
Abstract
Interleukin-1β (IL-1β) induces cyclooxygenase-2 (Cox-2) expression in many of its cellular targets resulting in production and release of prostaglandins. Although IL-1β-induced Cox-2 expression most likely requires activation of nuclear transcription factor kappa B (NFκB) pathway, this has never been formally demonstrated in vivo. We tested this using a specific inhibitor of NFκB activation, the NEMO binding domain (NBD) peptide, that has been shown previously to be effective in various in vivo models of acute inflammation. Incubation of rat glioma cells with the NBD peptide blocked IL-1β-induced NFκB nuclear translocation. Furthermore, after injection of a biotinylated version of the NBD peptide into the lateral ventricle of the brain, we found that it readily diffused to its potential cellular targets in vivo. To test the effects of the peptide on NFκB activation and Cox-2 expression in the brain, we injected it intracerebroventricularly (36 μg/rat) into rats before intraperitoneal injection of IL-1β (60 μg/kg). Treatment with NBD peptide completely abolished IL-1β-induced NFκB activation and Cox-2 synthesis in microvasculature. In contrast, the peptide had no effect on constitutive neuronal Cox-2. These findings strongly support the hypothesis that IL-1β-induced NFκB activation plays a major role in transmission of immune signals from the periphery to the brain.
Introduction
Prostaglandins (PGs) largely account for interleukin-1β (IL-1β)-mediated responses to immune challenge like fever, alteration in cerebral blood flow and activation of the hypothalamic-pituitary-adrenal axis because these responses are alleviated by administration of nonsteroidal antiinflammatory drugs (O'Banion, 1999). Prostaglandins synthesis occurs through activation of cyclooxygenase 1 (Cox-1) and 2 (Cox-2). Cyclooxygenase-1 appears to be maximally expressed under basal conditions whereas inflammatory agents as IL-1 usually induce Cox-2. Our previous in vivo findings indicate that when released into the bloodstream, binding of IL-1β to its type 1 receptor (IL-1RI) activates the nuclear transcription factor kappa B (NFκB) after a well-described kinetic (Nadjar et al, 2003). Nuclear transcription factor kappa B is then susceptible to bind the promoter of various genes including Cox-2 (Laflamme et al, 1999). However, this scenario is largely based on indirect neuroanatomical and biochemical data and therefore remains correlative. Cox-2 expression in the rat brain has been widely studied. Pyrogenic doses of IL-1β or lipopolysaccharide (LPS) injected intraperitoneally or intravenously induce Cox-2 mRNA synthesis in leptomininges and cells of the brain vasculature within 1 h after treatment (Cao et al, 1995, 1996; Lacroix and Rivest, 1998; Laflamme et al, 1999). Anatomical localization of this effect is compatible with expression of type I IL-1 receptor (IL-1RI), inducible IκBα mRNA synthesis and NFκB activation in most of the rat brain vasculature 15 mins after intraperitoneal injection of IL-1β (Nadjar et al, 2003; Konsman et al, 2004).
The evidence is much clearer in in vitro conditions because putative NFκB motifs are found on Cox-2 promoter (Sorli et al, 1998; Nakao et al, 2000). However, the direct link between NFκB activation and Cox-2 synthesis has not yet been evidenced in vivo during the acute phase reaction. Therefore, we studied, in vivo, consequences of the inhibition of NFκB activation on Cox-2 synthesis by targeting NFκB in the brain.
The general scheme of NFκB pathway activation is well described: binding of IL-1β to IL-1RI is followed by a cascade of phosphorylation that results in NFκB nuclear translocation. This depends on the regulatory protein NEMO (NFκB essential modifier) associated with a complex containing two kinases, IKKα and IKKβ (Akira and Takeda, 2004; Hayden and Ghosh, 2004). Once within the nucleus, NFκB binds to its consensus sequence on target genes that promotes transcription of a variety of genes like IL-1β, IκB, other proinflammatory cytokines, Cox-2 and various chemokines (Miyamoto and Verma, 1995). To inhibit NFκB pathway, we used a short cell-permeable peptide, NEMO-binding domain peptide (NBD), spanning IKKβ. This peptide was already shown to disrupt the association of NEMO with IKKβ in vitro, to block TNFα-induced NFκB activation, and to effectively ameliorate responses to various inflammatory stimuli in vivo (May et al, 2000).
In the present experiment, we made sure that the NBD peptide was able to act on brain cells and then injected into the brain, to diffuse to its potential sites of action. We then showed that intracerebroventricular NBD peptide significantly attenuated IL-1β-induced translocation of NFκB and expression of Cox-2 in the brain vasculature. These findings show unequivocally that NFκB activation is a crucial step in the induction of Cox-2 in the brain in response to peripheral IL-1β.
MATERIALS AND METHODS
Cell Culture
C6 rat glioma were grown in Dulbecco's modified Eagle's medium (DMEM, Invitrogen SARL, Cergy-Pontoise, France) supplemented with 10% fetal calf serum, 2 mmol/L L-glutamine and 50 μg/mL gentamycine, at 37°C with 5% CO2. For immunocytochemistry, cells were pretreated with NBD peptide (30 μmol/L) for 2 h and then stimulated with recombinant rat IL-1β (rrIL-1β) (10 ng/mL) for 15 mins.
Immunocytochemistry
After treatment, cells were fixed with paraformaldehyde (PF) 4% and permeabilized in PBS/Triton 0.1% pH 7.4 for 2 mins. The p65-NFκB subunit was stained with a polyclonal goat primary antibody (1:500; Santa Cruz Biotech. Inc., CA, USA; SC 372-G, lot G016). This antibody recognizes both the inactive form of p65 subunit bound to p50 and IκB in the cytoplasm and the active monomeric form in the nucleus. After an incubation of 24 h at 4°C with the primary antibody, cells were incubated for 1 h with a FITC-coupled secondary anti-goat antibody (1:800) in PBS/BSA 2% at room temperature. Nuclear transcription factor kappa B staining was observed with a fluorescence microscope (Nikon Eclipse E 400).
Animals
The experiments were performed on adult rats male Wistar (Charles River, l'Arbresle, France) weighing 200 to 250 g. They were kept in individual polypropylene cages in temperature-controlled (23°C±1°C) and light-dark cycle-controlled animal room (lights off from 8.30 am to 8.30 pm) with access to food (Extralabo, Provins, France) and water ad libitum. First rats were used after an adaptative period of 2 weeks to our laboratory conditions, then to habituate rats to the handling procedure to be used during the experiments, rats were handled daily for 5 days before to initiation of treatments.
