
Abstract
Recent evidence suggests that reproductive steroids are important players in shaping stroke outcome and cerebrovascular pathophysiologic features. Although women are at lower risk for stroke than men, this native protection is lost in the postmenopausal years. Therefore, aging women sustain a large burden for stroke, contrary to a popular misconception that cancer is the main killer of women. Further, the value of hormone replacement therapy in stroke prevention or in improving outcome remains controversial. Estrogen has been the best studied of the sex steroids in both laboratory and clinical settings and is considered increasingly to be an endogenous neuroprotective agent. A growing number of studies demonstrate that exogenous estradiol reduces tissue damage resulting from experimental ischemic stroke in both sexes. This new concept suggests that dissecting interactions between estrogen and cerebral ischemia will yield novel insights into generalized cellular mechanisms of injury. Less is known about estrogen's undesirable effects in brain, for example, the potential for increasing seizure susceptibility and migraine. This review summarizes gender-specific aspects of clinical and experimental stroke and results of estrogen treatment on outcome in animal models of cerebral ischemia, and briefly discusses potential vascular and parenchymal mechanisms by which estrogen salvages brain.
STROKE IN WOMEN VERSUS MEN: RISKS AND OUTCOMES
Overall incidence of stroke is uniformly higher in men than in women in all countries (Sudlow and Warlow, 1997), but increases with age in both sexes (Prencipe et al., 1997) and strongly reflects ethnic/racial background (Kuhlemeier and Stients, 1994; Morgenstern et al., 1997) (Fig. 1). Stroke is rare in women during the reproductive years, and in young women (15 to 44 years of age) is predominately embolic in origin (Carolei et al., 1993) or is caused by subarachnoid hemorrhage (Petitti et al., 1996, 1997). The latter is the only subtype of stroke more common in women than men. In longitudinal studies, women sustain fewer strokes of all types than age-matched men (incidence ratio as high as 1:1.35) (Kurtzke, 1985). The importance of female sex steroids to this relative protection is not known, but male-female differences weaken in aged subjects (Manolio et al., 1996). For example, gender effects in the Framingham Study are most striking in subjects within the 45- to 54-year age cohort (combined incidence of stroke and transient ischemic attack 2.1 versus 1.2 per 1,000 in men and women, respectively), but equalize in the 55- to 64- year-old cohort (Wolf, 1990). Alternatively, others report higher incidence of intracerebral hemorrhage and cortical infarction in men versus women, trends which persist well beyond the menopause (Giroud et al., 1991).
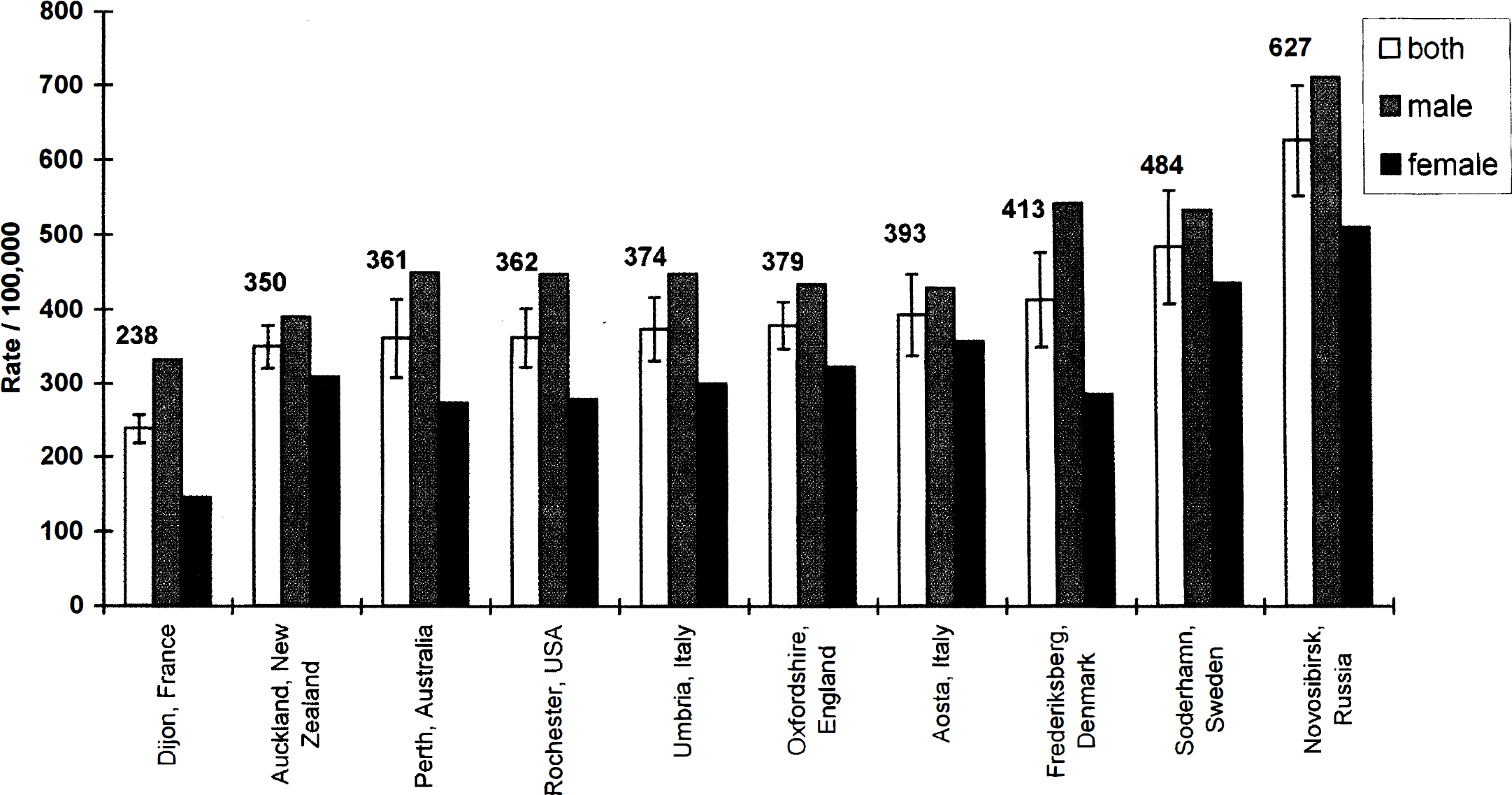
Age- and sex-standardized annual incidence per 100,000 of all types of stroke combined for the group aged 45 to 84 years and with men and women considered separately. Studies are arranged in ascending order of age- and sex-standardized incidence rates. The numbers shown are point estimates, and the error bars are 95% confidence intervals. Reprinted with permission from Sudlow and Warlow, 1997.
Known risk factors for ischemic stroke are similar in men and women; most epidemiologic studies do not identify significant differences in relative risks. Recent studies found no sex differences in the importance of hypertension as a risk factor (Hayes and Taler, 1998), although diabetes and atrial fibrillation may be larger risk factors in women (Wolf et al., 1991). These statements do not include relative risks to women younger than 50 years of age, including pregnancy, migraine, and oral contraceptives (for review, see Bousser, 1999). Furthermore, atherosclerotic disease may progress more slowly and to a lesser degree in women versus men. In a study of 5,033 consecutive autopsies, women demonstrated strikingly less cerebral atherosclerosis relative to men until age 65 years; then lesion frequency equaled that of men (Flora et al., 1968). Consistent sex-linked differences also have been reported in carotid and vertebral arterial disease (Caplan et al., 1986).
Gender may have an influence not just on the risk of having a stroke (stroke proneness or latency to stroke) but also on the severity of damage induced by the stroke (stroke sensitivity). Although lifetime risk of stroke is higher in men, mortality associated with stroke is higher in women. However, this is most likely because of their 10-year-longer life expectancy and the older age at which clinical disease is manifested. Interactions with ethnic background may influence these data (Morgenstern, 1997). However, findings from the World Health Organization Monitoring of Trends and Determinants in Cardiovascular Disease (WHO MONICA project) demonstrate that average case-fatality rates are higher in women than men in 12/17 European and Asian countries studied (WHO MONICA Project, 1997). Further, current projections suggest a higher likelihood of death from stroke for women (16% of the population) than for men (8%) (Bonita, 1992). Gender-based recovery from stroke has not been heavily studied, but functional impairment (Wyller et al., 1997) and depression (Kotila et al., 1998) appear greater in women.
Since most strokes occur in postmenopausal women, there has been much interest in determining whether the potential for ovarian hormone replacement to improve coronary heart disease is equally valid in reducing cerebrovascular disease. Although detailed analysis is beyond the scope of the present review, the efficacy of estrogen replacement therapy (ERT) or combined hormone therapy (HRT) with estrogen and a progestin remains unproven for stroke. In part, there are large difficulties in interpreting or comparing results from previous studies because they (1) are cohort or case-control observational studies, not randomized trials; (2) use a variety of estrogen and progesterone preparations or combinations; (3) vary in sampling women who are “ever users” or “current users” of HRT; (4) may be unavoidably biased by the observation that women likely to seek HRT tend to be healthier in general; and (5) vary in the stratification for subtype of stroke, such as subarachnoid or intracerebral hemorrhage. Some clarity may be obtained as the results of ongoing randomized studies emerge, including the Women's Heath Initiative to be reported at approximately year 2006 and a study of secondary prevention, the Women's Estrogen for Stroke Trial (WEST) to be published in the year 2000.
Most studies examine the effects of unopposed conjugated equine estrogen, and more recently, effects of estrogen plus a progestin, for example, medroxyprogesterone acetate (Falkeborn et al., 1993; Grodstein et al., 1996, 1997; Schairer et al., 1997; Petitti et al., 1998). Grady et al. (1992) found little consistent evidence in an early and comprehensive literature review that ERT reduced stroke risk, although estrogen use was associated with reduced death from stroke. Subsequent studies provide further data for effect of treatment on risk and mortality, but the overall picture remains inconsistent. Likewise, ERT has been reported to increase (Wilson et al., 1985), decrease (Finucane et al., 1993; Lafferty and Fiske, 1994) or have no effect (Pederson et al., 1997; Petitti et al., 1998) on stroke risk. Combined estrogenprogestin treatment also has produced varied results: some studies report risk reduction for intracerebral hemorrhage (Schairer et al., 1997) or stroke of multiple etiologies (Falkeborn et al., 1993), whereas others show no effect (Grodstein et al., 1996), particularly in nonfatal thromboembolic, hemorrhagic (Pederson et al., 1997), and ischemic stroke (Petitti et al., 1998). In contrast, available data suggest that ERT (Henderson et al., 1991; Grady et al., 1992; Sourander et al., 1998) and HRT (Finucane et al., 1993; Grodstein et al., 1997) reduce mortality from stroke in women when it does occur.
In summary, women may be “protected” from stroke, but this is restricted by factors that involve reproductive age and ethnic or genetic background. When stroke occurs in the postmenopausal woman, mortality may be higher relative to the male, although this observation is based on highly limited data. The value of exogenous hormone replacement in improving risk and outcome is therefore unclear. However, most reports suggest that fatal strokes are reduced.
GENDER AND EXPERIMENTAL STROKE
Given the complexity of the clinical observations, several laboratories have used animal models to study gender influences on ischemic brain injury. In addition, rat strains developed by selective breeding programs to display increased stroke proneness and stroke sensitivity (spontaneously hypertensive rat-stroke prone [SHR-SP]) have been used to evaluate gender-genetic interactions. These data establish a foundation or rationale for subsequent studies of ovarian steroids as neuroprotective agents.
