repeated measures analysis of variance with Student-Newman-Keuls post hoc tests for multiple comparisons).
Total Fractalkine was measured by ELISA in the brain homogenates of traumatized mice at 1, 24, 48, 72 hours, and 1 week postinjury. The brains of sham-operated animals that underwent identical treatment except for the trauma and the brains of untreated normal mice were used as controls. Interestingly, high levels of total Fractalkine were detected in brain homogenates of normal mice (2,717 ± 250 pg/mg protein, range 2,348 to 3,124 pg/mg protein). At any time point assessed, there was no difference between brains of traumatized mice (2,027 ± 673 pg/mg protein, range 1,009 to 2,988 pg/mg protein) and normal control brains or sham brains (2,092 ± 526 pg/mg protein, range 1,201 to 2,630 pg/mg protein; P > 0.05, data not shown).
Expression of total Fractalkine and Fractalkine receptor mRNAs is differentially regulated after closed head injury in mice
Expression of total Fractalkine and CX3CR1 mRNAs was analyzed by Northern blot hybridization of brain homogenates from sham operated and traumatized mice up to 1 week postinjury. Changes in mRNA levels were expressed using arbitrary density units calculated as a percentage of constitutive GAPDH values. In brains of mice subjected to experimental CHI, total Fractalkine mRNA levels ranged between 10.8% and 18.1% and in sham-operated control brains between 13.0% and 14.2% (Figure 3A). Whereas a statistically significant (P < 0.05) increase in total Fractalkine mRNA expression between the 24-hour and 72-hour time-point was found in traumatized mice, no difference in total Fractalkine mRNA expression was found between traumatized and sham-operated controls at any time point assessed and therefore, corroborating the above findings of a lack of increase in total Fractalkine protein levels as determined by ELISA.
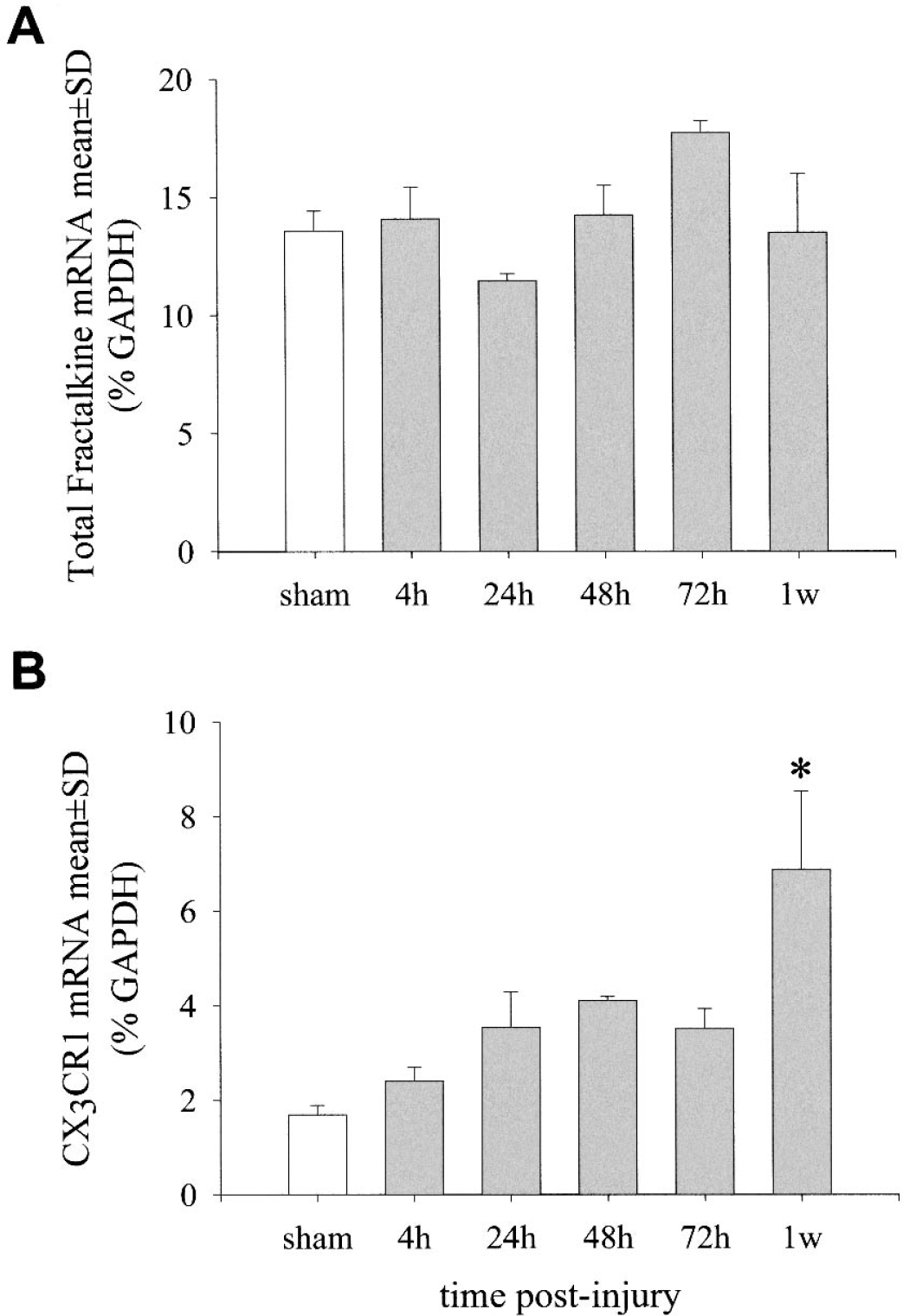
Expression of total Fractalkine and CX3CR1 mRNAs after closed head injury in mice. Levels of total (soluble and membrane-bound) Fractalkine
In contrast, the levels of CX3CR1 mRNA expression in sham-operated animals were found to be relatively low (range 1.5% to 1.8%; Fig. 3B). Interestingly, CX3CR1 mRNA levels in traumatized animals (range 2.1% to 8.5%) increased steadily over time and were found to be elevated fourfold at 1 week after trauma (6.9 ± 1.7) compared with sham-operated controls (1.7 ± 0.2; Fig. 3B).