Surgical Procedures
Surgery and treatments were carried in accordance with the Animal Protection Association ethical standards for the humane treatment of animals defined by the guidelines for experimentation that were approved by the Institutional Animal Care and Use Comittee. For central injection, a stainless-steel guide cannula (23-gauge, 7 mm length) was implanted unilaterally 1 mm above the lateral ventricle. Rats were anesthetized with a mixture of ketamine and xylasine and secured in a Kopf stereotaxic instrument. Coordinates for the guide cannula were 0.6 mm posterior to bregma, 1.5 mm lateral, and 3.1 mm below the skull surface at the point of entry (Swanson, 1998). A 10-day recovery period was allowed before testing. Substances were administered intracerebroventricularly by gravity using a 30-gauge needle inserted into the guide cannula and protruding 1 mm below the tip of the guide. At the end of the experiment, cannula placement was verified by injecting a solution of black ink into the guide cannula and then slicing the brain after removal from the skull.
Treatments
Recombinant rat IL-1β (biological activity: 317 IU/mg, NIBSC, Potters Bar, UK) was dissolved in saline 0.9%. whereas NBD peptide (given by Dr MJ May, University of Pennsylvania, Philadelphia, USA; May et al, 2000) was dissolved in undiluted dimethylsulfoxide (DMSO) (Sigma-Aldrich Corporation, St Louis, MI, USA). The dose of rrIL-1β (60 μg/kg, intraperitoneally) was selected on the basis of previous experiments as appropriate to induce the whole spectrum of clinical signs of sickness (Anforth et al, 1998). The dose of NBD peptide (36 μg, intracerebroventricular) was selected on the basis of previous in vivo data (May et al, 2000) and of pilot experiments using different doses of rrIL-1β and inhibitor. NBD peptide (36 μg) was injected intracerebroventricularly as a pretreatment 1 h before intraperitoneal injection of rrIL-1β (60 μg/kg) for all the in vivo experiments of this study. Animals were killed 1 h after the peripheral injection of the cytokine. All substances were administered in a volume of 600 μL (intraperitoneally) or 2 μL (intracerebroventricularly) to vigil animals.
Tissue Preparation
For immunohistochemical study, animals were killed with a lethal dose of pentobarbital at specified time shown in the figure legends after intraperitoneal and intracerebroventricular injections. Brains were fixed by intracardiac perfusion of 4% PF in 0.1 mol/L NaH2PO4/Na2HPO4 buffer, pH 7.5 (phosphate buffer, PB). Brains were removed and postfixed in the same fixative solution overnight at 4°C and cryoprotected in 30% sucrose in 0.1 mol/L PB. Coronal 30 μm cryostat sections were kept in antifreeze solution (30% ethylene glycol, 30% glycerol, 25% Tris Buffer (TB) sterile, and 15% sterile water) at –20°C until processing for immunohistochemistry.
Immunodetection of NFκB
The polyclonal goat primary antibody generated against NFκB p65 subunit (Santa Cruz Biotech. Inc., CA, USA; SC 372-G, lot G016) was used at a final dilution of 1:500 (Nadjar et al, 2003). Immunohistochemical processing was performed on free-floating sections using the streptavidin-biotin-immunoperoxidase technique. After washing off the cryoprotectant solution, sections were incubated 2 mins in PBS containing 0.3% Triton X-100. Endogenous peroxidase activity was quenched by treating the sections in 3% H2O2 to PBS for 30 mins. After 30 mins in blocking buffer (PBS containing 0.2% gelatin and 5% nonfat milk), the first antibody was added for 48 h at 4°C (1:500) in PBS and 0.25% BSA. After three washes in PBS, sections were incubated for 2 h at room temperature with biotinylated donkey anti-goat IgG (Amersham Biosciences, Buckinghamshire, UK for rats; Biosys, France for mice) at 1:800 in PBS and 3% BSA. After rinses, avidin-biotin-peroxidase complex (Vectastain ABC kit, Vector laboratories, Burlingame, CA, USA) was added for 2 h at room temperature. The peroxidase reaction product was developed using diaminobenzidine and the nickel-enhanced glucose oxidase method (Shu et al, 1988). Sections were then mounted onto gelatin-coated glass, dried, dehydrated through alcohol to xylene and coverslipped for light microscopic analysis.
To ensure uniformity of NFκB immunostaining conditions for quantitative analysis of immunointensity, all sections were processed at one time.
Immunodetection of Cox-2
Free-floating sections were incubated for 1 h in TBS 1 × containing 0.4% casein, 0.4% Triton X-100, and 0.5% hydrogen peroxide. Sections were then incubated at 20°C overnight in a goat polyclonal anti-Cox-2 (sc-1747; Santa Cruz Biotechnology; 1:1,000) in TBS 1 × with 0.4% casein, 0.4% Triton X-100. Tissue sections were rinsed in TBS 1 × and incubated with biotinylated donkey anti-goat antibodies IgG (Amersham Biosciences, Buckinghamshire, UK; 1:2,000) for 2 h. After 2 h of incubation in ABC kit, the peroxidase reaction product was developed as above. We obtained a dark and concentrated labeling in the perinuclear space of the cells for Cox-2 immunoreactivity that allowed the measure of intensity of staining by Scion image software (see below the Microscopy section).
To ensure uniformity of Cox-2 immunostaining conditions for quantitative analysis of immunointensity, all sections were processed at one time.
Colocalization of Cox-2 and NFκB with the Nucleic Acid Marker DAPI
Sections were first incubated with Cox-2 or NFκB antibody and immunohistochemistry was performed as described above but revealed in absence of Ni ions, thus giving a brown precipitate. DAPI (4,6-diamidino-2-phenylindole, dihydrochloride; Molecular probes, Interchim, Eugene, OR, USA), diluted at 1.5 nmol/L, was applied for 1 min to obtain nuclear counterstain.
Colocalization of Cox-2 with PECAM-1, a Marker of Endothelial Cells
Double labeling immunohistochemistry was performed by incubating sections for 24 h with primary anti-Cox-2 antibody. The tissue was then treated as aforementioned and the reaction was revealed in absence of Ni ions, thus giving a brown precipitate. After several rinses, the same sections were incubated for 1 h in TBS 1 × containing 0.4% casein, 0.4% Triton X-100, and 0.5% hydrogen peroxide. Sections were then incubated at 4°C overnight in a goat polyclonal anti-PECAM-1 (sc-1505; Santa Cruz Biotechnology; 1:100) in TBS 1 × with 0.4% casein, 0.4% Triton X-100. Tissue sections were rinsed in TBS 1 × and incubated with biotinylated donkey anti-goat IgG (Amersham Biotechnology; 1:2,000) for 2 h. PECAM-1 antibody was then revealed with an Alexa 488-conjugated anti-goat antibody for 2 h at room temperature (1:1,000) (Molecular Probes) (for technical procedure, see Gautron et al, 2003).
In all, 50% glycerol was added to the slides that were coverslipped for light and fluorescent microscopic analysis.
Despite the generation of PECAM-1 and Cox-2 antibodies in goat, colocalization is still possible because the markers are expressed in very distinct compartments of the cell, that is, nuclear or perinuclear for Cox-2 and at the intercellular junctions of the endothelial cell for PECAM-1.