Numerous studies demonstrate gender-specific responses to experimental cerebral ischemia, head trauma (Emerson et al., 1993; Roof et al., 1993), and hypoxic brain damage (Stupfel et al., 1979; Saiyed and Riker, 1993). Early studies of forebrain ischemia in gerbils indicate that females sustained lower mortality and less neuronal damage after unilateral carotid occlusion than males (Payan and Conard, 1977; Hall et al., 1992). The severity of cerebral ischemia and the quality of reperfusion were not different between the sexes. However, preservation of endogenous vitamin E was observed in females, suggesting an enhanced antioxidant capacity (Hall et al., 1992). Although gonadectomized animals were not evaluated in these studies, female resistance to ischemia was present only in animals receiving carotid ligation after sexual maturity (Payan and Conard, 1977).
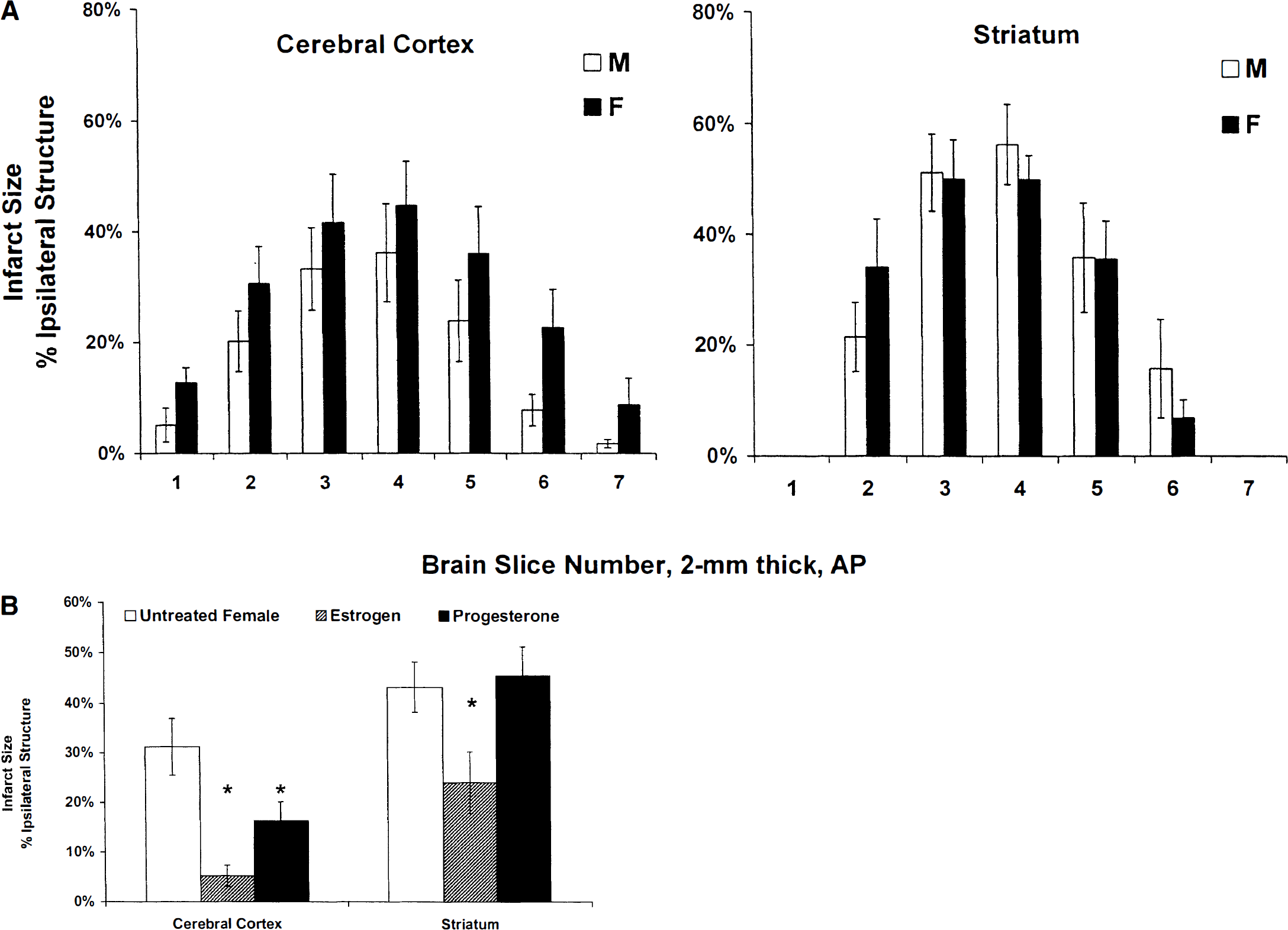
Subsequent studies in a range of different rat stroke models (direct or indirect middle cerebral artery occlusion [MCAO]) have uniformly confirmed that histologic damage is less in female animals, sampled randomly across estrous cycle, than in age-matched males (Li et al., 1996; Alkayed et al., 1998a; Zhang et al., 1998) (Fig. 2A). In an animal model of embolic stroke induced by photochemical irradiation of the common carotid artery, thromboembolism produced greater inflammatory cellular infiltrate, but less severe infarction, in female compared with male rats (Li et al., 1996).
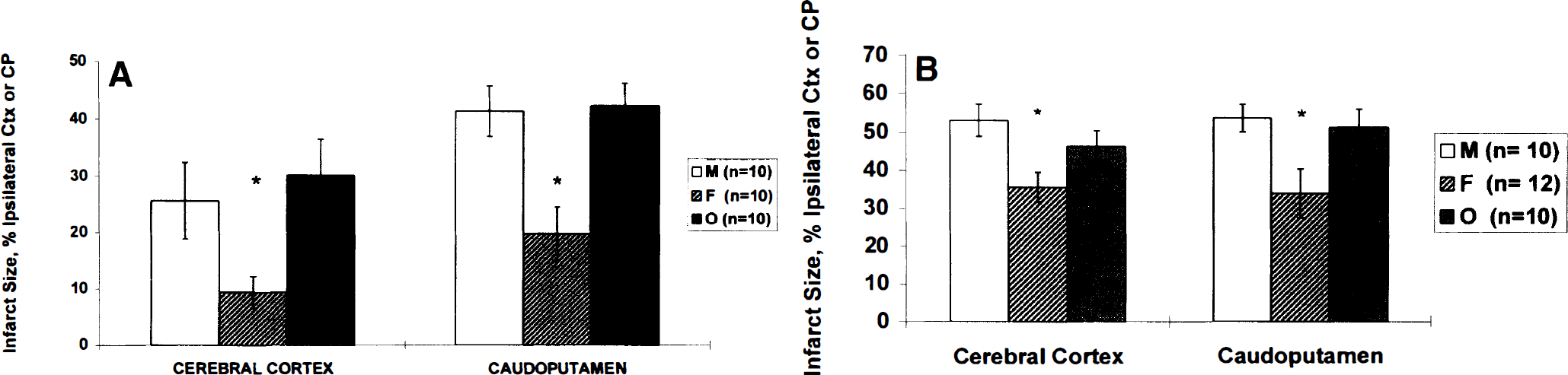
Cerebral infarct after focal cerebral ischemia in Wistar rats
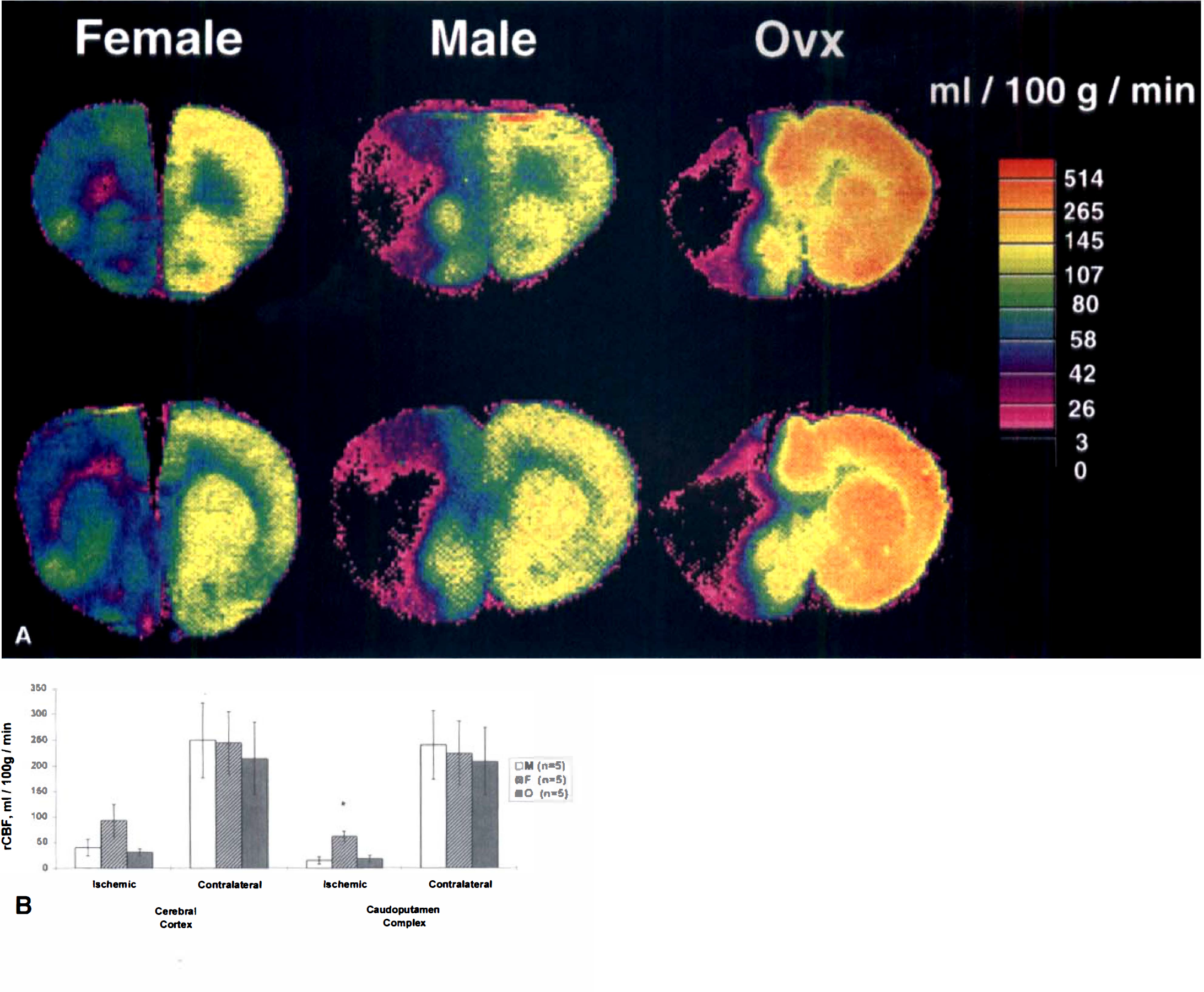
Evidence that reproductive steroids are key to the gender differences comes from studies in gonadectomized animals. Using a reversible MCAO model, we have demonstrated that both cortical and striatal infarct volumes are smaller in sexually mature female rats relative to their male or ovariectomized (OVX) female counterparts (Alkayed et al., 1998b) (Fig. 2A). Estrogen seems to be the major hormonal player in these experiments because (1) estrogen levels were reduced in OVX female rats compared with those of the male, and (2) although progesterone was reduced relative to normal female values, plasma values remained higher than in the male.