DISCUSSION
In the present study, the potential role of the chemokine Fractalkine has been assessed choosing a dual approach based on a clinical investigation on patients with TBI and on a standardized mouse model of CHI.
The most important findings are as follows: (1) control values assessed in healthy humans indicated that sFractalkine was detectable at low concentrations in the CSF and at much higher concentrations in serum; (2) in patients with severe TBI, increased concentrations of sFractalkine were found in the CSF but not in serum; (3) changes in the profile of sFractalkine in CSF closely paralleled BBB dysfunction and higher concentrations corresponded to maximal QA values, suggesting the passage of sFractalkine from the periphery into the brain; (4) after experimental CHI in mice, no changes in total Fractalkine mRNA and protein levels were found as compared to sham-operated controls, whereas mRNA expression of the Fractalkine receptor CX3CR1 increased continuously over time and was found to be significantly elevated at 1 week after trauma.
In healthy humans, Fractalkine mRNA levels were reported to be highest in the heart and the brain, whereas no Fractalkine mRNA was found in peripheral blood (Bazan et al., 1997). This is unlike most chemokines, which are abundantly expressed by leukocytes (Bazan et al., 1997). Of the different cell types in the brain, Fractalkine has been primarily localized on neurons and endothelial cells, whereas CX3CR1 is expressed by microglia, astrocytes, and neurons (Beck et al., 1999; Harrison et al., 1998; Meucci et al., 2000; Nishiyori et al., 1998; Pan et al., 1997). Expression of Fractalkine on endothelial cells has been observed in organs such as the lung (Balabanian et al., 2002) and heart (Harrison et al., 1999). Both in coronary and larger cerebral blood vessels, Fractalkine staining was reported to be restricted to the luminal side of the endothelium, suggesting a pivotal role in mediating cell adhesion and extravasation of leukocytes (Harrison et al., 1999; Pan et al., 1997). Release of sFractalkine from the membrane-bound molecule can occur by the action of different proteases, such as tumor necrosis factor-α–converting enzyme (Tsou et al., 2001). Thus, the high concentrations measured in the peripheral circulation may be caused by the proteolytic cleavage of Fractalkine from endothelial cells.
In our study, control sFractalkine serum levels in humans ranged between 21,288 and 74,548 pg/mL. Apparently, the abundance of Fractalkine in the brain tissue reported in previous studies is not reflected by its CSF levels, because control sFractalkine CSF concentrations were very low, ranging between 12.6 and 57.3 pg/mL. These findings corroborate the results of a recent work on other neuropathologies showing CSF sFractalkine levels being approximately 500-fold below serum levels in normal subjects (Kastenbauer et al., 2003). However, it cannot be excluded that traumatic injury increases the cleavage rate of membrane-bound Fractalkine into the soluble molecule within the brain tissue, contributing to the elevation of CSF concentrations.
In TBI patients, we observed an early and significant increase of secreted sFractalkine in the CSF as compared to control liquor, but no increase in serum levels (Fig. 1). Elevated CSF levels were strongly associated with BBB dysfunction (R = 0.706; P < 0.01). Besides a possible passage from the periphery, an increased expression and/or cleavage of Fractalkine from activated cerebral endothelial cells can be postulated, as shown in experimental studies on experimental autoimmune encephalomyelitis (Fischer et al., 2000; Pan et al., 1997) as well as in vitro in response to challenge with inflammatory mediators known to be increased during inflammatory CNS diseases, such as interleukin-1, tumor necrosis factor-α, or interferon-γ (Fraticelli et al., 2001). Previously, we found comparable results in the production of the anti-inflammatory cytokine transforming growth factor-beta (TGF-β) in patients with severe TBI (Morganti-Kossmann et al., 1999). Similarly to Fractalkine, TGF-β was present in high levels in the serum of both normal and TBI individuals, but it was almost undetectable in control CSF. Although increased levels of TGF-β in CSF of TBI patients were associated with BBB dysfunction suggesting a passage from the periphery, a partial intrathecal production was assumed, possibly modulated by elevation of interleukin-6.