Colocalization of Cox-2 with a Neuronal Marker, NeuN
Sections were incubated simultaneously with anti-Cox-2 antibody (dilution 1:200) and a mouse polyclonal anti-NeuN antibody (dilution 1:200) (Chemicon International, Euromedex, Souffelweyersheim, France) overnight at room temperature. After rinses, sections were incubated with an anti-mouse secondary antibody Alexa 594-conjugated (red staining) (Molecular Probes), diluted at 1:500 for 2 h at room temperature, to reveal NeuN. Tissue sections were rinsed and incubated with biotinylated donkey anti-goat antibodies IgG as above (1:500 for 2 h), followed by an incubation in presence of streptavidin Alexa 488-conjugated (green staining) (1:500; Molecular Probes) for 2 h at room temperature, to reveal Cox-2.
Biotin-Tagged NBD Peptide Localization
To determine NBD peptide diffusion into the brain parenchyma and the brain microvasculature, a biotin-tagged NBD peptide (36 μg) was injected by intracerebroventricular route. Rats were killed 2 h after treatment and brains were prepared as previously mentioned for histochemistry. The biotin-tagged NBD peptide was revealed by incubating the sections in avidin-biotin-peroxidase complex (ABC kit) for 2 h and then revealed with the nickel-glucose-oxidase diaminobenzidine method. Small black precipitates, interpreted as fixation of NBD peptide on NEMO protein in the cytoplasm of the cells were visible. Brain sections were counterstained with Cresyl violet to confirm subcellular localization of the biotin-tagged NBD peptide.
Microscopy
Brain sections were examined under a light and fluorescent microscope (Nikon Eclipse E 400) and the images were captured by a high-resolution video camera image (Digital camera Nikon DXM 1200). Camera aperture, magnification, light power, and exposure time were fixed for all images. The ACT-1 software (Nikon Corporation, Champigny-sur-Marne, France) generated images fed into a personal computer. Image editing software (Adobe PhotoShop, Adobe Systems, San Jose, CA, USA) was used to adjust size and mode for counting and brightness, focus, and contrast for photographs.
Image analysis was performed using a PC version of NIH-imaging software (Scion, Frederick, MD, USA). Gray images of some representative sections were compared with light microscopic views of these same sections before quantification to establish which gray levels and stained surfaces corresponded to immunolabeled cellular nuclei. Once established, these parameters remained unchanged throughout the analysis of a particular brain structure. The image was then converted to a binary image and the intensity of staining measured. Labeling was quantified bilaterally in at least five sections per animal for a particular brain structure.
Statistical Analysis
Data from immunohistochemistry experiments were expressed as means±s.d. and analyzed by a two-way analysis of variance (two factors: pretreatment and treatment). Post hoc comparisons of distinct groups means were performed using the Newman-Keuls test. In all cases, a level of P<0.05 was considered as statistically significant.
RESULTS
NBD Peptide Inhibits p65-NFκB Translocation in Rat Glial Cells
Because the NBD peptide has never been used in neurobiological experiments, we first checked that it is active in brain cells. C6 rat glioma cells were used for this purpose because astrocytes are target cells of IL-1β (Pousset et al, 2001; Gautron et al, 2003; Nadjar et al, 2003). No p65-NFκB nuclear translocation was observed in DMSO/saline or NBD peptide/saline-treated cells (Figures 1Aa and 1Ab, respectively). Addition of rrIL-1β for 15 mins in the culture medium induced a strong translocation of p65-NFκB into the nucleus of the cells (Figure 1Ac), which was strikingly prevented by a 2 h pretreatment with the NBD peptide (Figure 1Ad).
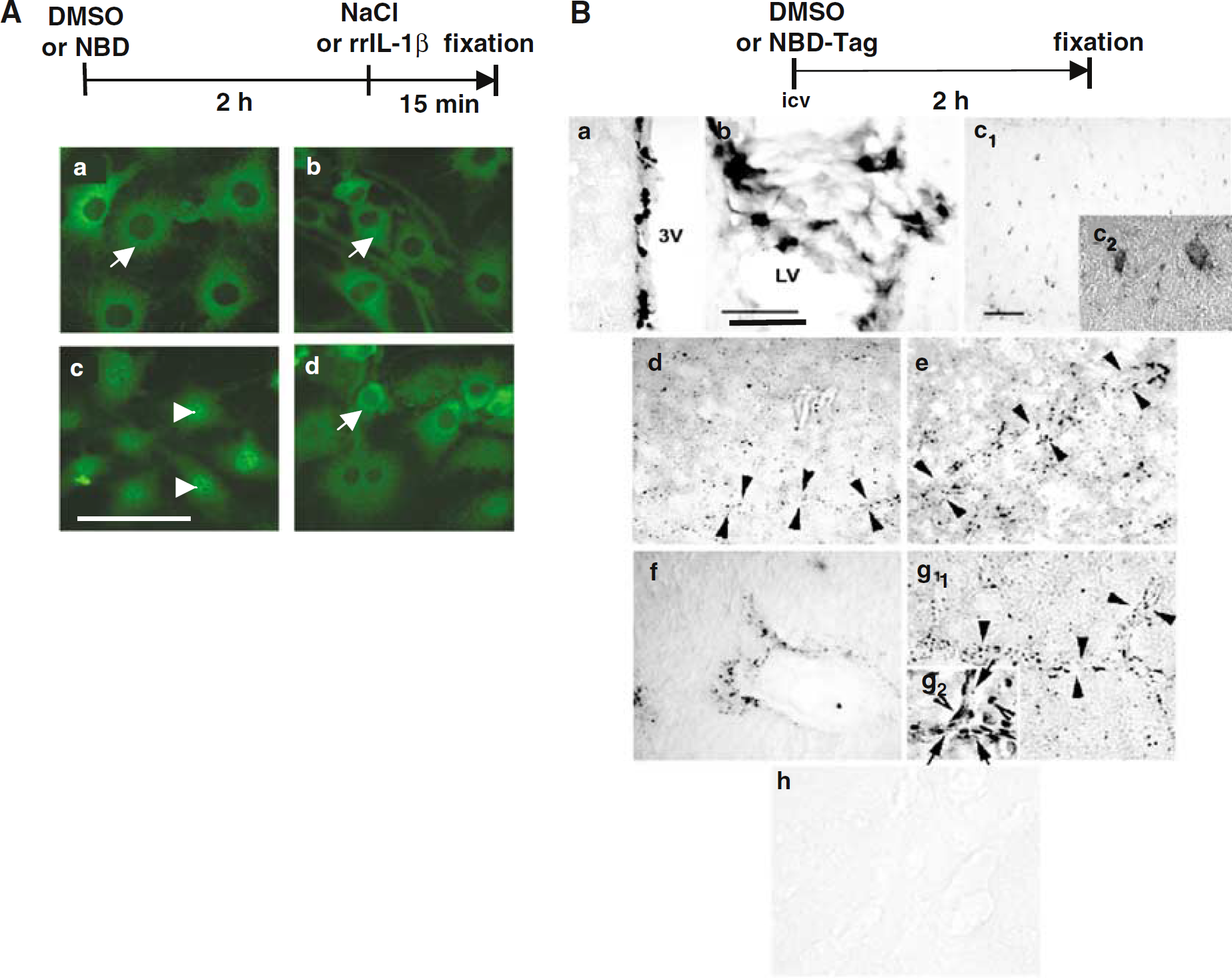
(
When Injected Intracerebroventricularly, NBD Peptide Can Diffuse into the Brain and Enters Cells
To test whether the NBD peptide was able to diffuse into the brain, we injected a biotin-tagged NBD peptide into the lateral ventricle of the rat brain and revealed its localization after 2 h of diffusion. Cytoplasmic staining of biotin-tagged NBD peptide was observed in ependymal cells lining the ventricles (Figure 1Ba), choroid plexus (Figure 1Bb), and cell bodies of a few neuron-like cells, for example, neurons of the cortex (data not shown) and the lateral reticular nucleus (Figure 1Bc1). A higher magnification of Figure 1Bc1 (Figure 1Bc2) shows labeling in cytoplasm of neuron-like cells of the lateral reticular nucleus. It is important to note that the most remarkable staining was observed along the blood vessels (Figure 1Bd-g1) where we observed small black precipitates of DAB. To confirm the subcellular localization of NBD peptide, brain slices were counterstained with Cresyl violet. This allowed to show staining in the cytoplasm of the cells lining the blood vessels rather than in their nucleus (Figure 1Bg2), which is in accordance with the cytoplasmic localization of NEMO and IKKβ. These findings indicate that 2 h after its injection, the NBD peptide has diffused into the brain and entered several types of cells including endothelial-like cells along the blood vessels. It is therefore strategically located to inhibit rrIL-1β-induced p65-NFκB activation.