The influence of male sex hormone testosterone on cerebral ischemic damage is less clear. Like estrogen, testosterone is a vasodilator of some vascular beds, possibly by a common mechanism involving vascular smooth muscle potassium channels (Yue et al., 1995; Chou et al., 1996). Further, androgen receptors are present in brain regions not associated with reproduction (Simerly et al., 1990; Takeda et al., 1990). Loss of testosterone (castration) has been studied in relation to MCAO in two studies. In one, castration did not influence outcome to MCAO, suggesting a lack of influence of testosterone on outcome (Toung et al., 1998). The other report demonstrated a reduction in lesion size of 59% with castration, which was partially reversed by testosterone replacement, and concluded that testosterone had an adverse effect on outcome to ischemia (Hawk et al., 1998). Testosterone has been implicated as a risk factor for acute myocardial infarction (Phillips et al., 1994), altered lipid metabolism in young men (Hromadova et al., 1991), and in cardiovascular collapse after trauma and hemorrhagic shock in animals (Remmers et al., 1997). Further study of the effects of testosterone on functional or histologic outcome and on intraischemic CBF are needed to resolve the importance of this hormone in stroke.
Gender-genetic interactions
The SHR-SP strain, originally derived by selective breeding from the normotensive reference strain, the Wistar Kyoto rat (Okamoto et al., 1974), has a genetically determined increased sensitivity to experimental stroke (Coyle and Jokelainen, 1993; Gratton et al., 1998; Alkayed et al., 1998a; Carswell et al., 1999a) (Fig. 2B), as well as an increased incidence of spontaneous stroke (stroke proneness) and hypertension. In an early study, Yamori et al. (1976) report that the incidence of spontaneous strokes was greater in male SHR-SP compared with female SHR-SP up to age 25 weeks. Female SHR-SP also had a longer life span and later onset of spontaneous stroke than males. Furthermore, estrogen pretreatment in castrated males decreased the incidence of spontaneous stroke, increased life span, and delayed hypertension relative to intact males, whereas androgen treatment in gonadectomized females reduced life span relative to intact females.
Female SHR-SP also demonstrate reduced infarct volumes compared with SHR-SP males when taken at random during the 4- to 5-day estrus cycle (Fig. 2B). This situation is reversed when stroke is induced during metestrus (Carswell et al., 1999a) (Fig. 3), the stage in the cycle when estrogens and progesterone are at their lowest levels in the circulation (Fig. 4), suggesting that changes in the levels of sex hormones, even within the physiologic range of the estrus cycle, can alter stroke sensitivity. This seems particularly applicable in the SHR-SP since, in this strain, rats that have experimental stroke induced during metestrus sustain significantly greater infarct volumes compared with those occluded during proestrus (plasma estrogen at peak levels) (Carswell et al., 1999b) (Fig. 5). However, this was caused by improvement in CBF (see later section, “Vasodilation and Improved CBF?”)
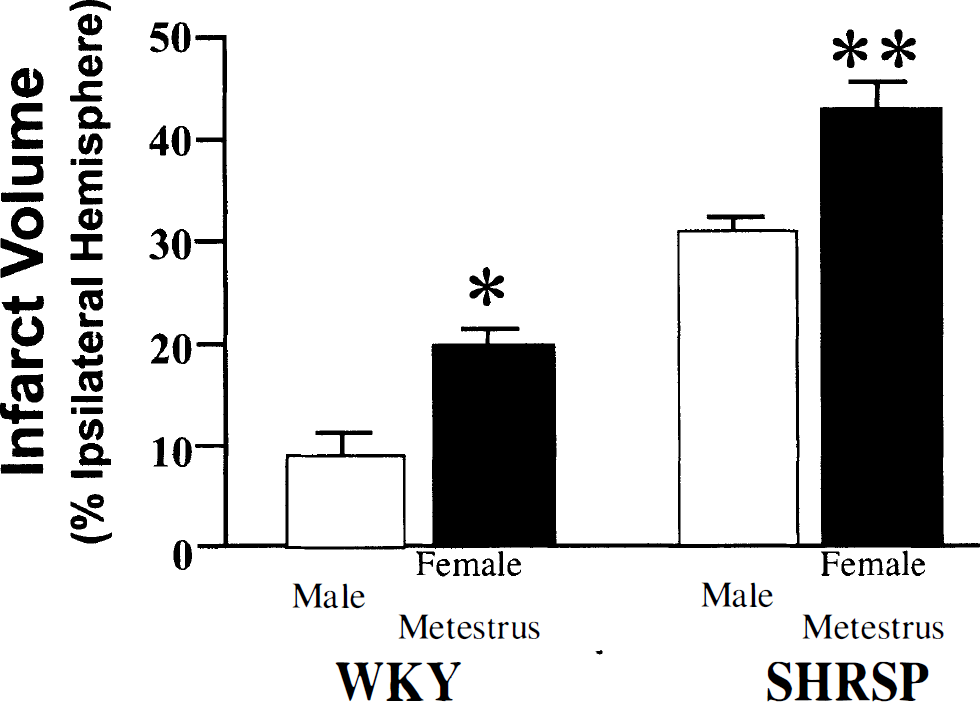
Focal cerebral ischemia induced in males and females during metestrus in Wistar Kyoto (WKY; males n = 9, females n = 8) and spontaneously hypertensive stroke-prone rats (SHR-SP; males n = 8, females, n = 7). Cerebral ischemia was induced by a distal diathermy occlusion of the middle cerebral artery for 24 hours. Infarct volumes were measured in hematoxylin and eosin-stained brain sections and expressed as a percentage of the ipsilateral hemisphere. Data are represented as mean ± SEM; *significant difference from male WKY (P < 0.05); **significant difference from male SHR-SP (P = 0.01). Reprinted with permission from Carswell et al., 1999a.
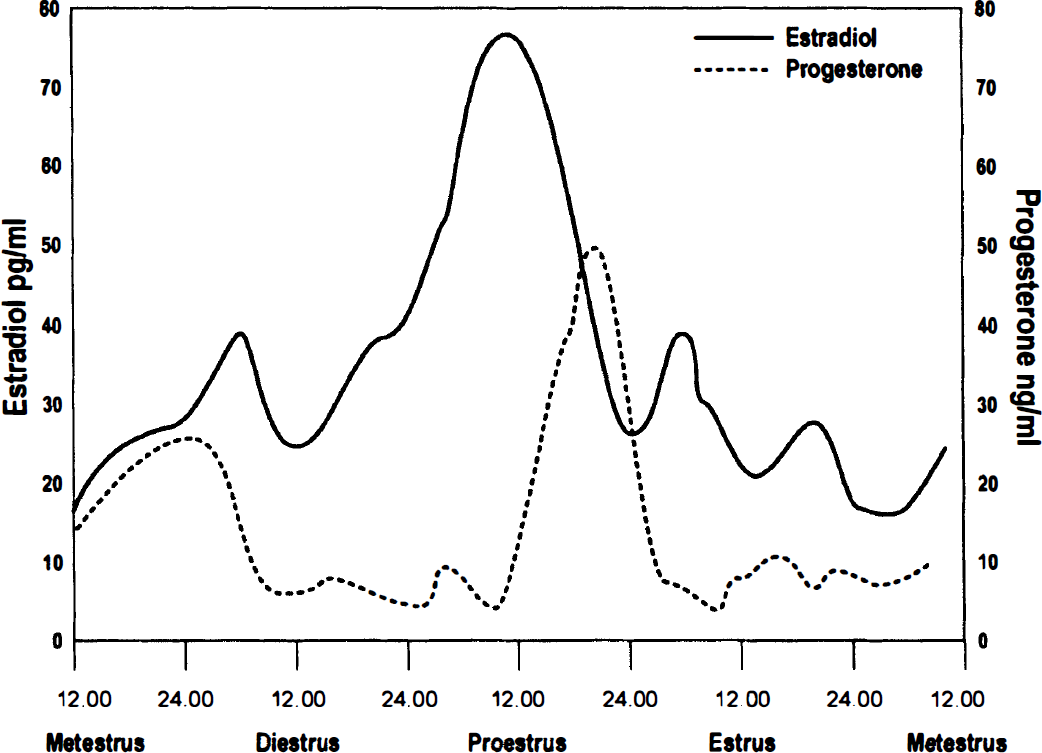
Representative plasma levels of 17β-estradiol (pg/mL) and progesterone (ng/mL) during the 4-day rat estrus cycle.
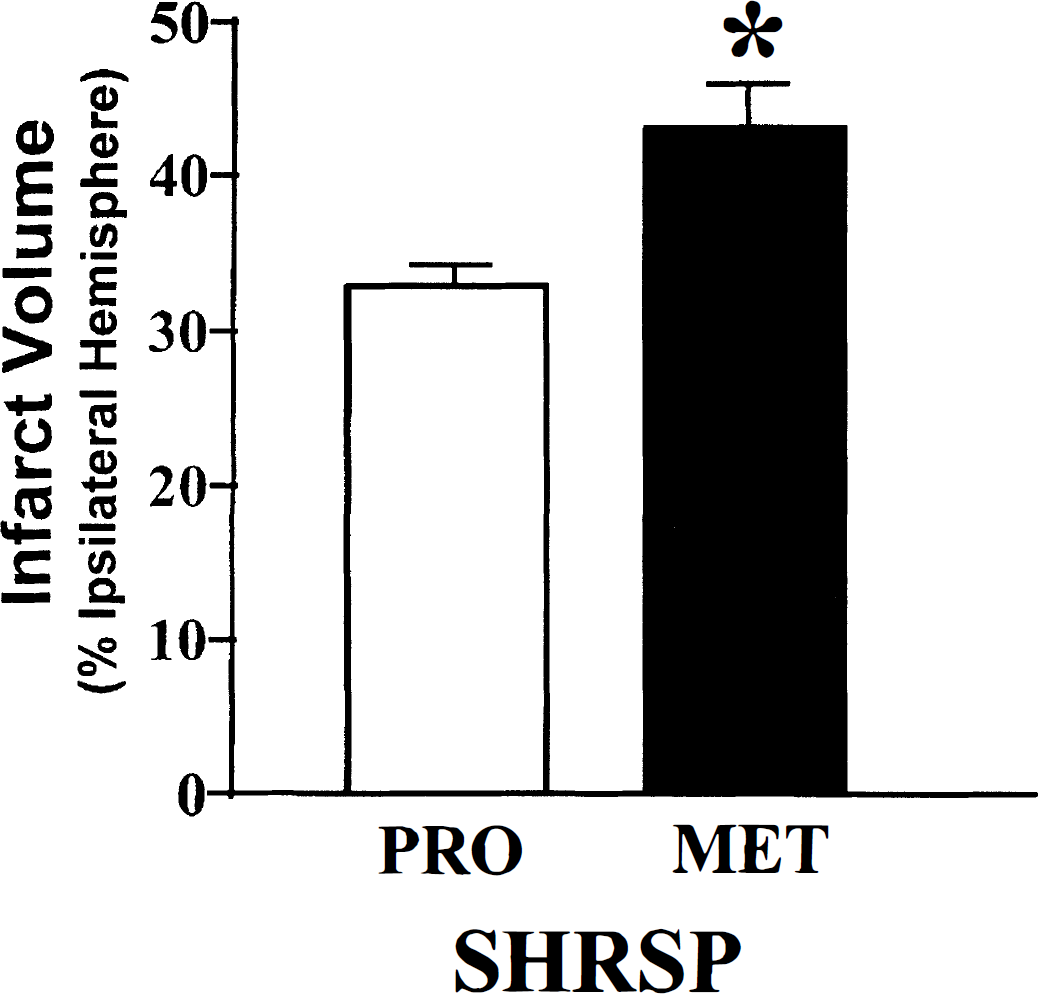
Focal cerebral ischemia induced in female SHR-SP during proestrus (PRO, n = 7) or metestrus (MET, n = 7). Cerebral ischemia was induced by a distal diathermy occlusion of the middle cerebral artery for 24 hours. Infarct volumes were measured in hematoxylin and eosin-stained brain sections and expressed as a percentage of the ipsilateral hemisphere. Data are represented as mean ± SEM; *significant difference from proestrus group (P < 0.01), Student's unpaired t-test. Reprinted with permission from Carswell et al., 1999b.