The elevation of sFractalkine in human CSF seems to contradict the lack of increase in mouse brain homogenates after experimental CHI, in which total Fractalkine concentrations did not differ between trauma and sham mice at any time-point assessed. However, the data are based on two distinct types of measurements: on the one hand the soluble protein released into human CSF, and on the other hand the complete membrane-bound and soluble Fractalkine in mouse brain homogenates. The latter may not reveal any difference in concentration measured after CHI, which may possibly reflect the increase of soluble Fractalkine rather than the degree of total Fractalkine synthesis. However, crucial in determining the function of Fractalkine may be the variations in the expression of CX3CR1 rather than Fractalkine itself, since in our study, a significant increase of CX3CR1 was found in mouse brains after experimental CHI (Fig. 3). This would be supported by a study of Meucci et al. (2000), who showed in vitro that neuronal CX3CR1 expression mediates the neurotrophic effects of Fractalkine, because Fractalkine protected hippocampal neurons from the neurotoxicity induced by the human immunodeficiency virus-1 envelope protein gp120IIIB, an effect blocked by anti-CX3CR1 antibodies.
The pathophysiological significance of elevated Fractalkine or CX3CR1, or both, within the brain remains controversial. One of the first described functions of Fractalkine is its chemotactive activity towards leukocytes (Bazan et al., 1997). However, the responses of Fractalkine-deficient mice to a variety of inflammatory stimuli were indistinguishable from those of wild-type mice (Cook et al., 2001). Similar findings were obtained for mice deficient for the only murine Fractalkine receptor CX3CR1 (Jung et al., 2000), where absence of CX3CR1 did not interfere with monocyte extravasation in a peritonitis model. In the same study, a prominent response of CX3CR1-deficient microglia to peripheral nerve injury indicated unimpaired neuronal-glial crosstalk in CX3CR1 knockout mice.
Recent evidence against Fractalkine as a chemoattractant in the CNS came from an experimental study that reported only modest microglial activation, but no leukocyte infiltration after intracerebral injection of mouse or rat Fractalkine into both species (Hughes et al., 2002). On the contrary, in a mouse model of human atherosclerosis, the Fractalkine-CX3CR1 pair seemed to play a direct and critical role in monocyte recruitment and atherosclerotic lesion development, because CX3CR1/apolipoprotein(apo)E double knockout mice showed a 59% reduction of lipid-stained lesion in the thoracic aorta compared with their CX3CR1(+/+)/apoE(–/–) littermates, and the development of atherosclerosis in the aortic sinus was markedly altered in the double knockout mice with a 50% reduction in macrophage accumulation (Combadiere et al., 2003). Also, after transient focal cerebral ischemia, Fractalkine-deficient mice had a 28% reduction in infarction size and lower mortality rate, when compared to wild-type littermates (Soriano et al., 2002). Zujovic et al. (2001) showed that intracerebral neutralization of endogenous Fractalkine with specific antibodies potentiates the effects of intracerebroventricular lipopolysaccharide injection. In a stroke model in the rat, Fractalkine was increased at 12 hours on necrotic neurons of the cortex and was expressed at 24 and 48 hours in morphologically intact cortical neurons of the ischemic penumbra. Furthermore, CX3CR1 expression was detected in the activated microglial cells of the ischemic tissue 24 and 48 hours after ischemia and became strongly upregulated in macrophages/phagocytic microglia inside the infarcted tissue 7 days after ischemia (Tarozzo et al., 2002). The latter data support our findings of significantly increased CX3CR1 mRNA expression after experimental CHI in mice 1 week postinjury (Fig. 3) and suggest that Fractalkine and its receptor participate in the activation and chemotaxis of microglia into the damaged tissue, contributing to the control of leukocyte trafficking from blood vessels into the damaged brain parenchyma. However, it cannot be ruled out that increased CX3CR1 levels after TBI may be a consequence of massive infiltration of macrophages and neutrophils occurring in the early days postinjury, which are known to express the receptor and consequently leading to an enrichment of its mRNA in the brain.
Taken together, our results show that sFractalkine was found to be increased in the CSF of patients with severe TBI over the whole study period, and correlation with the BBB dysfunction suggests a passage from the bloodstream. Interestingly, no elevation of total Fractalkine was found in mouse brain homogenates after experimental CHI, but a significant increase in CX3CR1 mRNA was demonstrated, implicating that the expression of CX3CR1 rather than Fractalkine itself might be crucial for determining the function of Fractalkine after TBI. There is experimental evidence in vitro for neuroprotective activities of Fractalkine mediated through neuronal expression of CX3CR1 (Meucci et al., 2000). However, the exact role played by Fractalkine and CX3CR1 after TBI requires further experimental work.
Footnotes
Abbreviations used
Acknowledgments:
We gratefully acknowledge Emerita Ammann for excellent technical assistance in the establishment of the Fractalkine ELISA.