NFκB and Cox2 Immunoreactivity are both Localized in Endothelial-Like Cells of Cerebral Blood Vessels
Figures 2A, 2B and 2C show a typical labeling of IL-1RI, p65-NFκB, and Cox-2 respectively, in blood vessels. It is worth noticing the similarity between p65-NFκB localization and Cox-2 localization on the wall of the blood vessel. Photomicrographs of Figures 2A and 2B have previously been published (Nadjar et al, 2003). The phenotype of cells displaying Cox-2 immunoreactivity in the brain of rats injected with intraperitoneal rrIL-1β was further characterized by double labeling. Cox-2 immunoreactivity was revealed by a brown precipitate visible in transmitted light in a nuclear or perinuclear compartment (Figure 2E). On the same brain section, PECAM-1 labeling, used to label endothelial cells, was revealed by a green fluorochrome detected under fluorescence microscope and delineated the vessel lumen (Figure 2D). Brain cells displaying inducible Cox-2 were PECAM-1 positive.
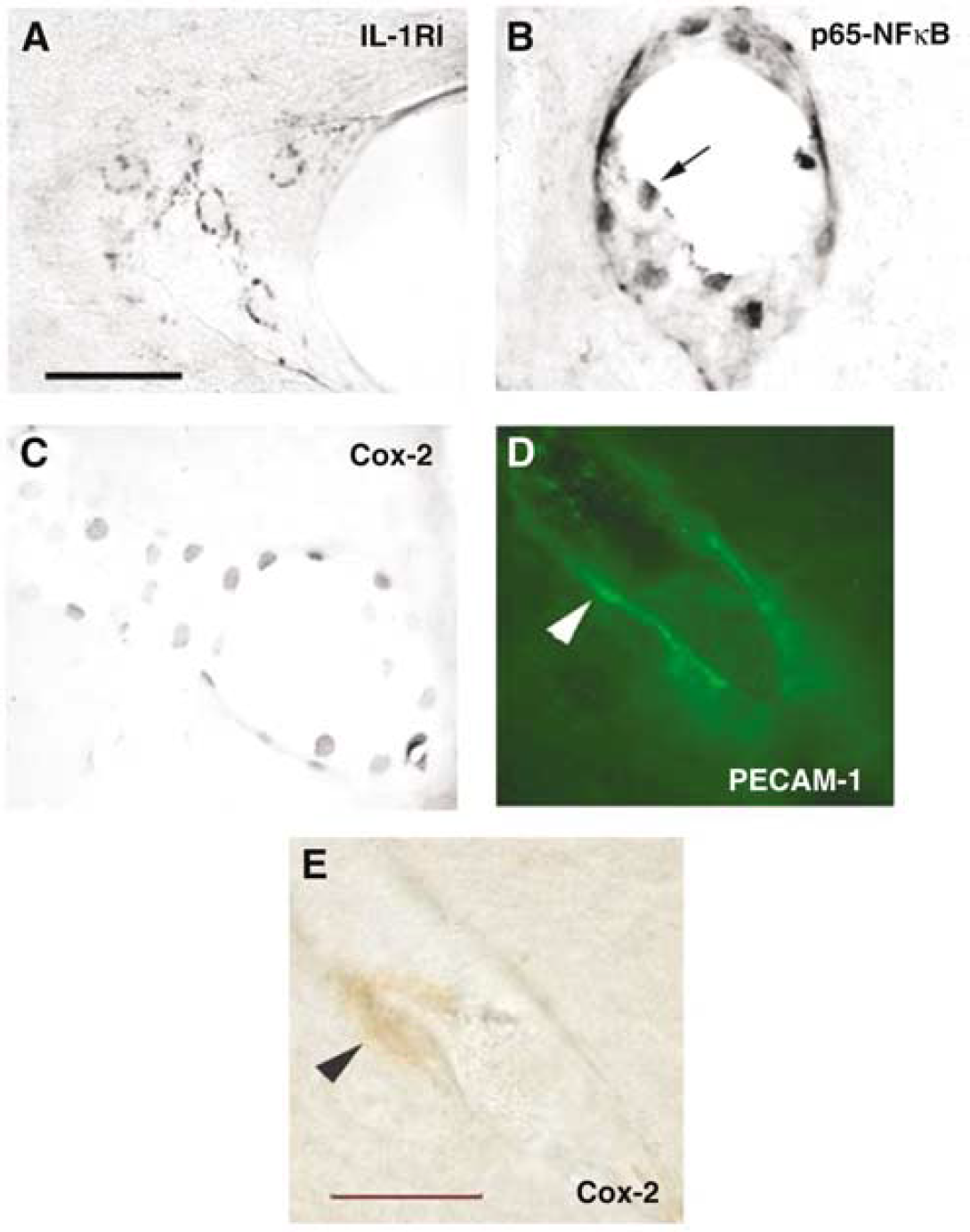
Nuclear transcription factor kappa B (NFκB) and Cyclooxygenase-2 (Cox-2) immunoreactivity are localized in endothelial-like cells of cerebral blood vessels. In pictures (
NBD Peptide Inhibits p65-NFκB Translocation in Blood Vessels of the Brain Vasculature
To test the effect of the NBD peptide on p65-NFκB translocation in the brain vasculature and to validate the method of p65-NFκB immunoreactivity measure, we analyzed a typical brain nucleus, the medial septum nucleus (MSN) because it contains a large number of blood vessels. A bar graph representative of the intensity of p65-NFκB staining in blood vessels of MSN is shown in Figure 3A. Pretreatment with the NBD peptide inhibited the p65-NFκB translocation, which is usually visible in endothelial-like cells 1 h after rrIL-1β injection. The effects of the pretreatment and treatment factors were significant (pretreatment: F (1,16) = 10.41, P = 0.005; treatment: F (1,16) = 8.3, P = 0.011) as well as their interaction (F (1,16) = 10.54, P = 0.05). In control conditions, low basal p65-NFκB staining was observed in blood vessels of the MSN and was not inhibited by NBD pretreatment (Figures 3Ca and 3Cb). The basal p65-NFκB staining was certainly because of cannulation, as observed in previous studies, in brain capillaries, ventricular ependyma, choroid plexus, and meninges (Proescholdt et al, 2002; Nadjar et al, 2003).