Notice that although gender-specific outcome after cerebral ischemia has been repeatedly demonstrated in rodents, there is a paucity of studies in higher order animal species where no data are available in models of ischemic, hemorrhagic, or thromboembolic stroke. A single study evaluates female versus male neurologic recovery at 24 hours after cardiac arrest in the dog (Zwemer et al., 1997). Systemic organ function was more impaired in females, but neurologic score was not different between sexes. Histologic injury was not measured, and one confounding variable in this study was the use of female dogs of varying reproductive states, including postpartum and partially gonadectomized animals.
ESTROGEN TREATMENT IN EXPERIMENTAL STROKE
If estrogen is a naturally occurring neuroprotectant, then treatment with physiologically relevant amounts of the steroid should improve outcome in estrogen-deficient brain. A mounting number of in vivo investigations suggests that this hypothesis is correct (Table 1) (Hurn et al., 1995; Kondo et al., 1997; Shi et al., 1997; Sudo et al., 1997; Chen et al., 1998; Green et al., 1998; Pelligrino et al., 1998; Shi et al., 1998; Wang et al., 1999; Zhang et al., 1998). Critical considerations include the therapeutic range and treatment duration for efficacy, determination of effect in both sexes, estrogen's actions on the level of ischemic insult (i.e., flow-mediated effects) and on reperfusion, and efficacy in young mature versus postmenopausal animals.
The effect of 17β-estradiol treatment on infarct size in rodent models of experimental stroke
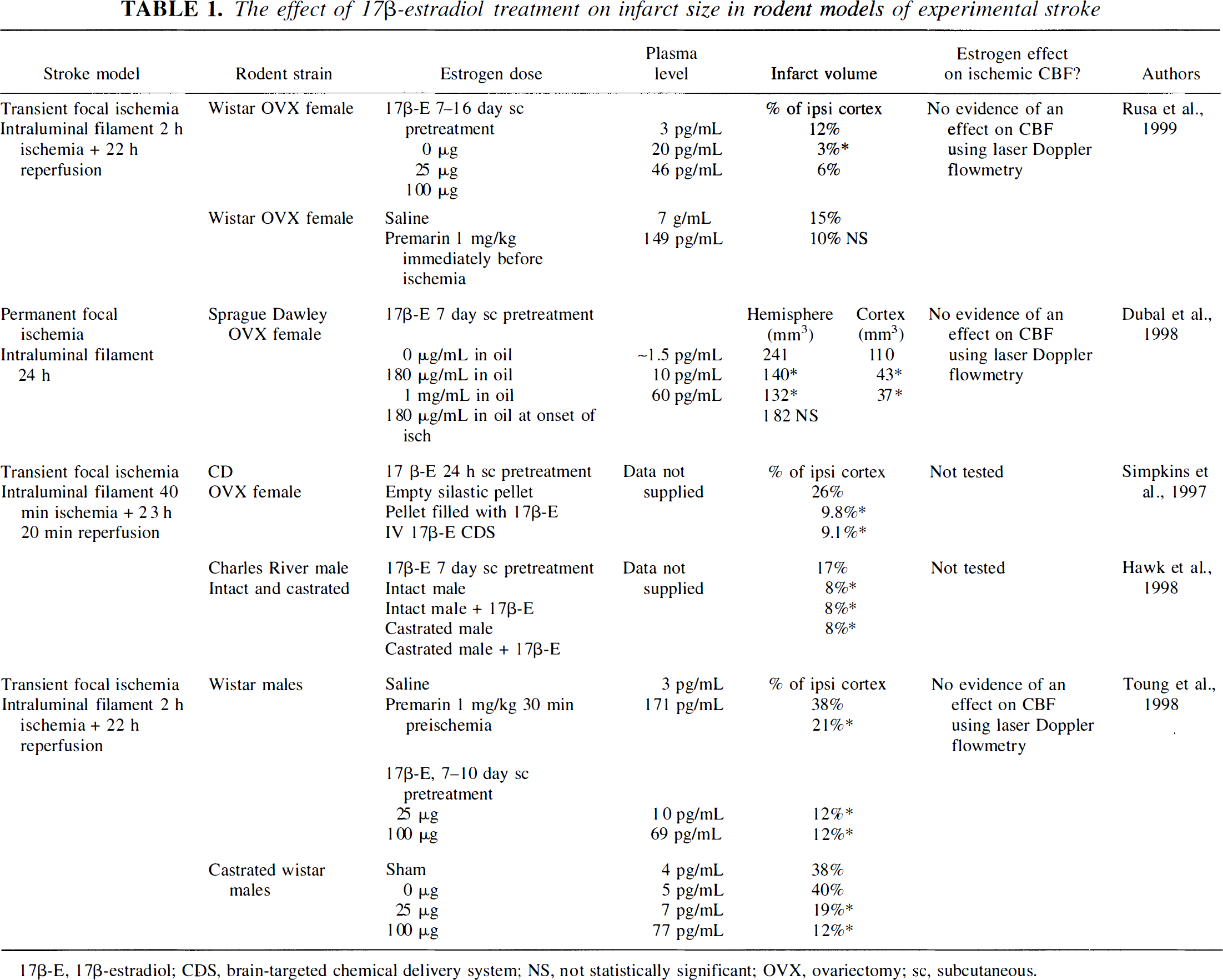
17β-E, 17β-estradiol; CDS, brain-targeted chemical delivery system; NS, not statistically significant; OVX, ovariectomy; sc, subcutaneous
In most studies, 17β-estradiol has been the agent chosen for the replacement paradigm, in part because the steroid is the predominant, biologically active estrogen in mammals. Since systemic estrogen levels were not measured or reported in all articles reviewed, some inferences are needed to compare treatment effects from physiologic versus supraphysiologic doses. For point of reference, physiologic plasma 17β-estradiol levels, as reported in the rat, range between 10 and 30 pg/mL with peak levels during proestrus of 80 to 140 pg/mL. (Smith et al., 1975; Butcher et al., 1974; Nequin et al., 1979).
Effects of estrogen restoration have been most comprehensively studied in OVX female rodents (Table 1). Chronic treatment (with subcutaneous implants or daily injections) and acute treatment (with one-time infusion or injection) have been used. Investigations address efficacy in improving histologic, hemodynamic, or behavioral outcomes after transient MCAO (Simpkins et al., 1997; Zhang et al., 1998; Dubal et al., 1998; Rusa et al., 1999) or global forebrain ischemia (Hurn et al., 1995; Kondo et al., 1997; Pelligrino et al., 1998; Wang et al., 1999).
Histologic outcome
Women. In studies of chronic estrogen replacement after ovariectomy (approximately 1 week before ischemia), which produced physiologic plasma steroid levels, tissue damage was uniformly decreased in global forebrain ischemia (Pelligrino et al., 1998) and focal ischemia (Table 1) (Dubal et al., 1998; Rusa et al., 1999). However, the therapeutic dose range was narrow, with beneficial effects decreasing as the dose and plasma estradiol level increased upward within the physiologic range (Pelligrino et al., 1998; Rusa et al., 1999). In contrast, high-dose implants of 17β-estradiol placed 24 hours before MCAO reduced infarction volume, suggesting that short-term exposure to pharmacologic doses also can be beneficial (Table 1) (Simpkins et al., 1997). In addition, high doses of estrogen (plasma levels up to 25 to 28 times greater than in OVX females) have been shown to improve cognitive performance in the Morris water maze in OVX gerbils after bilateral carotid artery occlusion (Kondo et al., 1997).
Two studies indicate that estrogen must be delivered chronically before ischemia, rather than as a single injection or subcutaneous implant at ischemic onset, to be effective in OVX females (Table 1) (Dubal et al., 1998; Rusa et al., 1999). However, the study by Simpkins et al., (1997) illustrates that a high dose 17β-estradiol injection before MCAO or at the onset of reperfusion can effectively reduce infarction (Table 1). A subsequent study from this same group demonstrates that 17β-estradiol injection within a more physiologically relevant range also produced a dose-dependent reduction in infarction, even when given at the onset of reperfusion (Zhang et al., 1998).
Men. An important question is whether exogenous estrogen also protects the male brain, interacting with tissue not ordinarily exposed to cyclically high concentrations of female reproductive steroids. Interactions between native testosterone and estrogen treatment have been examined, although in a limited manner, with some conflicting results. Several investigations in the male rat and gerbil confirm that preischemic estrogen, administered either chronically or as a single injection, reduces tissue damage and functional deficits after both global (Chen et al., 1998; Sudo et al., 1997) and focal (Hawk et al., 1998; Toung et al., 1998) ischemic insults.
After transient forebrain ischemia, 17β-estradiol, given over a wide range of concentrations, protects CA1 neurons and improves performance in passive avoidance tasks in male gerbils (Sudo et al., 1997). However, this study demonstrates a ceiling and loss of efficacy at extreme doses (1.25 μg/day for 7 days through the lateral ventricle) and increasing mortality in some cases (Sudo et al., 1997), but Chen et al. (1998) report no mortality problems with high doses (30 μg, intracerebroventricular) and suggest that high doses of 17β-estradiol are required for neuroprotection (3 and 10 μg were not protective).
Doses yielding plasma estradiol levels that are physiologic or supraphysiologic in the female rat are equally efficacious in males exposed to MCAO. Both chronic (7- to 10-day subcutaneous) and acute (30-minute intravenous) pretreatments significantly reduce infarct volume in intact male rats (Table 1) (Toung et al., 1998; Hawk et al., 1998), suggesting that nontranscriptional mechanisms of action may be important in the male brain.
Estradiol treatment combined with castration is more complex. In one study, where castration did not influence infarct size, the addition of estrogen dose-dependently reduced the lesion to a similar extent as reported in non-castrated males with normal testosterone levels (Toung et al., 1998). However, in another study, castration per sereduced infarct size significantly, with estrogen pretreatment producing no further reduction in infarct size (Table 1) (Hawk et al., 1998). Simultaneous testosterone and estradiol treatment in castrates resulted in an ischemic lesion intermediate between animals treated with either hormone alone (Hawk et al., 1998). Because estradiol treatment was accompanied by a decrease in testosterone levels in intact males, the authors conclude that estrogen protects the male brain by this means.
Further studies are required to evaluate estrogen-testosterone interactions in cerebral ischemia and to reconcile the experimental differences in the literature. The efficacy of estrogen administered during or after the ischemic insult has not been reported in male animals. Nevertheless, the data clearly show that estradiol reduces postischemic brain injury and functional deficits in intact male and in female rats.
Senescent animals. We also have examined stroke outcomes in 16-month-old reproductively senescent male and female Wistar rats to test the hypothesis that gender differences are lost as endogenous hormone levels wane (Hurn et al., 1998; Alkayed et al., 2000). In aged animals, damage to the striatum and cortex after MCAO (2 hours of occlusion, 22 hours of reperfusion) was equivalent in females and males (Fig. 6A). This observation is consistent with the concept that endogenous ovarian steroids are neuroprotective in stroke and the finding of higher stroke rates in postmenopausal versus premenopausal women. When estrogen was restored in aged females using subcutaneous 17β-estradiol implant (25 μg, 7 to 10 days' duration), infarction size was strikingly reduced in all brain regions examined, although the effect was particularly prominent in cortex (Fig. 6B). However, the neuroprotective mechanism is unlikely to be flow enhancement, since intraischemic regional CBF, as measured by 14C iodoantipyrine, was not altered by estrogen treatment. These experiments in aged female Wistar rats demonstrate that the benefit of estrogen replacement is not restricted to young animals artificially deprived of ovarian steroid, since physiologic doses of 17β- estradiol also are neuroprotective in the postmenopausal female brain.
To summarize these animal studies, estrogen is an effective neuroprotectant in both male and female brain, although the requisite dose and duration of steroid exposure may differ between the sexes. Although these findings suggest a plausible and novel clinical use for estrogen, numerous experiments are needed to determine fully the steroid's therapeutic window of activity and optimal dose-response relationships in stroke.
POTENTIAL MECHANISMS OF ESTRADIOL-MEDIATED NEUROPROTECTION
Vasodilation and improved CBF?
Native estrogens or exogenous 17β-estradiol could act as direct vasodilators or through vasodilatory mediators during ischemic stress. Numerous studies document vasomotor effects of estrogen under nonischemic conditions, and 17β-estradiol is a dilator of most vascular beds (for reviews see Mendelson and Karas, 1994; White et al., 1995). Early studies using 133Xe inhalation methodologies show that absolute regional CBF was higher in women versus men, particularly in gray matter (Davis et al., 1983; Shaw et al., 1984; Rodriguez et al., 1988), and that gender differences disappeared by 50 to 60 years of age (Davis et al., 1983; Shaw et al., 1984). A recent functional magnetic resonance imaging study in normal volunteers demonstrates a significantly greater rise in regional CBF in women compared with men during focal physiologic neural activity (Kastrup et al., 1999). In normal subjects aged 21 to 58 years, women also demonstrate greater cerebral vasodilatory capacity to CO2 than men assessed by bilateral transcranial Doppler sonography (Kastrup et al., 1997). Similarly, enhanced cerebral vasodilation to acetazolamide has been reported in women versus men (Karnik et al., 1996). Estrogen replacement may improve perfusion in postmenopausal women with and without known cerebrovascular disease. In one study of 14 postmenopausal women, ERT with oral conjugated estrogens increased whole cerebral and cerebellar flow, as measured by single-photon emission computed tomography (Ohkura et al., 1995). A larger, retrospective study of women 40 to 90 years of age used 133Xe inhalation to assess gray matter blood flow (Funk et al., 1991). The ERT modestly improved perfusion and cognitive test performance among women with a history of transient ischemic attack or reversible ischemic neurologic deficit (but had no effect on postmenopausal women with no history of cerebrovascular disease).
In experimental research, direct evidence of estrogenic vasoactivity in vivo appears to be model specific and in some cases difficult to demonstrate. In basal, nonischemic conditions, little evidence supports a tonic influence for estradiol on CBF. Animal studies demonstrate similar CBF values between the sexes in both anesthetized and conscious states (Hurn et al., 1995; Alkayed et al. 1998a; Carswell et al., 1999c; Goldman et al., 1976). In conscious rodents, ovariectomy per se does not influence basal CBF (Holshneider and Scremin, 1998), and under anesthesia, similar basal CBF values are reported in intact, OVX, and OVX plus estradiol treatment groups (Wang et al., 1999). Estradiol supplements did not influence basal CBF in intact female rabbits (Hurn et al., 1995), and in a study on OVX sheep, infusions of physiologic doses of 17β-estradiol did not influence CBF acutely (2 hours), although improved flow was evident after 3 to 6 days of treatment (Magness et al., 1998). Finally, high-dose (10 μg) intravenous estradiol has been measured at one time point (10 minutes after administration) in conscious rats and was found to produce global increases in CBF in both sexes (Goldman et al., 1976).
In ischemic conditions, estrogen-mediated improvement in CBF has been most clearly identified in models of global or forebrain ischemia (Table 2). In near-complete global cerebral ischemia, residual intraischemic blood flow was greater in cerebral hemispheres and cerebellum of 17β-estradiol-treated rabbits compared with untreated females or males (Hurn et al., 1995). In addition, hyperemia was less during early reperfusion. However, the significance of enhanced perfusion to improved tissue outcome was not established in this non-survival protocol. Pelligrino et al. examined a range of 17β-estradiol repletion doses in OVX rats exposed to right carotid occlusion with hemorrhagic hypotension and 72 hours of reperfusion (Pelligrino et al., 1998). Here, 17β-estradiol produced a dose-dependent preservation of intraischemic cortical perfusion as assessed by laser Doppler flowmetry (LDF), accompanied by preserved EEG power, as well as improved histopathologic and neurologic outcome. Importantly, efficacious effects were lost at supraphysiologic doses (0.5 mg/kg/day) of 17β-estradiol. This study offers insight into mediators by which estrogen could act in the ischemic cerebral circulation. First, 17β-estradiol's ability to preserve the intraischemic flow signal was blocked by treatment with a selective neuronal nitric oxide synthase (NOS) antagonist. Second, cortical Ca2+-dependent NOS activity was markedly suppressed in OVX animals and restored by estrogen treatment at all doses. However, restoration of NOS activity with high-dose 17β-estradiol treatment did not result in preservation of intraischemic vasodilation, suggesting that potential estrogen-NOS interactions are specific to low estrogen concentrations.
Consequences of ovariectomy and/or 17β-estradiol treatment on basal and ischemic CBF
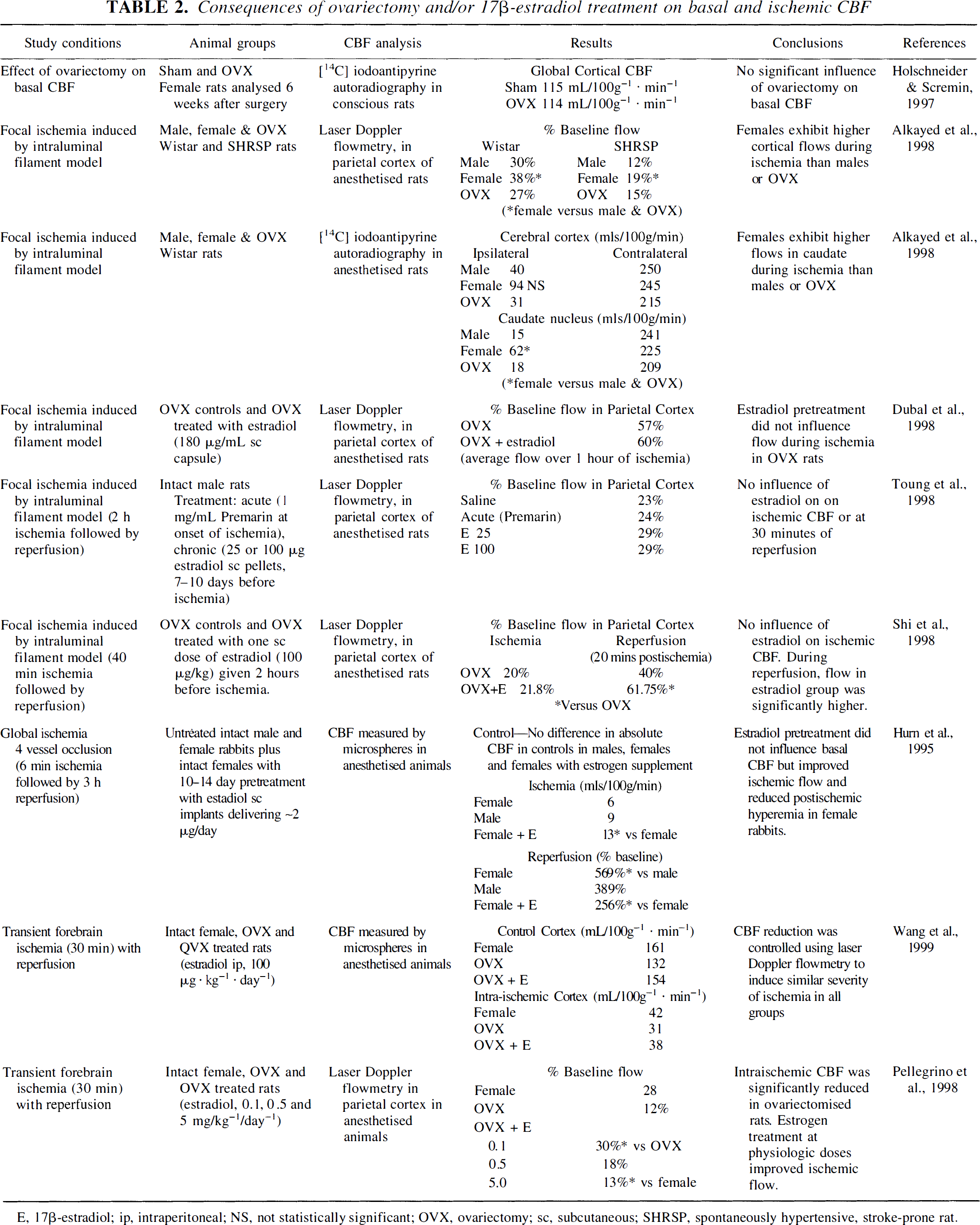
E, 17β-estradiol; ip, intraperitoneal; NS, not statistically significant; OVX, ovariectomy; sc, subcutaneous; SHRSP, spontaneously hypertensive, stroke-prone rat.
The situation is much less clear for focal cerebral ischemia. Three studies monitored CBF by cortical LDF during ischemia in OVX and 17β-estradiol-treated OVX rats (Rusa et al., 1999; Dubal et al., 1998; Shi et al., 1998). Although 17β-estradiol significantly reduced infarct volume, there was no evidence of 17β-estradiolmediated improvement in ischemic CBF in these studies. However, caution must be exercised in interpreting these data, since flow is monitored from only a single discrete point on the cortical surface with this technique and, therefore, flow-mediated effects of estrogen may have been missed. Shi et al. (1998), however, report a significant improvement in CBF at 1 hour of reperfusion in the 17β-estradiol-treated animals (Table 2). Toung et al., (1998) examined the influence of acute and chronic (low- and high-dose) 17β-estradiol treatment on ischemic CBF in male rats, again with LDF. Neither acute nor chronic treatments influenced CBF during ischemia or at 30 minutes of reperfusion.
Turning to native estradiol, Alkayed et al. (1998) measured intraischemic CBF in males and in intact and OVX females in both normotensive (Wistar) rats and SHR-SP (Table 2). In both rodent strains, intraischemic CBF (measured by LDF) was significantly higher in females compared with males or OVX females. Further, using [14C] iodoantipyrine autoradiography in Wistar rats to provide absolute values of flow throughout the middle cerebral artery territory, a significant elevation in ischemic flow was demonstrated in the caudate nucleus of intact females compared with the other two groups (Table 2, Fig. 7). Carswell et al. (1999d) also examined intraischemic CBF autoradiographically in conscious SHR-SP to explore the mechanism of reduced infarct volume in proestrus (highest estradiol levels) compared with metestrus (lowest estradiol levels) in this strain. However, neither measurement of CBF in specific neuroanatomic regions nor frequency distribution analysis throughout the middle cerebral artery territory revealed any difference in ischemic CBF between the two stages of the estrus cycle (Carswell et al., 1999c). Therefore, in this instance, protection of brain tissue in proestrus was not mediated by flow enhancement.