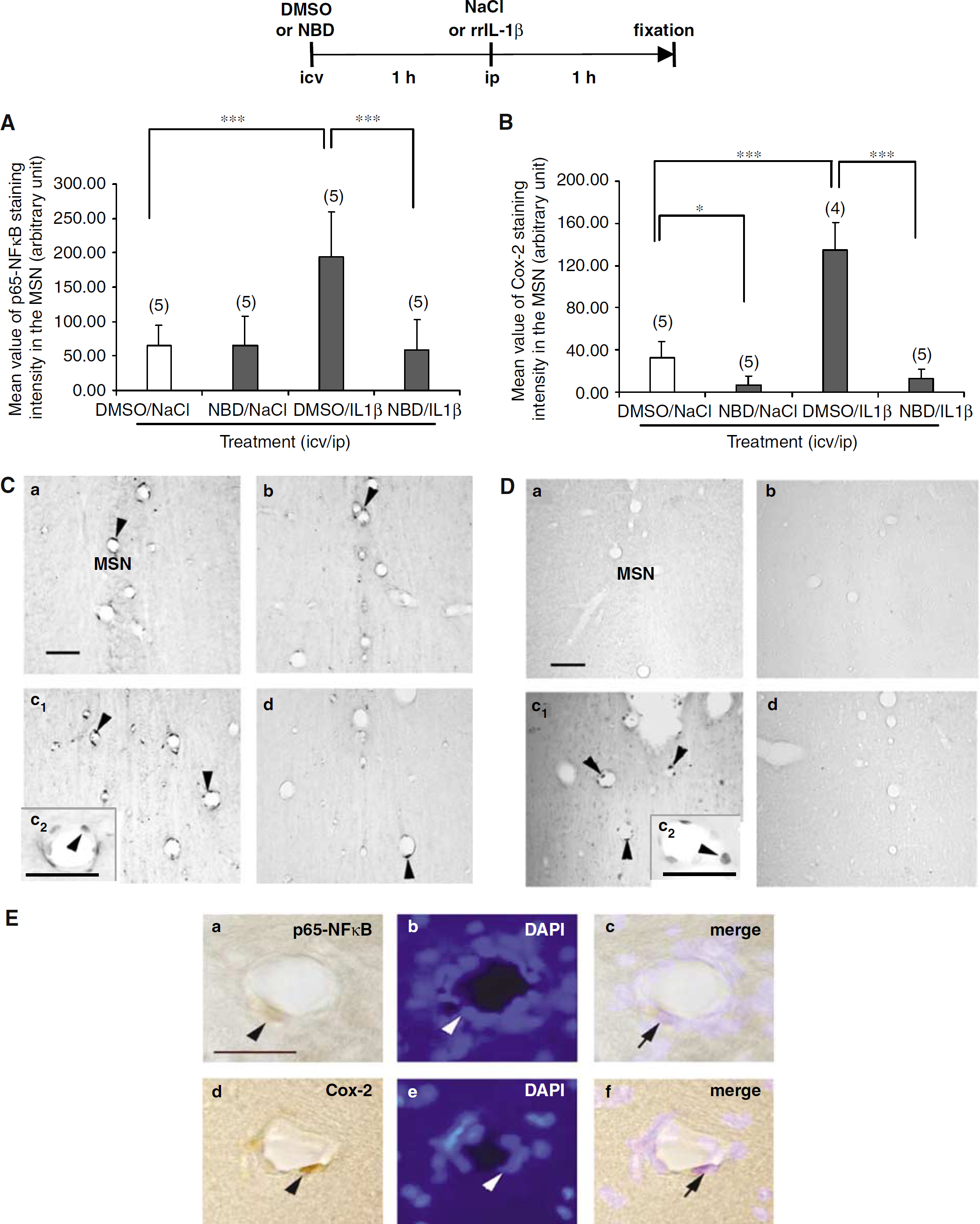
An intracerebroventricular injection of NEMO-binding domain (NBD) peptide inhibits recombinant rat IL-1β (rrIL-1β)-induced p65-NFκB translocation and cyclooxygenase-2 (Cox-2) immunoreactivity in endothelial cells of the medial septum nucleus (MSN). (
Figure 3C shows representative photomicrographs of immunohistochemical detection of p65-NFκB in blood vessels of the MSN. Nuclear transcription factor kappa B staining (Figure 3Ca) increased after a peripheral injection of rrIL-1β (Figure 3Cc) but was reduced to basal level in presence of NBD peptide (Figure 3Cd). Injection of NBD peptide alone (Figure 3Cb) had no significant effect on basal p65-NFκB nuclear staining.
The colocalization of p65-NFκB with the nucleic acid marker DAPI is presented in Figure 3Ea-c. These pictures confirm that after the peripheral administration of rrIL-1β p65-NFκB is translocated into the nucleus of endothelial cells of the brain vasculature.
Inhibition of NFκB Pathway by NBD Peptide Decreases rrIL-1β-Induced Cox-2 Protein Synthesis in Blood Vessels
As for p65-NFκB detection, Cox-2 immunostaining intensity was first measured in endothelial-like cells of the MSN (Figure 3B). The NBD peptide had a small significant inhibitory effect on the low basal level of Cox-2 protein. Moreover, NBD peptide significantly decreased Cox-2 synthesis induced by a peripheral injection of rrIL-1β (135.08±25.41 cells for DMSO/rrIL-1β compared with 12.58±8.96c ells for NBD/rrIL-1β; P<0.001). The effects of the pretreatment and treatment factors and their interaction were significant (pretreatment: F (1,15) = 114.7, P<0.001; treatment: F (1,15) = 60.33, P<0.001; interaction: F (1,15) = 47.15, P<0.001).
Figure 3D shows representative photomicrographs of immunohistochemical detection of Cox-2 in blood vessels of the MSN. Almost no Cox-2 staining was observed in basal conditions (Figure 3Da) or after NBD peptide injection (Figure 3Db) whereas rrIL-1β injection at the periphery induced a strong and typical perinuclear staining of Cox-2 protein in endothelial-like cells (Figure 3Dc). The increase of Cox-2 immunoreactivity was almost totally inhibited by pretreatment with NBD peptide (Figure 3Dd). As for p65-NFκB, Figure 3Ed-f shows that Cox-2 is colocalized with DAPI.
Cox-2 immunoreactivity was measured in most rat brain structures displaying a clear Cox-2 induction in response to rrIL-1β injection compared with saline injection. Figure 4 shows that rrIL-1β induced a significant increase of Cox-2 immunoreactivity in the medial preoptic nucleus (MEPO) (P<0.001) (Figure 4A), paraventricular nucleus (PVN) (P<0.001) (Figure 4B), nucleus of the tractus solitarius (NTS) (P<0.01) (Figure 4C) and supraoptic nucleus (SON) (P<0.001) (Figure 4D). Pretreatment with NBD peptide significantly decreased rrIL-1β-induced Cox-2 protein levels in endothelial-like cells of the blood vessels in MEPO (P<0.005), PVN (P<0.001), NTS (P<0.01), and SON (P<0.001). In all these structures, the effects of the pretreatment, treatment, and their interaction were significant.
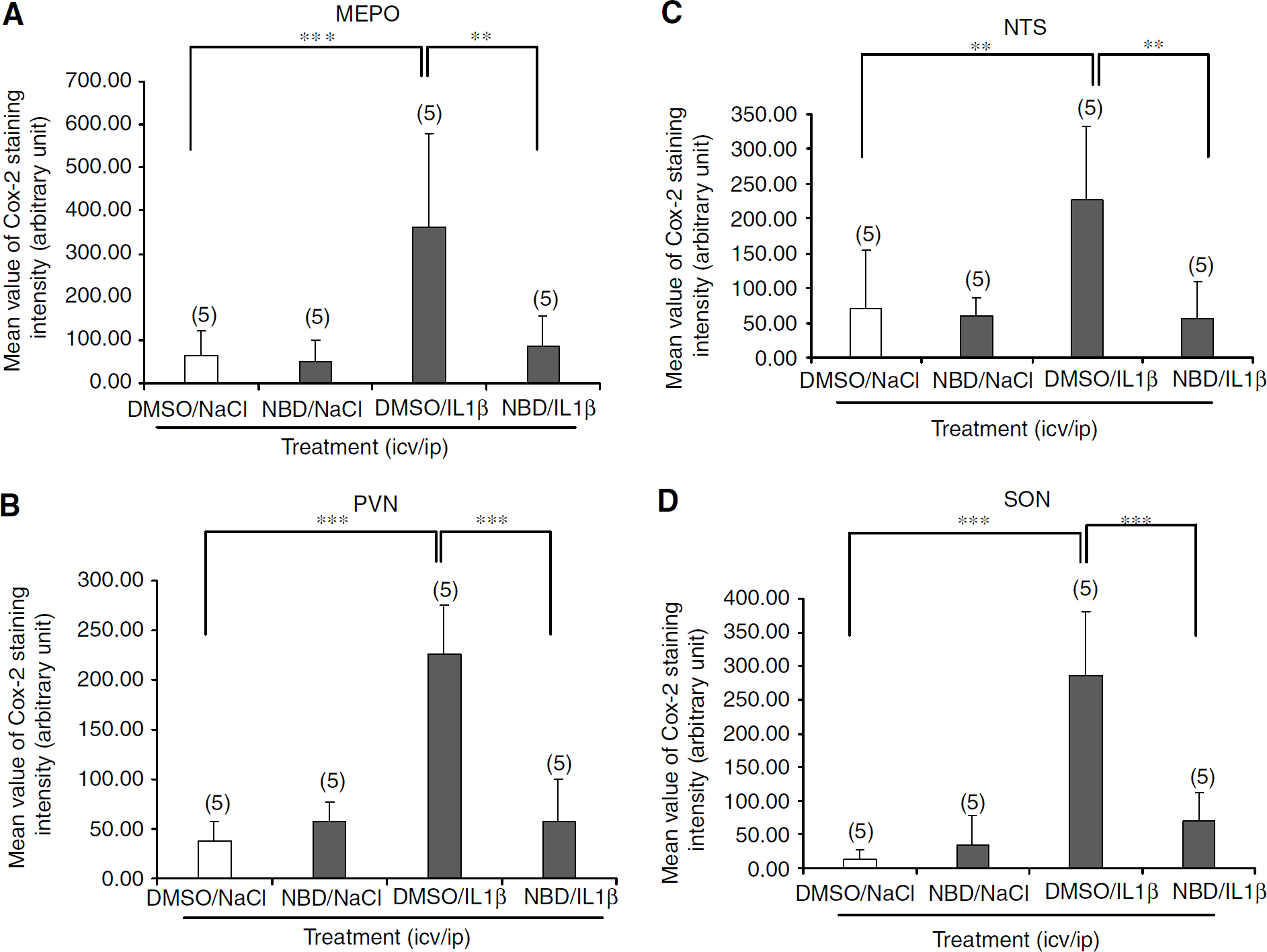
An intraperitoneal injection of recombinant rat IL-1β (rrIL-1β) induces a nuclear transcription factor kappa B (NFκB)-dependant cyclooxygenase-2 (Cox-2) synthesis in medial preoptic nucleus (MEPO), paraventricular nucleus (PVN), nucleus of the tractus solitarius (NTS) and supraoptic nucleus (SON). Bar graphs summarize the intensity of Cox-2 immunoreactivity in MEPO, PVN, NTS and SON. Note that injection of rrIL1β significantly increases Cox-2 staining intensity in the MEPO (***P<0.001), PVN (***P<0.001), NTS (**P<0.01) and SON (***P<0.001) compared with control groups and this effect is reversed by a preliminary intracerebroventricular injection of NEMO-binding domain (NBD) peptide, in the MEPO and NTS (**P<0.005), PVN and SON (***P<0.001). Data were analyzed by a two-way ANOVA and Newman-Keuls post hoc test. MEPO: median preoptic nucleus (number of rats are specified on each plot).