In summary, the flow enhancement effects of estradiol have been demonstrated convincingly in global and forebrain ischemia, but only limited evidence is available in focal cerebral ischemia. This may result from limitations of the CBF methodologies or may suggest that for focal cerebral ischemia, the protective effects of estradiol are nonvascular. Whether endogenous estrogens or pharmaceutical preparations alter CBF or microvascular dysfunction during reperfusion from an ischemic insult has not been well studied. Furthermore, there are numerous potential mechanisms by which estrogen acts directly on neurons or glia. Available data for vascular and selected parenchymal mechanisms are summarized in the subsequent sections, as well as potential links between the steroid hormone's cognate receptors and neuroprotection.
ESTROGENIC SIGNALING AND ROLE OF ESTROGEN RECEPTORS IN CEREBRAL ISCHEMIA
Early autoradiographic studies using [3H]-estradiol injections clearly demonstrate numerous sites of nuclear accumulation and retention of radioactivity in brain and spinal cord of both sexes (Stumpf, 1968). Furthermore, estradiol binding sites have been demonstrated in vascular endothelium, and functional estrogen receptors (ER) are present in vascular smooth muscle. Consequently, it is not surprising that estrogen exhibits significant biological actions in blood vessels, neurons, and glia, many of which are likely relevant to its antiischemic properties. Recent evidence emphasizes that estrogenic steroids use diverse signaling mechanisms to produce biological effects. These include the following: (1) ER-linked genomic actions; (2) non-ER-linked transcriptional mechanisms that use generalized signaling molecules; (3) ER-dependent, nontransciptional mechanisms; and (4) cell membrane associated activity, which clearly does not depend on messenger RNA (mRNA) transcription and protein synthesis (Fig. 8). Classically described estrogen signaling occurs through a nuclear ER that acts as a ligand-activated transcription factor, a protein complex that binds to a hormone-responsive element within genomic DNA and alters transcription of mRNA and cognate protein expression. Therefore, activation of classic ER signaling results in enhanced or suppressed modulation of target gene transcription efficiency, and there are numerous examples of estrogen's genomic actions in steroid-responsive tissue. Furthermore, hormone response elements for estrogen, or consensus palindromic DNA sequences, are present on many mammalian genes not necessarily associated with reproductive function, for example, the gene for constitutive NOS (Venema et al., 1994).
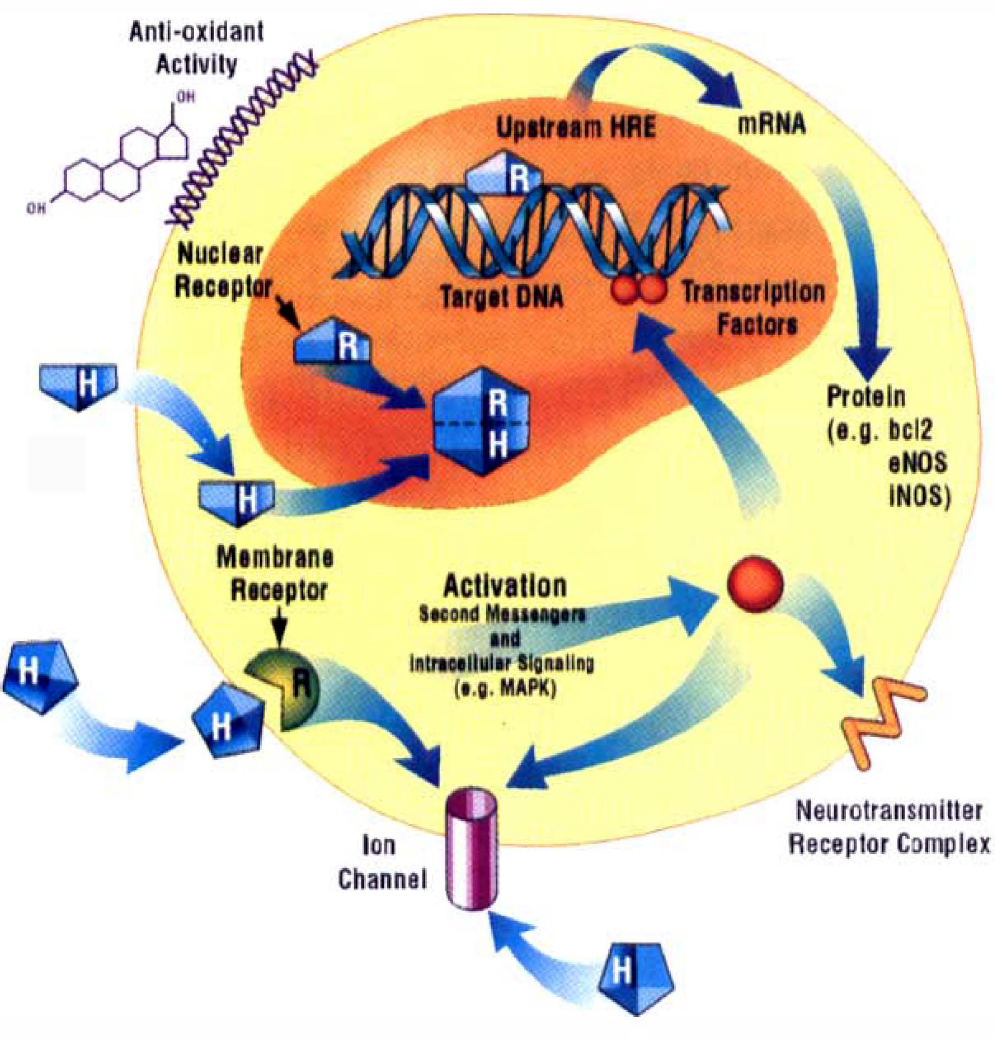
Estrogen's cellular signaling targets and transduction mechanisms. Estrogenic steroids use diverse mechanisms to produce biological effects, and its neuroprotective properties probably are multifactorial. Classic estrogen signaling (H) occurs through a nuclear receptor that acts as a ligand-activated transcription factor (RH), a protein complex that binds to a hormone-responsive element (HRE) within genomic DNA and targets transcription of mRNA and cognate protein expression. The result is enhanced or suppressed efficiency of gene transcription. There are numerous examples of estrogen's ability to upregulate gene expression, including selected members of the nitric oxide synthase (NOS) and bcl-2 families. Recently, estrogen's activation of plasma membrane hormone receptors and downstream intracellular signaling has been postulated (e.g., through mitogen-activated protein kinase). Such events initiate generalized signaling pathways to the nucleus or to membrane-localized effectors. Evidence also indicates that the hormone can act directly at neurotransmitter receptor complexes or at ion channels (e.g., voltage-dependent calcium channels), resulting in altered neuronal current conductance or transcellular ion flux. Crosstalk between nuclear activation and membrane-associated events is likely. Last, estrogen has important non-cell type-specific actions, such as antioxidation and conservation of endogenous free radical scavenging agents.
Other signaling mechanisms have been described that differ from this classic schema. For example, estrogen may act through the cAMP pathway to regulate cAMP-mediated gene transcription or to alter transcription of estrogen-regulated genes (Katzenellenbogen, 1996; Watters and Dorsa, 1998). Furthermore, although estrogen has been depicted as a transciptional hormone, there are known nontransciptional effects that occur through rapid cell membrane-associated activity with (Shaul et al., 1999; Goetz et al., 1999) or without input from known ER (Tischkau and Ramirez, 1993; Mermelstein et al., 1996). Some of these rapid actions involve membrane and intracellular receptors, which are coupled by second messengers to ion channels and to neurotransmitter receptors (Mermelstein et al., 1996; Ross et al., 1999), or involve membrane-localized enzymes such as endothelial NOS (Goetz et al., 1999; Chen et al., 1999). Comprehensive reviews of these topics are found elsewhere (Farhat et al., 1996; Ross et al., 1997; Revelli et al., 1998; Woolley and Schwartzkroin et al., 1998).
Few data distinguish which of these diverse signaling mechanisms are used by estrogen in its protective actions during stroke and cerebral ischemia. Limited data address the relative importance of ER or transcriptional dependence. Two ER subtypes have been characterized: ERα and, recently, an ERβ subtype (Kuiper et al., 1996, 1997; Mosselman et al., 1997). These isoforms differ in the C-terminal ligand binding domain and in the N-terminal trans-activation domain. There are no known subtype-selective antagonists; however, general ER antagonists, such as tamoxifen and the steroid ICI family (e.g., ICI 182,780), block estrogen's actions in a tissue-specific manner by inhibiting ER-initiated gene activation (Kangas, 1992). Most clues as to the role of ER come from paradigms that used general ER antagonists or 17α-estradiol, a relatively inactive estrogen that does not bind to known ER. Furthermore, some inferences about lack of ER relevance have been drawn from positive results obtained only in the presence of pharmacologic estrogen doses and concentrations that saturate receptor binding (e.g., micromolar concentrations). With regard to estrogen and cerebral ischemia, several in vitro investigations use ER antagonists or protein synthesis inhibitors to evaluate ER-initiated transcriptional activity. These reports are summarized in subsequent sections addressing specific vascular or parenchymal mechanisms. In addition, several in vivo studies address the issue of whether estrogen's neuroprotective effects in stroke are mediated by ER ((Simpkins et al., 1997; Hurn et al., 1998, Sawada et al., 1999). Using a rat MCAO model, Simpkins et al. report that pretreatment with subcutaneous implants containing 17α-estradiol reduced infarction volume equivalently to the same dose of the 17β isomer. Notice that large doses of both 17α- and β-estradiol were used, and although plasma steroid levels were not reported, it is likely that supraphysiologic concentrations resulted. Nevertheless, the efficacy of 17α-estradiol suggests that estrogen's neuroprotection was not likely mediated by nuclear ER signaling. This is an important finding when taken in context of pharmacologic neuroprotection. Further studies are needed to demonstrate if estrogen's effects are receptor independent over the range of estradiol doses currently shown to be effective in experimental stroke. Two further studies evaluate whether endogenous estrogen acts through ER mechanisms in transgenic and wild-type mice treated with MCAO (Hurn et al., 1998a, 1998b; Sawada et al., 1999). First, chronic treatment with the pure ER antagonist, ICI 182,780, increased infarction volume in striatum by approximately 30% in female C57B1/6J mice compared with vehicle-treated controls. No effects on end-ischemic CBF were observed in any brain region. The deleterious effect of ICI 182,780 was notably restricted to striatum, and the distribution of ER, either subtype α or β, within this region is under active investigation (Couse et al., 1997; Shughrue et al., 1997; Osterlund et al., 1997). Further, loss of estrogen-ER signaling was limited to intact females with circulating native estrogen, and no effect was observed in age-matched males. In a companion study, outcome from MCAO was evaluated in transgenic mice deficient in the α receptor subtype (Korach, 1994; Hurn et al., 1998). Infarction volume was not exacerbated in these mice compared with wild-type females, suggesting that ERα is not important in estrogen's antiischemic properties. Whether the recently identified ERβ subtype plays a role in stroke remains to be tested.
In summary, in vivo data examining the role of ER subtypes and the efficacy of pharmacologic estrogen preparations are limited, particularly at doses that simulate the physiologic condition. Estrogen receptors may play a regionally restricted role in protecting the female brain with endogenous estrogen during experimental stroke; however, the relative receptor subtype distribution within the region of interest is likely a determining factor.