Constitutive Expression of Cox-2 is not Influenced by Administration of NBD Peptide
In baseline conditions, a strong Cox-2 immunoreactivity was observed in neuron-like cells of the hippocampus (HPC) and basolateral amygdala (BLA). To study the possible effects of rrIL-1β and the involvement of NFκB pathway on this constitutive neuronal expression, the intensity of Cox-2 staining was quantified in these structures in the various experimental conditions. Recombinant rat IL-1β injection or impairment of the NFκB pathway by NBD pretreatment had no effect on constitutive neuronal Cox-2 expression in BLA and HPC (Figures 5A1 and 5A2, respectively). Figure 5B shows representative photomicrographs of immunohistochemical detection of Cox-2 in neuron of the BLA (Figures 5Ba and 5Bb) and HPC (Figures 5Bc and 5Bd) as confirmed by colocalization with the neuronal marker NeuN (Figure 5C). The strong staining observed in control conditions (data not shown) was not modified after rrIL-1β injection alone (Figures 5Ba and 5Bc) or in combination with the NBD peptide (Figure 5Bb and 5Bd).
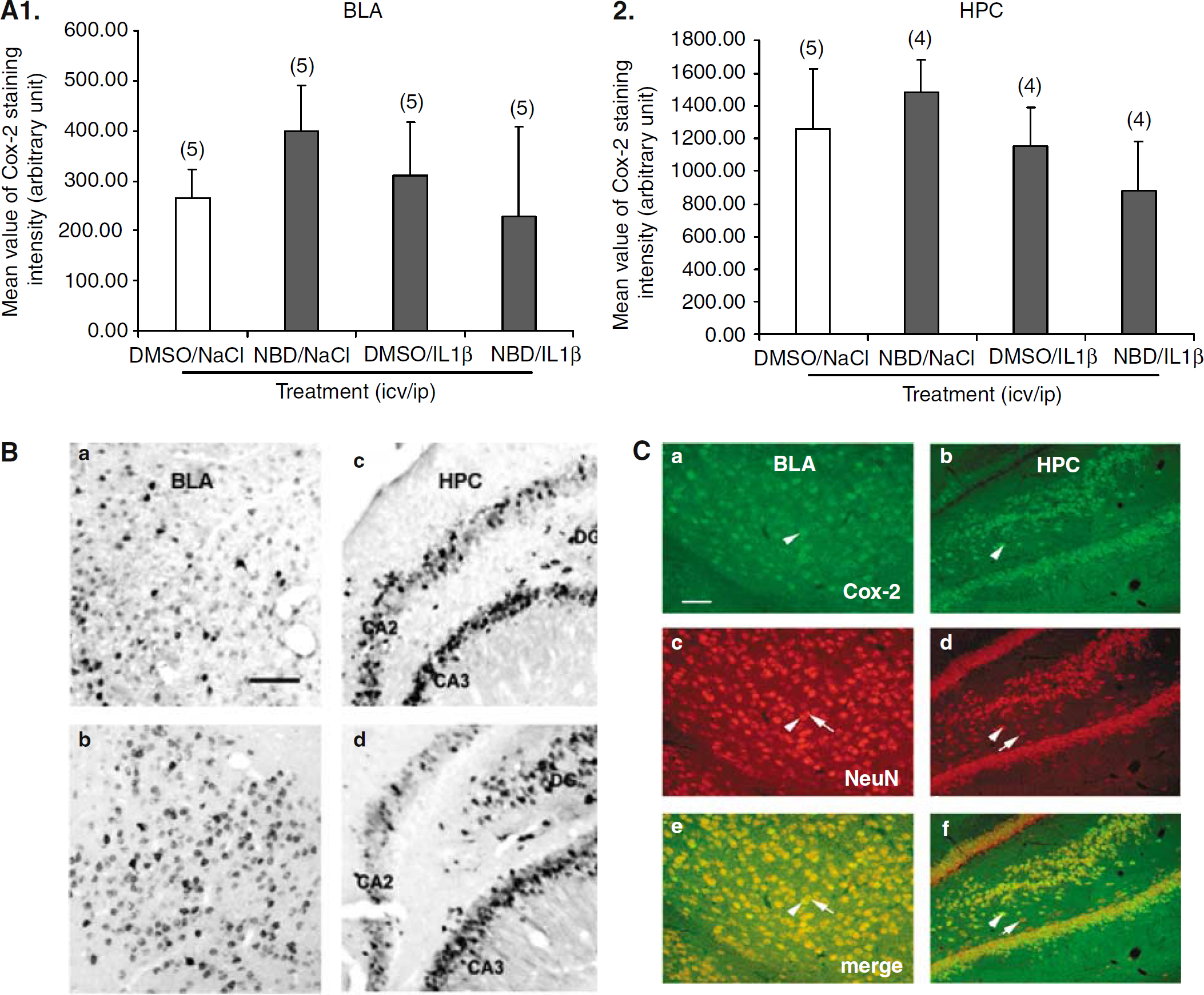
Constitutive neuronal cyclooxygenase-2 (Cox-2) in the basolateral amygdala (BLA) and the hippocampus (HPC) is regulated neither by intraperitoneal recombinant rat IL-1β (rrIL-1β) injection nor by blockade of the nuclear transcription factor kappa B (NFκB) pathway. (
DISCUSSION
Based on a functional in vivo approach using a well-described inhibitor of NFκB pathway, NBD peptide, the present findings show that p65-NFκB translocation mediates the induction of Cox-2 in endothelial-like cells of the rat brain in response to IL-1β stimulation. This result was obtained after having checked the efficiency of the NBD peptide in vitro and in vivo and visualized its brain distribution once injected centrally. Two important and new findings are presented here: (1) regulation of activation of cerebral NFκB can negatively influence the expression of the IL-1β-inducible Cox-2 in vivo and (2) neuronal constitutive Cox-2 is not altered by the impairment of NFκB activation.
An extensive collection of drugs that range widely in structure and activity are known to inhibit NFκB activity and alleviate inflammation (D'Acquisto et al, 2002). A novel approach to the development of selective antiinflammatory drugs comes from the prospect of using recombinant proteins or peptides to target signal transduction pathways by developing cell-penetrating peptides to block NFκB activity (D'Acquisto et al, 2002). Few peptide inhibitors are available as SN50 or NLS (for nuclear localization sequence) that effectively inhibit IL-1-induced nuclear translocation of NFκB in different cell lines (Lin et al, 1995; Kolenko et al, 1999; Koulich et al, 2001; D'Acquisto et al, 2000; Fujihara et al, 2000). However, subsequent analysis has shown that they are also able to block the nuclear localization of other transcription factors as STAT, AP-1, or NAFT (Torgerson et al, 1998) and therefore minimized their specificity. The NBD peptide used in the present study and described previously (May et al, 2000) consists of a hexapeptide sequence within the extreme C-termini of both IKKα and IKKβ fused to penetratin, a membrane translocation sequence. The resulting peptide blocks the association of NEMO with IKKβ and IKKα and has been shown to effectively inhibit TNF-α-induced NFκB activation in HeLa cells, to inhibit E-selectin expression in HUVEC and NO production in macrophages. Moreover, it has been shown now in various models of inflammation as phorbol ester-induced ear edema, zymosan-induced peritonitis or LPS-induced septic shock, to function as a potent antiinflammatory agent (May et al, 2000, 2002; Dai et al, 2004). The present study is the first to show that NBD peptide injected directly into brain ventricle is able to penetrate cells of the central nervous system, to enter cells of the brain vessels, and to diffuse into the brain parenchyma, although it has been previously used to protect mice from spinal cord mononuclear cell invasion in a model of experimental allergic encephalomyelitis (Dasgupta et al, 2004). Therefore, it is a useful tool to investigate NFκB-dependent gene regulation in situ. In consequence, we present here the first data of the use of NBD peptide to control the synthesis of brain endothelial Cox-2, essential for the production of PGs, in response to IL-1β stimulation.