VASCULAR MECHANISMS
Much evidence obtained in noncerebral vessels suggests that estrogen improves vasodilatory capacity and alters vessel responses to several well-studied vasodilator and vasoconstrictor agents (for previous reviews see Mendelsohn and Karas, 1994; White et al., 1995; Duckles et al., 1996; Farhat et al., 1996). Data in cerebral vessels are limited, but those available support this concept. The precise signaling mechanisms by which estrogen alters blood flow or vascular reactivity are potentially diverse. Estrogen receptors have been located in vascular smooth muscle (Harder and Coulson, 1975; Karas et al., 1994) and in endothelial cells (Colburn and Buonassisi, 1978). Therefore, transcriptional mechanisms may be involved. One prevailing hypothesis is that estrogen amplifies release of vasodilators, in part because there are estrogen response elements on the genes for endothelial and inducible NOS and for prostacyclin synthetase (Xie et al., 1992, Chartrain et al., 1994; Venema et al., 1994; Mendelsohn and Karas, 1994). Rapid vasomotor effects, independent of gene activation, have been reported both in vivo and in vitro, mediated by intracellular second messengers such as Ca2+ and the cyclic nucleotides (for review, see Farhat et al., 1996a, 1996b). Crosstalk between membrane-mediated events and nuclear receptor activation also is likely within the vasculature (for reviews, see Mendelsohn and Karas, 1994; Baysal and Losordo, 1996).
The coronary, uterine, aortic, and peripheral vessel literature provides evidence that estrogen influences multiple aspects of vessel function and disease by targeting vascular smooth muscle cells, endothelium, and platelets. There is evidence for the following: reduction of vascular smooth muscle proliferation under baseline conditions (Moraghan et al., 1996) and in response to balloon injury (Sullivan et al., 1995); prevention of neointimal thickening after vessel injury (White et al., 1997; Oparil et al., 1997) and atheromatous changes over time (Baron et al., 1998); maintenance of endothelial structure, elaboration of growth factors (Schnaper et al., 1996; Albuquerque et al., 1998), and reduction of endothelial apoptosis (Alvarez et al., 1997); reendothelialization after injury (White et al., 1997); and activation of vascular smooth muscle K+ channels (White et al., 1995; Wellman et al., 1996; Darkow et al., 1997) and inhibition of vascular smooth muscle contractility by blocking Ca2+ influx through voltage-dependent calcium channels and enhancing efflux (Collins et al., 1993; Shan et al., 1994; Freay et al., 1997; Prakash et al., 1999). Increased production and activity of vasoactive endothelial-derived products with estrogen exposure have been widely reported in cultured endothelium, intact vessels, and in systemic plasma assays. The most prominent products are nitric oxide (NO) (Hayashi et al., 1992; Rosselli et al., 1994; Hayashi et al., 1995; Hishikawa et al., 1995; Bell et al., 1995; MacRitchie et al., 1997; Caulin-Glaser et al., 1997; Vagoni et al., 1998; Knot et al., 1999) and eicosanoids, specifically prostacyclin (Mikkola et al., 1995, 1996; Myers et al., 1996; Bolego et al., 1997; Jun et al., 1998). Most of these reports agree that the elaboration of these vasodilators is mediated through the ER. Lastly, estrogen availability reduces leukocyte adhesion in cerebral venules in vivo(Santizo and Pellegrino, 1999), alters platelet aggregation in cerebral microvessels (Rosenblum et al., 1985), and reduces vasomotor responsivity to autogenous platelets in coronary vessels (Miller et al., 1999). There are gender differences in platelet release of cyclooxygenase metabolites of arachidonic acid and serotonin; estrogen deficiency results in enhanced platelet release of serotonin, prostacyclin, and thromboxane A2(Miller et al., 1999).
Lack of endogenous estrogen impairs vascular reactivity, at least in noncerebral vessels. For example, endothelium-dependent vasodilation is impaired in aortic and femoral tissue from OVX animals and restored with chronic exogenous estrogen (Gisclard et al., 1987). Furthermore, estrogen partially restores dilator responses to acetylcholine in atherosclerotic monkey coronary arteries, whereas ovariectomy produces paradoxic constriction (Williams et al., 1990). Ethinyl estradiol administration acutely attenuates abnormal coronary artery responses to acetylcholine in women (Reis et al., 1994), and estrogen also modulates selected vasoconstrictor stimuli and may therefore alter the net balance of vasomotor signals toward dilation under some conditions. Examples include estradiol's depression of vasoconstriction to endothelin-1 (Jiang et al., 1992) and to the thromboxane mimetic U46619 in coronary vessels (Thompson and Weiner, 1997). Similarly, estrogen depresses adrenergic responsiveness, that is, vasoconstriction to norepinephrine in noncerebral vascular beds by mechanisms involving alterations in adrenergic receptor affinity and expression, modification of catecholamine release, uptake, and neural efflux (Hamlet et al., 1980; Colucci et al., 1982; McNeill et al., 1996; Li et al., 1997; McNeill et al., 1999), or by decreasing adrenergic sensitivity by increasing the basal release of relaxing factors from the endothelium (Meyer et al., 1997). Whether similar effects occur in the cerebral circulation is unclear. Although estrogen had no effect on noradrenergic perivascular innervation in rabbit basilar artery, chronic treatment reduced serotonin immunofluorescence in this preparation, and estrogen withdrawal increased vasoreactivity to serotonin in basilar artery rings (Dhall et al., 1988; Futo et al., 1992).
Although there are substantial data that estrogen increases eicosanoid production in the vasculature, it is not clear if this is an important vasodilator mechanism under physiologic or ischemic conditions. Physiologic concentrations of 17β-estradiol increase prostacyclin production and biosynthetic activity in aortic smooth muscle culture (Chang et al., 1980; Wakasugi et al., 1989) and in intact aortic rings as measured by elevated levels of 6-ketoprostaglandin F1α (Wakasugi et al., 1989; Bolego et al., 1997). Similarly, estradiol increases 6-ketoprostaglandin F1α production in human and animal aortic endothelial cells specifically at physiologic concentrations (Myers et al., 1996), and the effect can be prevented by tamoxifen treatment (Mikkola et al., 1995, 1996). Further, cyclooxygenase-1 is induced by estrogen in fetal pulmonary artery endothelium and by pregnancy in uterine artery, again inhibitable by ER antagonists (Jun et al., 1998; Janowiak et al., 1998). However, estradiol also may stimulate production of contractile prostanoids, since the steroid augments endothelium-dependent contraction to arachidonic acid in rabbit aorta (Miller and Vanhoutte, 1990). The possibility that estrogen acts on more than one eicosanoid pathway has not been well studied, and it is likely that interaction among endothelium-derived mediators is complex (Barber and Miller, 1997). Cytochrome P-450 products and free radicals may be linked as mediators of estrogen's effects on vascular behavior (Harder et al., 1994; Hayashi et al., 1992).
In a variety of experimental models, gender and estrogen effects on the endothelium have been linked to NO. Estrogen increases NO availability, possibly by up-regulating Ca2+ activated, endothelial NOS (Weiner et al., 1994) or inducible Ca2+-independent NOS (Binko and Majewski, 1998), by increasing substrate availability for NO formation (Kauser and Rubanyi, 1997), or by limiting the inactivation of NO as a substrate in other reactions (Arnal et al., 1996; Kauser and Rubanyi, 1997). In noncerebral vessels, at least one of estrogen's vasomotor mechanisms involves NO/cGMP/protein kinase G signaling (Stice et al., 1987; Magness et. al, 1991; White et al., 1995; Wellman et al., 1996; Darkow et al., 1997). Further, data from coronary vessels suggest that estrogen's downstream effector in this signal transduction pathway is vascular smooth muscle voltage-activated K+ channels. White et al. have shown that 17β-estradiol relaxes coronary arteries in vitro in an endothelium-independent manner, opening large conductance, calcium and voltage-activated K+ channels (BKCa) through cGMP-dependent phosphorylation (White et al., 1995). Further, gender-specific myogenic tone in coronary vessels is estrogen- and endothelium-dependent and can be inhibited by ER antagonists such as tamoxifen (Wellman et al., 1996). This latter study also offers evidence that the signaling pathway involves activation of BKCa in vascular smooth muscle cells by protein kinase G. Notice that the protein kinase G-BKCa mechanism is a known signaling pathway in brain (Yao et al., 1995) and cerebral vascular smooth muscle (Braydon and Nelson, 1992; Robertson et al., 1993) and can serve as a “vasomotor effector” common to both endothelium and smooth muscle derived vasodilators. From the limited data available, high conductance, calcium-activated K+ channels did not mediate estrogen's effect in the cerebral circulation, in contrast to findings reported in coronary vessels (Wellman et al., 1996). Passive mechanical properties were similar among vessels, regardless of estrogen availability, suggesting that estrogen did not alter vessel wall composition in these young, adult animals. Myogenic tone differences induced by estrogen also may be vessel size dependent (Geary et al., 1996).
Ovariectomy does reduce the pial microvascular response to topical acetylcholine in situ but does not alter vasodilation to the direct NO donor S-nitroso-N-acetyl-penicillamine, suggesting a lack of direct effect on vascular smooth muscle (Pelligrino et al., 1997). Chronic 17β-estradiol treatment also elevates basal cerebral microvascular cGMP content, although the effect is restricted to high doses of pharmacologic rather than physiologic relevance (Palmon et al., 1998). Geary et al. examined pressurized cerebral resistance vessels (280- to 300-μm diameter) and documented essential gender differences in myogenic tone (Geary et al., 1998). Furthermore, 17β-estradiol treatment was associated with reduced tone and increased vascular distensibility, acting by a NO-dependent mechanism. In the presence of NG-nitro-L-arginine methyl ester, middle cerebral artery response to increased transmural pressure in female or estrogen-treated tissue mimicked that of estrogen-deficient tissue from males or OVX females. In summary, estrogen is well established as a vasodilator in many regional beds, and available data suggest that the steroid has vascular activity within the cerebral circulation. Such activity may include direct vasodilation, enhanced vasodilatory capacity through increased elaboration of NO, prostacyclin- or other endothelium-derived mediators, and reduced sensitivity to selected vasoconstrictor stimuli such as endothelin-1, norepinephrine, and thromboxane. Although the mechanisms remain to be elucidated, in certain circumstances endogenous estrogen can partially restore intraischemic CBF. Limited data are available that evaluate interactions between pharmacologic estrogen preparations and vascular mechanisms of neuroprotection.
PERFUSION-INDEPENDENT MECHANISMS OF PROTECTION
The ability of estrogen to protect neurons by perfusion-independent mechanisms is illustrated in both tissue culture experiments (Goodman et al., 1996; Regan et al., 1997) and in in vivo studies, where the ischemic insult is controlled to produce the same level of ischemia in control, OVX, and estrogen-treatment groups (Wang et al., 1999). Estrogen has direct and rapid effects on neuronal tissue. For example, synaptic architecture within areas such as hippocampus changes with the estrous cycle and can be altered in less than 24 hours by exogenous estradiol (Woolley et al., 1992). Complementary fluctuations in the volume of astrocytic processes and synaptic numbers also occur in response to ovarian steroids (Klintsova et al., 1995), and ER have been localized on glial cells (Azcoitia et al., 1999). Therefore, perfusion-independent mechanisms involving neurons or glia are likely involved in estrogen's ability to reduce ischemic brain injury. Potential mechanisms include antioxidant actions, induction of neuroprotective gene products such as bcl-2 and neurotrophic growth factors, and nontranscriptional modulation of excitatory neurotransmission and consequent glutamate toxicity (Fig. 8).