The present observation that Cox-2 is induced in endothelial cells of the brain vasculature in response to IL-1β administration is consistent with a number of previous data showing that pyrogenic doses of IL-1β or LPS injected at the periphery induce Cox-2 mRNA synthesis in endothelial cells of the brain vasculature, together with a robust activation of the NFκB pathway in the endothelium of the brain capillaries (Cao et al, 1995, Cao et al, 1996, Cao et al, 1997; Lacroix and Rivest, 1998; Laflamme et al, 1999). Recently, evidence for colocalized expression of IκBα transcript and Cox-2 protein in the brain vascular cells in LPS-treated rats has also been shown (Konsman et al, 2004). This spatio-temporal coincidence agrees with a role for NFκB in the regulation of Cox-2 gene expression. Various in vitro studies have implicated NFκB in the transcriptional regulation of Cox-2 using inhibitors (D'Acquisto et al, 1997), anti-sense (Roshak et al, 1996), or decoy oligonucleotides targeting NFκB pathway (Schmedtje et al, 1997) since two putative NFκB motifs from the Cox-2 promoters are able to bind p50/p65 NFκB heterodimers in an IL-1β-dependent manner (Crofford et al, 1997; Sorli et al, 1998). However, the demonstration of a direct relationship between cerebral NFκB activation and Cox-2 induction had not yet been achieved in in vivo conditions. In the first part of this work, we show that in response to systemic IL-1β, NFκB activation takes place in endothelial-like cells of the cerebral vasculature bearing IL-1RI on their membrane and that nuclear translocation of p65-NFκB and Cox-2 immunoreactivity occur in the same endothelial-like cells. This demonstration was necessary because the exact cellular source of Cox-2 and therefore of prostaglandins production is still controversial. In 1999, Laflamme hypothesized that the differences visible in IL-1β-induced Cox-2 production in various brain structures were related to the degree of vascularization (Laflamme et al, 1999). Whether this is because of the involvement of endothelial cells of brain vessels (Cao et al, 1996, 1999; Quan et al, 1998; Matsumura et al, 1998) or perivascular cells (Elmquist et al, 1997; Schiltz and Sawchenko, 2002) remains unclear. In the present study, for the dose of IL-1β injected and at the time period under consideration, endothelial cells were found to be the major source of IL-1β-induced Cox-2.
The rapid activation of the NFκB pathway that is responsible for the Cox-2 induction in endothelial cells of the brain vasculature is in favor of a humoral route of communication between peripheral accessory immune cells and the brain. Although IL-1β induces Cox-2 quite ubiquitously across the cerebral blood vessels, some areas are more densely immunoreactive for Cox-2, as shown in the present study. Accordingly, it is tempting to suggest that the neuronal activities of PGE2 are probably limited to selective nuclei bearing specific prostaglandin receptors such as the PVN, MEPO, SON, NTS where we were able to show the negative effect of NBD peptide on Cox-2 induction. Whether this same inhibitory peptide modulates neuronal activities attributed to PGE2 on these precise brain structure needs now to be further investigated.
In rodents, many converging data support the hypothesis that fever evoked by peripheral injection of IL-1β is Cox-2 dependent. The levels of Cox-2 mRNA and Cox-2-like immunoreactivity rise in brain after the peripheral administration of LPS or IL-1β (Breder and Saper, 1996; Cao et al, 1996). Moreover, Cox-2 knockout mice fail to produce a febrile response to an immune challenge, but the increase in setpoint for thermoregulation is restored by intracerebroventricular injection of PGE2 (Li et al, 2001). Induction of Cox-2 in vascular endothelial cells participates to the cerebral vasodilatation effect of IL-1β (Osuka et al, 1997) through accumulation of cyclic nucleotides and increase of NO production (Bonmann et al, 1997). Interleukin-1β is also a potent activator of the hypothalamo-pituitary-adrenal (HPA) axis (Schobitz et al, 1994; Besedovsky et al, 1991) through PGs and this is consistent with the strong hybridization signal for Cox-2 mRNA (Watanobe and Takebe, 1994) and Cox-2 immunoreactivity observed along the microvessels of the parvocellular PVN in the present study. The use of the selective Cox-2 inhibitor (NS 398) totally abolished the effect of IL-1 on plasma ACTH and POMC transcripts at the pituitary level although the effect on corticosterone secretion was mitigated (Parsadaniantz et al, 2000).
In contrast to inducible endothelial Cox-2, very little is known about constitutive Cox-2. As the inducible form, neuronal Cox-2 appears to be involved in the response to inflammation where it plays a major role in nociceptive mechanisms (Samad et al, 2001). Cyclooxygenase-2 mRNA is constitutively expressed in neurons of the cortex, hippocampus and amygdala, and absent in endothelial cells of blood vessels (Quan et al, 1998). Recently, Kaltschmidt et al, (2002) showed in cultured hippocampal slices, that neuronal Cox-2 expression is essentially dependent on NFκB activity. Moreover, aspirin inhibited neuronal NFκB, which resulted in strongly reduced Cox-2 activity in the same preparation. That result seems to be in total discrepancy with our present in vivo study and others previous experiments performed in rats that did not mention any changes of neuronal Cox-2 mRNA expression after LPS treatment (Minghetti et al, 1999; Quan et al, 1998). A couple of explanations can account for the lack of effect of NBD peptide on neuronal Cox-2 expression in BLA and HPC of rat brain: (1) expression of neuronal Cox-2 is not dependent on NFκB pathway activation. Indeed, other signaling pathways are implicated in the regulation of neuronal Cox-2 because in vitro experiments show that IL-1β can induce p38 MAPK phosphorylation and activation of NFκB independently from each other and that p38 MAPK is predominantly involved to increase Cox-2 expression in human neuroblastoma cells, presumably by increasing mRNA stability (Fiebich et al, 2000); (2) the demonstration of an effect on constitutive neuronal Cox-2 could required a long-term treatment with NBD peptide that we did not test.
In conclusion, the findings of the present study lead us to suggest that the NBD peptide provides a target for the development of drugs that would potentially block Cox-2-dependent effects of proinflammatory cytokines. Because no effect of NBD peptide was observed on neuronal Cox-2, additional experiments are required to determine whether other modalities of treatment can affect constitutive neuronal Cox-2. In case of negative results, this will allow to impair specific pathway leading to inflammation with no consequences on the homeostasis of the animal.