Antioxidant activity
Many estrogens have well-established and potent lipid antioxidant activity, particularly the catechol estrogens (2-hydroxy estrone and 2-hydroxy estradiol) and, to a lesser extent, phenolic estrone and 17β-estradiol (Nakano et al., 1987; Mukai et al., 1990; Mooradian, 1993). Culmsee et al. (1999) have demonstrated that Fe2+-induced levels of reactive oxygen species and neurotoxicity were significantly attenuated by low micromolar concentrations of 17β-estradiol and 2-hydroxy estradiol in chick embryonic neurones, even in the presence of the ER antagonist tamoxifen. Given these data, several investigators have hypothesized that estrogen decreases oxygen radical-induced brain injury by direct radical scavenging action or through preservation of endogenous antioxidants. The distinction between these two different mechanisms appears to be linked to steroid concentration within the experimental system. In vitro studies in hippocampal cell lines, primary hippocampal culture, and organotypic slices have demonstrated that estrogen treatment at supraphysiologic concentrations (i.e., greater than 10−9 mol/L) reduced neuronal death after prooxidant and amyloid β-peptide exposure (Goodman et al., 1996; Behl et al., 1997). Further, 17β-estradiol and equilin (an equine mixture of estrogens) protected against DNA damage induced by hydrogen peroxide or arachidonic acid (Tang and Subbiah, 1996). Direct radical scavenging action thus may predominate in estrogen's neuronal protection at pharmacologic concentrations, since this activity was observed only at high estradiol concentrations in male gerbils treated with forebrain ischemia. However, sparing of hippocampal neurons was observed at near-physiologic doses (Sudo et al., 1997). Others report good protection against trophic factor withdrawal and amyloid β-peptide by estradiol at micromolar concentrations in PC12 cells. This was accompanied by a suppression of mitochondrial reactive oxygen species and preservation of mitochondrial function (Mattson et al., 1997). Pretreatment of cortical synaptosomes with 17β-estradiol also suppressed membrane lipid peroxidation induced by amyloid β-peptide and iron (Keller et al., 1997). In contrast, superoxide anion production was reduced in aortic endothelial cells by ethinylestradiol treatment with a physiologically relevant EC50 of 0.1 nmol/L (Arnal et al., 1996). This effect resulted in significant preservation of endothelium-derived NO bioactivity and was completely prevented by ER antagonists. Therefore, the capacity of estrogen to prevent oxidative damage may differ among the different cell types exposed to the intense ischemia associated with a stroke.
Effective protection, associated with a requirement for phenolic A ring structure, also has been demonstrated at physiologically relevant concentrations (Green et al., 1997) and with decreased lipid peroxidation (Gridley et al., 1997) in neuroblastoma cell lines. Subsequent work from this same group indicates that estrogen's protection at physiologic levels in cortical neurons relies on synergism with reduced glutathione by an ER-independent mechanism (Gridley et al., 1998; Green et al., 1998). Estrogen or glutathione had no independent effect on cell survival; however, the EC50 for estradiol was favorably shifted in the presence of glutathione (Gridley et al., 1998). Conservation of endogenous antioxidants by estrogen is a recurring finding in animal studies. In the intact female animal or in females treated with physiologic hormone replacement, vitamin E and ascorbate are spared during ischemic stress, whereas enhanced oxidant stress and depletion of antioxidants was observed in males and OVX animals (Hall et al., 1991; Kume-Kick et al., 1996; Kume-Kick and Rice, 1998). Further, baseline regional brain ascorbate levels were higher in female versus OVX or male rats. The gender difference was apparent after onset of puberty and largely lost in aged animals (Kume-Kick and Rice, 1998).
Interaction with bcl-2
Estrogen has been linked to an important set of neuroprotective proteins, the protooncogene bcl-2 family, which has been extensively implicated in cell survival in models of cerebral ischemia and apoptosis. In normal reproductive tissue, the steroid induces and supports bcl-2 expression and suppresses cyclical cell death (Sabourin et al., 1994; Tilly et al., 1995). For example, estrogen induces bcl-2 in normal breast tissue in a phasic manner over the estrous cycle (Sabourin et al., 1994) and supports constitutive bcl-2 and bcl-x-1 expression during follicle development in the ovary (Tilly et al., 1995). Bax levels also are decreased, improving the bcl-2/Bax ratio (Tilly et al., 1995). In both tissues, estrogen suppresses cell degeneration and loss. Similar effects can be observed in injured or pathologic tissue. Human breast cancer cell lines strongly express bcl-2 in response to estrogen and ER activation, resisting cell death and chemotherapeutic agents (Teixeira et al., 1995). Apoptosis of aortic endothelial cells after injury is attenuated by exogenous estrogen (Spyridopoulos et al., 1996). If bcl-2 is richly expressed in steroid-sensitive brain under basal or postischemic conditions, then tissue sparing could result. Several reports support this hypothesis. First, estrogen treatment increases bcl-2 expression in NT2 cells with neuronal phenotype, a mechanism that may account for estradiol's neuroprotective activity when these cells are exposed to glutamate or hydrogen peroxide (Singer et al., 1998). Second, bcl-2 expression varies with the estrus cycle in rat hypothalamic neurons, as measured by in situ immunohistochemical study. Further, bcl-2 protein was decreased by ovariectomy but responsive to hormone replacement in the adult female brain (Garcia-Segura et al., 1998). Third, in experimental stroke, bcl-2 mRNA is enhanced in penumbral areas to a greater extent in female or estradiol-treated rats than in estrogen-deficient animals (Alkayed et al., 1998). A more recent report demonstrates that not only was estradiol treatment associated with increased bcl-2 mRNA in injured cortex, but its expression was paralleled by increased levels of ER transcripts (Dubal et al., 1999). These data suggest that one mechanism by which estrogen acts to reduce stroke damage in animals is through receptor-mediated, enhanced bcl-2 transcription. Whether this mechanism is brain region- or cell type-specific remains to be demonstrated.
Signaling through growth factors
Estrogen is a growth- or neurite-promoting factor, in its own right, in the mammalian brain (Lustig et al., 1991; Toran-Allerand, 1996a, 1996b). The hormone enhances growth and arborization of neuronal processes, both axonal and dendritic, primarily during development. In the mature brain, axotomy or deafferentation-type injuries in steroid-sensitive regions of the brain also respond to estrogen treatment (Matsumoto and Arai, 1981), and the loss of the trophic effects of estrogen with ovariectomy (Gould et al., 1990) can be reestablished by estrogen treatment. The result is formation of new synapses, dendrite differentiation, and increased dendritic spine density. Although the mechanism is controversial, it is likely that these actions occur in combination with endogenous growth factors (Chalbos et al., 1994; Toran-Allerand, 1996a, 1996b). One theory is that estrogen acts indirectly through increasing gene transcription or activity of estrogen-responsive peptide growth factors, which in turn act on neighboring cells through autocrine or paracrine mechanisms. There is also considerable evidence to implicate the ER in signaling crosstalk between hormones and growth factors in brain under physiologic conditions (Toran-Allerand, 1996b; Katzenellenbogen, 1996). Many of the growth factors linked to estrogen signaling also have clear neuroprotective and regenerative properties in brain. These include nerve growth factor (Gibbs et al., 1994; 1998), brain-derived neurotrophic factor and other neurotrophins (Singh et al., 1995; Sohrabji et al., 1995; Toran-Allerand, 1996a, 1996b; Murphy et al., 1998), insulin-like growth factor-I (Fernandez-Galaz et al., 1997), and the fibroblast growth factor family (in particular, bFGF; Takahashi and Nakagawa, 1997). Therefore, estrogen may act in concert with these peptides, or through their signaling pathways, to preserve neuronal survival after injury or even to promote neurogenesis in the adult brain (Tanapat et al., 1999).
Reducing excitotoxic-ischemic damage
A large body of literature indicates that estrogen alters both glutamatergic and γ-aminobutyric acid (GABA)-ergic neuronal activity in many steroid-sensitive brain regions. Therefore, the steroid may blunt excitotoxic cascades that are important mechanisms of injury in stroke. Behaviorally and morphologically, female animals are more sensitive than males to the adverse effects of N-methyl-D-aspartate (NMDA) receptor antagonists such as MK801, suggesting greater susceptibility to NMDA receptor blockade in females and sex-related differences in the endogenous modulation of this glutamate receptor subtype (Honack and Loscher, 1993; Akinci and Johnston, 1993; Auer, 1996). Estradiol prevents neuronal loss resulting from kainic acid injection into dentate gyrus (Azcoitia et al., 1998). Further, estrogen directly blocks kainate (Regan and Guo, 1997) and glutamate (Behl et al., 1995; Goodman et al., 1996; Singer et al., 1996, 1999) injury in vitro, as assessed in hippocampal and cortical neuronal cultures. Protective effects were hormone-specific in these latter studies; lesser protection was present with progesterone, and other steroids had no effect. The mechanism of protection may be linked to reduction of NMDA-induced calcium influx, since estrogen is known to modulate intracellular calcium through other types of ionotropic channels (Weaver et al., 1997; Mermelstein et al., 1996). Alternatively, estrogen may stimulate mitogen-activated protein kinase signal-transductive pathways to protect cells in a similar way as growth factors (Singer et al., 1999).
Female reproductive steroids also may ameliorate ischemic injury through promotion of GABAa receptor-mediated mechanisms, as well as through suppression of excitatory amino acid toxicity. Increases in extracellular glutamate, GABA, and glycine levels have been well documented in all models of cerebral ischemia, supporting the concept that it is the imbalance between excitation and inhibition that ultimately determines neuronal injury and death (Globus et al., 1991; Lin et al., 1992). Globus et al. demonstrated that an excitotoxic index derived from glutamate, GABA, and glycine concentrations can be a primary predictor of injury after transient cerebral ischemia (Globus et al., 1991). The potency of estrogen and progesterone on GABAergic signaling varies selectively across brain region and is under intense investigation (for reviews see Smith, 1989; Paul and Purdy, 1992; McCarthy, 1995). Both reproductive steroids have been reported to alter GABA conduction (Majewska, 1992), induce synaptosomal and hippocampal GABA release (Fleischmann et al., 1990; Tauboll et al., 1993), upregulate GABAa receptor number (Weiland and Orchinick, 1995; Canonaco et al., 1989; Juptner et al., 1991; Maggi and Perez, 1986), increase GABAa agonist binding affinity (Schumacher et al., 1989; Jussofie et al., 1995), and alter glutamic acid decarboxylase mRNA (Weiland, 1992; McCarthy et al., 1995; Grattan et al., 1996; Davis et al., 1996). Estrogen administration to OVX females increases brain GABA levels (Luine et al., 1997) and increases agonist binding at the GABAa receptor within hours, particularly in cortex and striatum (Maggi and Perez, 1986) and less so in hippocampus (Schumacher et al., 1989). The functional significance of these events to neuroprotective mechanisms in brain remains to be shown.
CONCLUSION
This review summarizes gender-specific aspects of clinical and experimental stroke, the favorable results of estrogen treatment in animal models, and potential vascular and parenchymal mechanisms of neuroprotection. Premenopausal women may thus be protected in part from stroke, with reproductive hormone status playing a significant role in this gender-linked protection. Stroke and cerebrovascular disease history should be considered in the individual's decision to use HRT during the postmenopausal years. Animal studies clearly indicate that gender is one additional factor that influences experimental stroke outcome and that estrogen is an effective neuroprotectant in both male and female brain. This new concept suggests that further dissection of the interactions between reproductive steroids and cerebral ischemia will continue to yield novel insights into generalized cellular mechanisms of stroke and therapy. Numerous experiments are needed to determine fully the steroid's therapeutic window of activity, optimal dose-response relationships, and flow-dependent and flow-independent cellular and molecular mechanisms.